

Abstract
JX-594 is a targeted and granulocyte macrophage-colony stimulating factor (GM-CSF)-expressing oncolytic poxvirus designed to selectively replicate in and destroy cancer cells through viral oncolysis and tumor-specific immunity. In order to study the mechanisms-of-action (MOA) of JX-594 in humans, a mechanistic proof-of-concept clinical trial was performed at a low dose equivalent to ≤10% of the maximum-tolerated dose (MTD) in other clinical trials. Ten patients with previously treated stage IV melanoma were enrolled. Tumors were injected weekly for up to nine total treatments. Blood samples and tumor biopsies were analyzed for evidence of transgene activity, virus replication, and immune stimulation. The β-galactosidase (β-gal) transgene was expressed in all patients as evidenced by antibody induction. Six patients had significant induction of GM-CSF-responsive white blood cell (WBC) subsets such as neutrophils (25–300% increase). JX-594 replication and subsequent shedding into blood was detectable in five patients after cycles 1–9. Tumor biopsies demonstrated JX-594 replication, perivascular lymphocytic infiltration, and diffuse tumor necrosis. Mild flu-like symptoms were the most common adverse events. In sum, JX-594 replication, oncolysis, and expression of both transgenes were demonstrated; replication was still evident after multiple cycles. These findings have implications for further clinical development of JX-594 and other transgene-armed oncolytic viruses.
Introduction
Targeted therapies for cancer with novel mechanisms-of-action (MOA) are needed. One strategy is the use of replication-competent viruses which self-amplify within the tumor causing lysis of infected cancer cells1,2,3 and a number of agents have entered phase 1 and 2 clinical trials.4,5,6 Engineered oncolytic poxviruses can replicate selectively in cancer cells, resulting in virus progeny production, tumor cell necrosis, release and spread within tumor tissues.3 These virotherapies can also be engineered to express multiple therapeutic, marker, and noninvasive imaging transgenes. We hypothesized that the poxvirus pharmacophore would result in rapid replication and motile spread, activation by the epidermal growth factor receptor-ras pathway (pathway activated in the vast majority of solid cancers), and efficient systemic delivery to metastatic tumors, secondary to intratumoral (IT) administration or after direct intravenous (IV) infusion. Safety attributes include the fact that vaccinia was used in smallpox vaccination programs in tens of millions of humans globally, and specific antiviral agents are available (e.g., cidofovir7 and vaccinia immune globulin8).
JX-594 is a targeted and transgene-“armed” oncolytic poxvirus engineered from the Wyeth vaccine strain (Dryvax; Wyeth Laboratories, Marietta, PA).9,10,11 JX-594 has both a disruption of the thymidine kinase (TK) gene and expression of human CSF2 [granulocyte macrophage-colony stimulating factor (GM-CSF)] complementary DNA under the control of a synthetic (i.e., engineered) early-late promoter, and the lac-Z marker transgene-expressing β-galactosidase (β-gal) protein under control of the p7.5 late promoter.10 JX-594 is tumor-selective due to epidermal growth factor receptor-ras pathway and elevated cellular TK protein dependency12,13 and tumor-resistance to interferons.14 Cellular TK is driven to high levels in cancer by cell cycle abnormalities.15 GM-CSF is effective in augmenting the tumor-specific immunity induced by oncolytic vaccinia.16,17 JX-594 caused complete tumor responses and enhanced survival after IV and IT administration in immunocompetent rat and rabbit tumor models, and product MOA were demonstrated.10
In a pilot clinical study of JX-594, seven melanoma patients were first revaccinated with wild-type vaccinia in normal skin, and subsequently received escalating doses of JX-594 injected into superficial melanoma skin metastases.9 No maximum-tolerated dose (MTD) was reported [up to the relatively low dose of 8 × 107 plaque-forming units (pfu)] regressions of small superficial tumors were documented. A phase 1 trial in patients with liver tumors was performed (n = 14 patients); whereas data on biological activity was reported, tumor histologies (n = 7) and doses (108–3 × 109 pfu) were heterogenous.18 The MTD within the liver was 109 pfu. Biological effects following repeated dosing and biopsy data on injected tumors were not reported from this trial.
In order to confirm and expand upon the preliminary findings from these two previous phase 1 trials, we performed a low-dose MOA-driven clinical trial of JX-594 in patients with metastatic melanoma injected weekly for up to nine total doses. The objectives of this trial were to assess the multiple MOA of JX-594 after repeated weekly IT injections. The results reported here differ from previously published trials because this trial assessed biological activity as follows: (i) after six to nine cycles (including JX-594 replication and shedding into the blood), (ii) without immediately preceding prevaccination, (iii) in a homogenous patient population, (iv) at a fixed dose, (v) with serial blood sample analysis for induction of white blood cells (WBC), and (vi) with detailed biopsy evaluation for replication, inflammation, and necrosis induction over time. The dose of 108 pfu was selected because this was similar to the highest dose in the melanoma pilot study and the lowest dose in the liver tumor phase 1 trial; this dose was 10% of the MTD in the liver. Detailed data were obtained to assess JX-594 replication, transgene expression (GM-CSF and β-gal) and pharmacodynamics (immune cell stimulation). Of note, expression of these transgenes from JX-594 is under the control of two distinct promoters; for both promoters, high transgene expression is dependent on virus replication.19 We believe the results reported herein have significant implications for future clinical trials with JX-594 and other products from this novel therapeutic class.
Results
Patient demographics
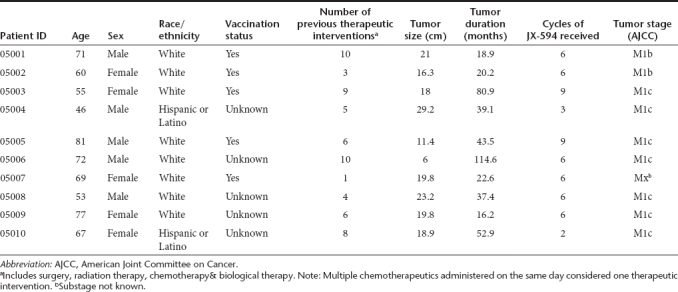
Ten patients were enrolled and treated with JX-594; 8 subjects (80%) completed the study. Two patients did not complete study. One patient requested to withdraw (after three treatments) and a second withdrew (after two treatments) due to investigator request. Patient baseline characteristics are listed in Table 1. Mean age was 65.1 years (range: 46–81 years). Gender was evenly distributed. All subjects had metastatic disease; mean duration of melanoma was 44.6 months. Patient prognosis at screening was evaluated using the American Joint Committee on Cancer staging system. Seventy percentage of patients belonged to the M1c category (visceral metastasis to sites other than skin, lymph nodes and lung with normal lactate dehydrogenase or distant metastasis with elevated lactate dehydrogenase).Patients were heavily pretreated, having received a mean of 6.2 prior therapeutic interventions (median 6, range 1–10). These could include prior surgery, radiation therapy, chemotherapy, or biological therapy. Prior biological therapies included interferon, interleukin-2, dendritic cell vaccines, and anti-CTLA-4 antibody treatment. Supplementary Table S1 provides a listing of prior biological agents by patient. Five patients were known to be previously vaccinated with vaccinia; five patients' vaccination status was unknown.
Table 1. Patient demographics.
Pharmacodynamics: induction of GM-CSF-responsive WBC subsets
Significant increases (>50% increase from baseline) in WBC subsets were detected in 60% of patients (Figure 1a). Changes in WBC were evaluated on days 5, 8, 12, 15, 22, 29, 36, 43, and 64. Since WBC generally peaked after the first injection, peak induction on days 5 or 8 was plotted in Figure 1a. Four subjects had an increase in neutrophils of >/=100% from baseline (Figure 1b) and an absolute neutrophil count increase of >/=2,000 cell/µl from baseline within 5–8 days of the first JX-594 treatment. Four subjects had significant increases in eosinophils (>100% increase from baseline). In a patient with detectable GM-CSF (9.4 pg/ml) in blood on day 5 (patient 05009), the absolute neutrophil count increased by 189% (day 5) and eosinophils increased by 1,300% (from 100 at baseline to 1,400 cells/µl on day 8) (Figure 1c).
Figure 1.

Induction of granulocyte macrophage-colony stimulating factor (GM-CSF)-responsive white blood cell (WBC) subsets by JX-594 treatment. (a) Absolute changes in WBC, neutrophil counts (ANC), eosinophils, and monocytes on day 5–8 post-JX-594. (b) Percentage changes from baseline in neutrophil counts (ANC), on day 5–8 post-JX-594. (c) Time course of WBC subset induction (absolute counts) during cycle 1 in patient 05009 (blood positive for detectable GM-CSF on day 5).
No significant change in hemoglobin/hematocrit was observed. No significant thrombocytopenia was detected. Three subjects had transient increases (>50% increase from baseline lasting </=14 days) in platelet counts at various time points during the study (data not shown).
Pharmacokinetics of JX-594 genomes in blood: two phases
Blood samples were obtained from each subject before the first treatment, at 15 minutes, 3 hours after infection and at multiple time points between days 5 and 64 (Table 2). During the initial clearance phase, low but detectable concentrations of JX-594 genomes were present in the blood immediately (15 minutes) after injection of superficial tumors in six subjects (60%), but rarely 3 hours after injection (Figure 2a). After clearance (15 minutes to 3 hours), delayed re-emergence of circulating JX-594 was detected in a subset of subjects, consistent with replication and shedding into the blood. Five subjects (50%) had detectable genomes from 5 to 7 days after the preceding injection (i.e., the 5–7 days following injections 1, 2, 3, 4, and/or 5) (Figure 2b). Two subjects received 3 additional treatments, and one had detectable JX-594 genomes in blood collected during this period as well on day 71 (7 days after 7th injection) and on day 85 (7 days after 9th injection) (Table 2). In some cases, subjects received JX-594 injections concurrently with detection of replication peaks in the blood (replicating JX-594 from a previous injection); no significant toxicity was noted in these patients.
Table 2. Patient treatment and initial plus delayed pharmacokinetics of JX-594.

Figure 2.

Pharmacokinetics of JX-594: acute clearance followed by delayed re-emergence associated with replication and shedding into the blood. (a) Acute genome concentrations in circulation at 15 minutes and 3 hours (patients with genomes below the limit-of-detection are not included). (b) Peak genome concentrations 5–7 days after treatments 1 through 5 (patients with no genomes detected during this time are not included). The delayed reappearance of JX-594 genomes provides evidence for replication and shedding into the blood.
Tumor biopsies: Assessment of JX-594 replication, inflammatory infiltration, and cell death
Tumor biopsies were optional based on patient consent. Tumor biopsies were obtained post-treatment in five patients; biopsies contained viable tumor tissue in three cases, and contained completely necrotic tumor tissue in two patients (not interpretable for immunohistochemistry). In addition, one of these patients had evaluable tumor tissue at baseline, day 5 and day 43. At baseline, no necrosis or inflammation was noted; rare scattered apoptotic tumor cells were noted (Figure 3a). In marked contrast, the day 5 biopsy of a noninjected tumor showed vaccinia replication, and day 5 and day 43 biopsies demonstrated progressive widespread necrosis from moderate (day 5) to intense (day 43) grade (Figure 3a,b). Similar necrosis was noted in tumor biopsies from two other patients on day 5 and day 12. Intense perivascular lymphocytic infiltration was noted on days 5–43 in two of three evaluable biopsies (Figure 4a–c).
Figure 3.

Biopsy evidence for JX-594 replication and necrosis induction. (a) JX-594 detection by immunohistochemistry (IHC) (red staining) in melanoma cells post-treatment (patient 05007): baseline (negative) and day 5 (positive). Examples of apoptotic and infected cells indicated with red and black arrows, respectively. (b) Hematoxylin and eosin (H&E) stain. Necrosis induction post-treatment (patient 05007): baseline (negative), day 5 (moderate), and day 43 (intense). Bar = 100 µm. Areas of necrosis circled in red.
Figure 4.

Biopsy evidence for JX-594-induced lymphocytic infiltration [hematoxylin and eosin (H&E) stain]. (a) Lymphocytic infiltration over time post-treatment (patient 05007): baseline (negative), day 5 (intense), day 43 (intense). Bar = 50 µm. Examples of perivascular lymphocytes indicated with white arrows.(b) High magnification of perivascular lymphocyte infiltration on day 43 (intense—patient 05007). Bar = 100 µm. Examples of perivascular lymphocytes indicated with white arrows. (c) High magnification of perivascular lymphocyte infiltration and day 12 (intense—patient 05004). Bar = 100 µm. Examples of perivascular lymphocytes indicated with black arrows.
Antibody development to JX-594 and to the β-gal marker transgene product
Neutralizing antibody (NAb) titers were assessed in serum samples taken at baseline, and at multiple time points between days 8 and 64. Vaccinia vaccination in childhood can result in residual antivaccinia antibodies in patients. At baseline, anti-JX-594 NAb titers were as follows: undetectable (n = 3), low (≤10; n = 4), or high (>10; n = 3). NAb titers increased as early as day 8 in 7 of 10 subjects. By day 15 all patients showed high titers, and titers had peaked by day 21 despite continued treatment for 6 weeks (Figure 5a). As a group, patients showed statistically significant induction (P value of 0.0141 using paired Student's t-test comparing baseline to D43 titers, n = 8 patients reaching D43).
Figure 5.

Antibody induction to JX-594 and to the β-gal marker transgene product. (a) Neutralizing antibody titers over time following treatment initiation; individual data points are circles and the line is the mean. (b) Anti-β-gal antibody titers over time; individual data points are circles and the line is the mean. All patients were tested on day 43 except two patients that withdrew early were tested on day 12 or day 22. Two patients had values above the upper limit of quantitation of 32,000—value is presented as 32,000.
Immunoglobulin G antibodies to the β-gal transgene product were detected by enzyme-linked immunosorbent assay. Antibodies to β-gal were negative at baseline in all patients, except one patient with a very low titer at baseline. Antibodies to β-gal were evident in all patients post-treatment (Figure 5b). One patient had not developed antibodies on day 43, but showed induction on day 64 (data not shown). Multiple additional treatments and/or time may increase titers—the two patients who withdrew from study early had lower titers on day 12 and 22 compared to patients completing study and being tested on day 43 (Figure 5b). As a group, patients showed statistically significant induction (P value of 0.0211 using paired Student's t-test comparing baseline toD43 titers, n = 8 patients reaching D43). Significant expression of β-gal is dependent on viral replication,19 thus development of antibodies to β-gal is dependent on JX-594 replication; antibodies therefore are markers of previous JX-594 replication in the patient.
Radiographic tumor assessment and survival duration
Radiographic tumor assessment was not the focus of this low-dose trial analysis. Anecdotal data was reported. Five subjects were evaluable for radiographic tumor assessments; protocol-defined radiographic assessments were not evaluable in five patients. Reasons included patient refusal, patient withdrawal from study and protocol violation. These patients clinically deteriorated before obtaining repeat tumor measurements, and they died soon thereafter. All five evaluable subjects had stable disease of injected tumors by RECIST criteria (n = 5). Stable disease (including a mixed response) was noted in injected tumors at week 6 in all patients. Noninjected tumors were present in all patients. Three patients had stable disease in noninjected tumors and two had progressive disease at week 6.
Durability of responses and progression-free survival were not assessable since most patients went off study within </=6 weeks. Two patients (05003 and 05005) with stable disease on the day 43 efficacy read received an additional three JX-594 treatments according to protocol. One patient (05003) had stable disease of injected and noninjected tumors on day 43 and on day 77 (after nine total JX-594 treatments). One noninjected tumor had a partial response on day 77. The second (05005) patient had stable disease in their injected tumors on days 43 and 77, whereas noninjected tumors progressed between days 43 and 77. Eight patients survived for at least 4 months, and four patients survived more than 7 months; one patient survived >20 months. Mean survival was 7.1 months (range 2.2–20.5).
Adverse events
All 10 subjects reported at least one treatment-emergent AE. JX-594 was generally well-tolerated. The most common treatment-related adverse events included nausea, chills, fever, fatigue, pain, myalgia, anemia, and headache. Most events were grade 1 or 2. One case each of hypoglycemia, fever, and anemia were reported as severe (grade 3) and deemed possibly or probably related to JX-594 treatment; all were transient and resolved without sequelae. Seven subjects reported a total of 55 treatment-related AEs (Table 3). No grade 4 or grade 5 (death) treatment-related events were reported. No patients died during the active study period (day 1–day 64). Two subjects had a serious adverse event (acute esophagitis, spinal cord compression due to tumor progression); both resolved (Note: The spinal cord compression resolved following a course of palliative radiation therapy) and both serious adverse events were assessed as not related to treatment.
Table 3. Treatment-emergent adverse events possibly related to JX-594 by grade.

No clinically significant treatment-emergent changes were detected in any serum chemistries (including alkaline phosphatase, albumin, aspartate aminotransferase, alanine aminotransferase, creatinine, potassium, sodium, total protein, calcium, bilirubin) or coagulation parameters (PT, PTT, or INR) while on study. Transient, treatment-emergent grade 1 hyperglycemia was noted in four subjects. Clinically significant cytopenias were not recorded (except a single transient case of anemia as described above).
Discussion
In this pharmacokinetics- and pharmacodynamics-focused clinical trial, we have shown that the multiple proposed JX-594 MOA were active in cancer patients, even at a low dose of ≤10% of the MTD in humans.18 JX-594 is a targeted, GM-CSF-expressing oncolytic poxvirus designed to selectively replicate in and destroy cancer cells through virus replication-dependent cytolysis and induction of antitumoral immunity. JX-594 replication was demonstrated reproducibly by several methods, including both serial analyses of blood and of tumor biopsies. In addition, the expression and/or pharmacodynamic activity of the two transgene products (GM-CSF, β-gal) were confirmed reproducibly. GM-CSF-responsive WBC subsets (neutrophils, eosinophils, and monocytes) were induced ~5–7 days after treatment as predicted. In addition, immune stimulation was also shown through intense perivascular infiltration of tumors by lymphocytes. Finally, associated tumor necrosis was evident on tumor biopsies. These data expand our understanding of this emerging product class in several important ways. First, the ability of JX-594 to replicate in the face of neutralizing antibodies, after multiple cycles, was demonstrated for the first time. Second, replication of JX-594 and shedding into the blood following the first treatment was studied concurrently with WBC stimulation and GM-CSF concentrations in blood. Third, these findings could be compared with the detailed evaluation of injected and noninjected tumor biopsies for replication, inflammation and necrosis induction over time. Fourth, the patient population studied here was of uniform tumor type and received a uniform dosage. Finally, no immediate prevaccination was carried out on this trial, in contrast to a previous study.9
Oncolytic viruses such as JX-594 must by definition replicate in tumors in order to cause direct oncolysis. In addition, for transgene-expressing (a.k.a. “armed”) viruses such as JX-594, transgene expression is also closely tied to virus replication since the transgene expression is under control of promoters whose activation is markedly higher and more sustained in late stages of virus replication. The evaluation of virus replication in cancer patients is therefore critical for the rational development of these agents. Unlike animal tumor models, in humans it is not feasible to directly assay for the precise amount of virus infectious units or genomes in all tumor masses over time. Innovative methods are required to assess this critical feature. Tumor biopsies can give a snapshot at a single point in time as long as histological markers of replication are available, although the sensitivity of this approach is likely low with a standard fine needle aspiration or core biopsy. For biopsies of tumors that were injected directly, replication markers must rely on detection of viral products that are not in the input dose and/or should be linked to cytopathic changes unique to viral infection. Furthermore, as demonstrated on this trial, valuable information regarding JX-594 replication can be gleaned from biopsies of noninjected tumors. Detection of JX-594 at noninjected tumor sites is evidence of infection at the primary site, spread of JX-594 to distant sites of disease and subsequent productive infection at the noninjected site. Another method for evaluating virus replication is to follow virus genome concentrations in blood serially over time. A biphasic replication-dependent pharmacokinetic pattern was demonstrated. Of note, this assay does not delineate between virus-incorporated or free DNA; in either case, however, the secondary peak should be indicative of viral replication. An initial peak concentration was typically detectable 15 minutes after dosing, and by 3 hours JX-594 was nearly always undetectable. Between days 5 and 8 of each cycle, however, as a result of replication there was a re-emergence of the agent into the blood. In order for JX-594 genomes to be detectable in blood at these late time points after injection, virus must replicate in cells and be released into the extracellular milieu, and subsequently gain access to the vasculature. Once in the blood, JX-594 is cleared with a half-life of ~30–60 minutes.18 Therefore, detection of JX-594 genomes in the blood 5–8 days after treatment appears to be a highly selective yet relatively insensitive marker for replication in humans. As predicted, in this trial JX-594 replication in tissues was demonstrated in biopsy material at the same time that a blood sample was negative for JX-594 (the converse did not occur). Another more durable and potentially more sensitive marker for JX-594 replication is the development of antibodies to β-gal protein; because β-gal protein expression only reaches significant levels during replication, antibody generation should also correlate with replication of JX-594 at some point in the past. Future products might express a secreted marker protein (e.g., CEA)20 and/ or a noninvasive imaging gene marker for real-time assessment and localization of replication21,22).
The immune response will very likely play a critical role in the clinical activity of JX-594 and other oncolytic virus products. Antiviral immunity, including NAb development, may inhibit virus replication and induce clearance. However, vaccinia viruses like JX-594 express numerous immune avoidance gene products,23 and the tumor milieu is often immunosuppressed24 and resistant to penetration by antibodies. Therefore, the outcome of the virus-immune system-tumor interaction is critical to evaluate and understand in humans. In this trial we demonstrated for the first time that JX-594 replication can occur repeatedly over time, despite high NAb titers, after as many as nine cycles. At the same time, immunostimulatory oncolytic viruses such as JX-594 also rely on the anticancer immune response for enhanced and complimentary efficacy. JX-594-associated WBC stimulation and lymphocytic infiltration of tumors were shown. GM-CSF transgene production was monitored by detecting GM-CSF in plasma of all patients. One patient exhibited detectable GM-CSF in the peripheral blood on this study. On a previous phase 1 trial studying IT injection of JX-594, in patients with evidence of high-level replication it was possible to occasionally detect GM-CSF in the systemic circulation at time points of peak replication (days 4–15),18 despite the short GM-CSF half-life in blood (<2 hours).25 Therefore, GM-CSF detection in blood is indicative of high-level GM-CSF expression; detection between 4 and 15 days after treatment (when all other inflammatory cytokines have returned to baseline values) may be a specific but insensitive marker of transgene expression. In addition, induction of immunoglobulin G antibodies to the β-gal transgene indicate Immunoglobulin class switching (requiring T-helper cells) has occurred. This proof-of-concept is critical for understanding this novel therapeutic approach. Nevertheless, future trials should explore the induction of functional anticancer immunity in treated patients (e.g., induction of cell-mediated and antibody-mediated immunity against tumor cells). Based on the data presented here, it does appear feasible for JX-594 to simultaneously avoid rapid immune clearance and to stimulate immunity in a fashion that could benefit the patient. The optimization of these two immune aspects will require further study. IV administration in the context of immunity, while feasible in animal tumor models, should be explored in humans.
Assessment of antitumoral activity was not the primary focus of this mechanistic proof-of-concept study upon localized injections of low-dose JX-594 into a subset of tumors accessible for injection. A follow-up study including a larger number of patients treated at the JX-594 MTD is needed to determine the potential efficacy of this approach in melanoma and other cancer types. Of note, published data demonstrated broad antitumoral activity and tumor necrosis in diverse cancer types; long-term survivors were reported.18 Responses were demonstrated in both injected and noninjected tumors in this published phase 1 trial.18 All evaluable patients exhibited stable disease at week 6 (RECIST criteria; n = 5), and a subset of patients exhibited prolonged disease stabilization. A RECIST partial response at later time points was observed in a noninjected tumor. Delayed responses have been noted with JX-594, as with other oncolytic viruses as well as other cancer therapeutics with immunostimulatoryproperties.6 Sze et al. reported a biphasic radiographic response pattern (initial tumor enlargement followed by regression of tumor size) which predicted for survival upon treatment with an oncolytic adenovirus Onyx-015.26 Furthermore, immune-related response criteria have been proposed to avoid premature discontinuation of immunotherapeutics, e.g., ipilimumab (anti-CTLA4 antibody). Authors report that patients treated with ipilimumab may exhibit acute “pseudoprogression” (progression upon use of standard RECIST criteria) followed by long-term responses.27 In summary, tumor response assessments are complex with immunostimulatory and proinflammatory agents. Given the results presented here, in combination with other published data18 future trials with JX-594 and other targeted oncolytic poxviruses are indicated. A phase 1 IV clinical trial was recently completed (data not shown). Two phase 2 studies are underway to evaluate the safety and efficacy of JX-594 in liver cancer by IT injection, and colorectal carcinoma trials are underway to explore IV safety and efficacy. Treatment of melanoma and other disseminated cancers in future trials of JX-594 should include IV administration in order to directly target multiple visceral metastases. Based on preclinical data, IV administration appears to be feasible for metastatic cancers. Previous results confirm that following IT injection, distant noninjected tumors can become infected after JX-594 travels through the blood.18 IV JX-594 can either be from the initial input dose (either IT or direct IV), or can result from replication in tumors with subsequent shedding into the blood. In summary, based on the data presented here and on previous findings, the multiple proposed JX-594 MOA are active in solid tumor patients, and further clinical development of JX-594 and other products from this class appears warranted.
Materials and Methods
Study approvals and registration. The final protocol was approved by the United States Food and Drug Administration, and the Institutional Review and Infection Control Committees at the University of California-Los Angeles, the Billings Clinic, and the Cancer Centers of the Carolinas (U.S. Oncology).
The protocol was registered at www.clinicaltrials.gov (NCT00429312).
Study design. This study was an open label, multicenter noncontrolled mechanistic proof-of-concept study. Study objectives included assessment of JX-594 transgene expression (both GM-CSF and β-gal) and pharmacodynamics (WBC induction), pharmacokinetics, replication, and shedding into blood, immune responses (neutralizing antibodies to JX-594) and antitumoral effects. Patients received IT injections (1–5 tumors) weekly at a dose of 108 pfu. Different tumors could be injected on different treatment days. This low dose was only 10% of the MTD on a previous liver tumor-based phase 1 trial with ITJX-594; this dose was selected in order to study virus replication-dependent pharmacodynamics and pharmacokinetics. If patients experienced a partial injected tumor response and/or tumor necrosis after completing six treatments, an additional three treatments administered weekly could be given.
Patient selection. Patients signed informed consent, according to Good Clinical Practice guidelines. Inclusion criteria included histologically confirmed, unresectable, metastatic, injectable stage IV melanoma that had progressed despite treatment with standard therapies, measurable disease by computed tomography or magnetic resonance imaging and/or physical examination, normal hematopoietic function (leukocyte count >3.5 × 109 cells/l, hemoglobin >10 g/dl, platelet count >125,000 cells/mm3 and organ function (including creatinine ≤2.0 mg/dl, aspartate aminotransferase/(alanine aminotransferase ≤2.0× of upper normal limit), life expectancy ≥16 weeks, and Karnofsky Performance Status ≥70. Exclusion criteria included increased risk for vaccination complications (e.g., immunosuppression, eczema requiring systemic therapy), significant cardiac disease, treatment with immunosuppressive or cancer treatment agents within 4 weeks, pregnancy or nursing.
Manufacturing and preparation of JX-594. JX-594 is a Wyeth strain vaccinia modified by insertion of the human CSF2 and LacZ genes into the TK gene region under control of the synthetic early-late promoter and p7.5 promoter, respectively. Clinical trial material was generated according to Good Manufacturing Practice guidelines in Vero cells and purified through sucrose gradient centrifugation. The genome-to-pfu ratio was ~70:1. JX-594 was formulated in phosphate-buffered saline with 10% glycerol, 138 mmol/l sodium chloride at pH 7.4. Final product quality control release tests included assays for sterility, endotoxin, and potency. Clinical trial material was also tested for GM-CSF protein concentration and was negative (lower limit-of-detection <14,000 pg/ml). JX-594 was diluted in 0.9% normal saline to a volume of ~2.5 ml, which was distributed into five syringes of 0.5 ml each.
Treatment procedure. All subjects were treated at either the Billings Clinic, the Cancer Center of the Carolinas, or at the University of California-Los Angeles by a physician trained in the injection protocol. JX-594 was administered by IT injection every week for a target total of six injections over 6 weeks (range 2–9 injections). JX-594 was injected by percutaneous injection (e.g., palpable skin nodules or lymph node metastases) or ultrasound-guided injection. Subjects received a dose of 108 pfu per treatment divided between ≤5 lesions. The Investigator determined the lesions to inject on each treatment day. Tumors were injected based on size; the largest lesions were injected at each treatment. An 18–22 gauge needle was used for injection. The entire syringe volume (0.5 ml) was injected along four equally spaced needle tracts per tumor radiating out from a central puncture site.
Patient monitoring. Patients were monitored after treatment in the hospital for at least 2 hours. Physical examination and interval medical history were performed at each visit. Safety monitoring included adverse event monitoring (NCI Common Toxicity Criteria, version 3) and standard laboratory toxicity grading for hematology, liver, and renal function, coagulation studies, serum chemistry, urinalysis.
Pharmacodynamics: WBC and differential counts induction. As a marker of biological effects of GM-CSF expression from JX-594, WBC count (including absolute neutrophil counts, eosinophils, and monocytes) evaluation was performed through routine laboratory testing on serial blood samples obtained every 7 days after cycles 1–6, in addition to days 4 after cycles 1 and 2.
NAb titers. NAb titers were determined by cytopathic effect inhibition assay. Heat-inactivated serum was serially diluted in media using half log dilutions. 50 µl samples were incubated with 1,000 pfu JX-594 for 2 hours, then inoculated onto A2780 cells. After 3 days, cell viability was determined using Cell Counting Kit-8 (Donjindo Laboratories, Kumamoto, Japan). NAb titer was defined as the reciprocal of the highest dilution of serum that resulted in ≥50% cell viability.
Antibody titers to the β-gal marker transgene. Human immunoglobulin G antibodies to β-gal were measured by enzyme-linked immunosorbent assay. Briefly, plates (NUNC MaxiSorp; Thermo Fisher Scientific, Waltham, MA) with wells containing β-gal (Sigma, St Louis, MO) orbicarbonate/carbonate buffer-only were incubated overnight at 4 °C and washed with phosphate-buffered saline-Tween before incubation with blocking buffer [phosphate-buffered saline with 1% bovine serum albumin (ELIS A grade; Sigma)]. Diluted plasma (1:50, 1:100, 1:200, in phosphate-buffered saline + 0.05% Tween + 1% bovine serum albumin) was added to β-gal-coated and control wells in duplicate and incubated at room temperature. Plates were washed and incubated with alkaline phosphatase labeled goat antihuman immunoglobulin G (AbCam, Cambridge, MA) diluted 1:2,000. After washing, colorimetric substrate pNPP (Sigma) was added, and NaOH was added to stop color development after 10 minutes. Absorbance was read at 405 nm, and absorbance of 630 nm was subtracted. Control well values were subtracted to account for nonspecific binding, and titers values were calculated by comparison to a standard curve of positive sera arbitrarily assigned a titer of 8,000.
Quantitative-PCR for JX-594 in blood samples. Quantitative-PCR was used to measure JX-594 genomes in whole blood over time due to its reproducibility and ability to detect product regardless of antibody and/or complement neutralization. JX-594 DNA was purified from samples using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Quantitative-PCR was run as described previously.18,28 The lower limits of JX-594 detection ranged from 667 to 3,333 copies/ml blood.
GM-CSF blood assay. GM-CSF was measured in plasma of all patients at predose day 1 and days 5, 8, 12, 15, 22, 29, 36, and 43.GM-CSF was detected in plasma by enzyme-linked immunosorbent assay kit (BioSource International; Carlsbad, CA) following the instructions of the vendor.
Tumor biopsy processing and analysis. Core biopsies or excisional biopsies were obtained in several patients (optional, if patient consent obtained separately) to evaluate viral replication, inflammatory, and immune cell infiltration, necrosis, apoptosis at injected tumor sites pre- and postinjection. Biopsies were collected from injected or noninjected tumors and were obtained at baseline (pretreatment), day 5–8, day 12 and/or day 43. Formalin-fixed, paraffin-embedded biopsies were stained with hematoxylin and eosin for routine histology (including assessment of viable tumor and/or normal tissues), including assessment of necrosis (graded based on proportion of necrotic area, pattern of necrosis and its location), apoptosis and immune/inflammatory cell infiltration (graded for severity and prominent cell type(s)). Viral replication in tumor and adjacent normal tissues was assessed by immunohistochemistry for viral proteins on the paraffin-embedded biopsies (graded based on expression intensity and percentage of positive tumor cells).
For immunohistochemistry, an antivaccinia polyclonal antibody (Quartett, Berlin, Germany) was used, followed by incubation with Levamizole block and permanent red chromagen secondary antibody kit (DAKO, Carpinteria, CA); negative controls were run without primary antibody and tumors from mice treated with JX-594 were included as positive controls. For immunohistochemistry detection of β-gal, an anti-β-gal polyclonal antibody (Abcam, Cambridge, MA) was used, though sections were not evaluable due to intense melanin deposition in tumor cells.
Radiographic assessment. All measurements were performed by physical examination and/or computed tomography or magnetic resonance imaging at baseline (within 14 days before first treatment with JX-594) and 1 week after the 6th dose was administered (day 43). At baseline, tumor lesions were categorized as target (injected) or nontarget. Maximum tumor diameters were obtained. Tumor assessments were performed according to RECIST criteria.29 For subjects who did not progress after discontinuing study drug, additional tumor assessments were to be performed approximately every 6 weeks until subject either met the criteria for progression or alternate therapy started. Nontarget (noninjected) tumor evaluation was the same as for target lesions.
Data quality assurance. Monitoring was conducted by the sponsor. The study was monitored at appropriate intervals to assure satisfactory enrollment rate, data recording, and protocol adherence. The frequency of monitoring varied depending on enrollment rate and the quality of the data collected.
Statistical issues. Study sample size was determined by safety and practical issues. The intent-to-treat population was assessed. This was a noncontrolled, nonrandomized clinical trial targeting a small number of subjects; no hypothesis testing was performed. Individual subject data listings as well as combined data are presented. The intent-to-treat population included all subjects who received at least one dose of JX-594. The evaluable population was the subset of the intent-to-treat population comprised of subjects who received at least one treatment and had undergone appropriate tumor measurement at protocol-specified time points.
Median NAb induction and antibody induction to β-gal was calculated using GraphPad Prism 5 software. Student's t-test was used to test the significance of induction of antibodies over time.
SUPPLEMENTARY MATERIAL Table S1. Listing of prior therapy with biologic anticancer agents, by patient.
Acknowledgments
We thank David Kerr, J. Andrea McCart, Peter Forsyth, and John Crowley for serving on the DSMB and reviewing the safety/toxicity data. This study was supported by the Bio-Scientific Research Grant funded by the Pusan National University (1. PNU, Bio-Scientific Research Grant) (PNU-2008-101-102) as well as by the Terry Fox Foundation. C.J.B., A.M., and D.H.K. are employees of JENNEREX, Inc., and J.C.B. is a consultant and shareholder in Jennerex, Inc. Jennerex, Inc. holds the license for JX-594.
Supplementary Material
Listing of prior therapy with biologic anticancer agents, by patient.
REFERENCES
- Parato KA, Senger D, Forsyth PA., and, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5:965–976. doi: 10.1038/nrc1750. [DOI] [PubMed] [Google Scholar]
- Cattaneo R, Miest T, Shashkova EV., and, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn DH., and, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- Liu TC, Galanis E., and, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4:101–117. doi: 10.1038/ncponc0736. [DOI] [PubMed] [Google Scholar]
- Msaouel P, Dispenzieri A., and, Galanis E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: an overview. Curr Opin Mol Ther. 2009;11:43–53. [PMC free article] [PubMed] [Google Scholar]
- Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G.et al. (2009Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma J Clin Oncol 275763–5771. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Cidofovir in the therapy and short-term prophylaxis of poxvirus infections. Trends Pharmacol Sci. 2002;23:456–458. doi: 10.1016/S0165-6147(02)02091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittek R. Vaccinia immune globulin: current policies, preparedness, and product safety and efficacy. Int J Infect Dis. 2006;10:193–201. doi: 10.1016/j.ijid.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA.et al. (1999Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma Cancer Gene Ther 6409–422. [DOI] [PubMed] [Google Scholar]
- Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE.et al. (2006Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF Mol Ther 14361–370. [DOI] [PubMed] [Google Scholar]
- Merrick AE, Ilett EJ., and, Melcher AA. JX-594, a targeted oncolytic poxvirus for the treatment of cancer. Curr Opin Investig Drugs. 2009;10:1372–1382. [PubMed] [Google Scholar]
- Gnant MF, Noll LA, Irvine KR, Puhlmann M, Terrill RE, Alexander HR., Jret al. (1999Tumor-specific gene delivery using recombinant vaccinia virus in a rabbit model of liver metastases J Natl Cancer Inst 911744–1750. [DOI] [PubMed] [Google Scholar]
- Buller RM, Smith GL, Cremer K, Notkins AL., and, Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317:813–815. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N.et al. (2000Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus Nat Med 6821–825. [DOI] [PubMed] [Google Scholar]
- Hengstschläger M, Pfeilstöcker M., and, Wawra E. Thymidine kinase expression. A marker for malignant cells. Adv Exp Med Biol. 1998;431:455–460. [PubMed] [Google Scholar]
- Thorne SH, Hwang TH, O'Gorman WE, Bartlett DL, Sei S, Kanji F.et al. (2007Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963 J Clin Invest 1173350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K.et al. (1993Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity Proc Natl Acad Sci USA 903539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC.et al. (2008Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial Lancet Oncol 9533–542. [DOI] [PubMed] [Google Scholar]
- Hammond JM, Oke PG., and, Coupar BE. A synthetic vaccinia virus promoter with enhanced early and late activity. J Virol Methods. 1997;66:135–138. doi: 10.1016/s0166-0934(97)00045-1. [DOI] [PubMed] [Google Scholar]
- Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC., and, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–4662. [PubMed] [Google Scholar]
- McCart JA, Mehta N, Scollard D, Reilly RM, Carrasquillo JA, Tang N.et al. (2004Oncolytic vaccinia virus expressing the human somatostatin receptor SSTR2: molecular imaging after systemic delivery using 111In-pentetreotide Mol Ther 10553–561. [DOI] [PubMed] [Google Scholar]
- Msaouel P, Iankov ID, Allen C, Aderca I, Federspiel MJ, Tindall DJ.et al. (2009Noninvasive imaging and radiovirotherapy of prostate cancer using an oncolytic measles virus expressing the sodium iodide symporter Mol Ther 172041–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GL, Symons JA, Khanna A, Vanderplasschen A., and, Alcamí A. Vaccinia virus immune evasion. Immunol Rev. 1997;159:137–154. doi: 10.1111/j.1600-065x.1997.tb01012.x. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- Hovgaard D, Mortensen BT, Schifter S., and, Nissen NI. Clinical pharmacokinetic studies of a human haemopoietic growth factor, GM-CSF. Eur J Clin Invest. 1992;22:45–49. doi: 10.1111/j.1365-2362.1992.tb01934.x. [DOI] [PubMed] [Google Scholar]
- Sze DY, Freeman SM, Slonim SM, Samuels SL, Andrews JC, Hicks M.et al. (2003Dr. Gary J. Becker Young Investigator Award: intraarterial adenovirus for metastatic gastrointestinal cancer: activity, radiographic response, and survival J Vasc Interv Radiol 14279–290. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C.et al. (2009Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria Clin Cancer Res 157412–7420. [DOI] [PubMed] [Google Scholar]
- Kulesh DA, Baker RO, Loveless BM, Norwood D, Zwiers SH, Mucker E.et al. (2004Smallpox and pan-orthopox virus detection by real-time 3'-minor groove binder TaqMan assays on the roche LightCycler and the Cepheid smart Cycler platforms J Clin Microbiol 42601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L.et al. (2000New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada J Natl Cancer Inst 92205–216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Listing of prior therapy with biologic anticancer agents, by patient.