

Abstract
Oncolytic adenoviruses are an emerging experimental approach for treatment of tumors refractory to available modalities. Although preclinical results have been promising, and clinical safety has been excellent, it is also apparent that tumors can become virus resistant. The resistance mechanisms acquired by advanced tumors against conventional therapies are increasingly well understood, which has allowed development of countermeasures. To study this in the context of oncolytic adenovirus, we developed two in vivo models of acquired resistance, where initially sensitive tumors eventually gain resistance and relapse. These models were used to investigate the phenomenon on RNA and protein levels using two types of analysis of microarray data, quantitative reverse transcriptase-polymerase chain reaction and immunohistochemistry. Interferon (IFN) signaling pathways were found upregulated and Myxovirus resistance protein A (MxA) expression was identified as a marker correlating with resistance, while transplantation experiments suggested a role for tumor stroma in maintaining resistance. Furthermore, pathway analysis suggested potential therapeutic targets in oncolytic adenovirus-resistant cells. Improved understanding of the antiviral phenotype causing tumor recurrence is of key importance in order to improve treatment of advanced tumors with oncolytic adenoviruses. Given the similarities between mechanisms of action, this finding might be relevant for other oncolytic viruses as well.
Introduction
Cancer remains one of the major causes of death and with aging of the population its relevance is predicted to increase. Several effective forms of treatment are available for most tumor types but these are nevertheless usually not curative and therefore new treatment approaches need to be developed. One promising experimental approach is the use of oncolytic viruses which selectively replicate in and kill tumor cells.1,2,3 In particular, oncolytic viruses are promising approach for treatment of tumors resistant to standard therapies. Initial trials have demonstrated the safety of the approach with a number of different viruses, including adenovirus, which is currently the most widely used agent in clinical oncolytic virotherapy.2,3,4 In addition to being a promising oncolytic platform, adenoviruses are commonly used as nonreplicating gene-delivery vectors due to efficient in vivo gene transfer.2 Since 1993, about 300 clinical trials based on adenoviral vectors have been performed.3 Also, the largest number of patients (>15,000 cancer patients) have been treated with adenovirus-based cancer gene therapy, and therefore a large body of safety data is available.5,6,7 Safety and gene transfer efficacy have also been validated in several randomized trials performed with oncolytic and non-oncolytic adenoviruses.5,6,7
The nature of advanced tumors entails a tremendous capacity for developing resistance to any therapeutic modality. One central aspect of adenoviruses as therapeutic agents is their immunogenicity.8 While this may be an important mediator of treatment efficacy,9,10,11 it might also play a role in developing resistance. Recently, with the molecular characterization of many tumors, transcriptional profiling has suggested the existence of two subgroups of cancer cells distinguishable by interferon (IFN) and inflammatory chemokine expression patterns.12,13,14
The interferon genes encode for a large family of multifunctional secreted small regulatory glycoproteins with important signaling roles during the innate immune response. There are two main types of interferons: type I or “viral” interferons include IFN-α, IFN-β, IFN-ω, and IFN-τ while type II interferons include IFN-γ.15 Transcriptional activation by interferon proteins binding to their specific cell surface receptors leads to interferon stimulated gene expression. Considerable progress has been made in describing the physiological role of interferon signaling components and subsequent antiviral activities.16,17,18 Gene targeting studies have distinguished four main effector pathways of the interferon-mediated antiviral response: the myxovirus (Mx) GTPase pathway, the ribonuclease L pathway, the protein kinase R pathway and the interferon stimulated gene 15 (ISG15) ubiquitin-like pathway. These pathways block viral transcription, degrade viral RNA, inhibit translation and modify protein function to control each replication step of most viruses.15
The myxovirus resistance protein A (MxA) is located at a critical intersection of these pathways. Therefore, it is rapidly induced to high levels when interferon signaling occurs and was one of the first antiviral mechanisms elucidated.19,20 Monsurrò et al. recently described two human pancreatic cancer cell lines characterized by different permissivity to viral vectors. The two phenotypes were characterized by differential expression of interferon stimulated genes and MxA protein expression.12 However, these in vitro studies did not evaluate the causality between actions of the virus and upregulation of interferon stimulated genes or MxA.
Therefore, in this study, we developed an orthotopic ovarian cancer mouse model by using a cell line that initially features mostly not only MxA negative but also some MxA positive cells. Despite the aggressive nature of the model, antitumor effects can be achieved with oncolytic adenoviruses such as Ad5/3-δ24.21 However, if the animals were followed long enough, all mice eventually relapsed after an extended period apparently disease-free. We found that interferon signaling was upregulated during escape from oncolytic adenoviral therapy.
Results
Therapeutic efficacy of Ad5/3-δ24 in ovarian tumors
Mice were xenografted intraperitoneally with SKOV3.ip1 ovarian carcinoma cells and treated with the oncolytic adenovirus Ad5/3-Δ24. From previous studies, we expected antitumor efficacy in this model with most mice rendered completely tumor-free for extended periods.21 However, in this experiment, mice were kept under observation for more than 100 days resulting in tumor relapse (Figure 1a). Thus, even though Ad5/3-Δ24 significantly increased median survival of the mice from 35 to 111 days (P < 0.01), eventually tumors relapsed in all mice.
Figure 1.

In vivo recurrence of ovarian cancer after treatment with oncolytic adenovirus. (a) Survival of oncolytic adenovirus treated mice: SKOV3.ip1 ovarian tumor bearing mice were treated intraperitoneally with oncolytic adenovirus Ad5/3-Δ24 [3 × 107 viral particle (VP)/mouse] or growth medium only (control). (b) Virus-resistant tumors relapse despite the presence of functional virus: Determination of viral content with 50% tissue culture infectious dose (TCID50) assay from susceptible tumors collected 4 days after infection and from tumors relapsing after more than a 100 days. Pfu, plaque forming unit.
Presence of virus in relapsing tumors
One possible hypothesis was that relapse occured because there was no more virus present in tumors. However, this was not the case as high titers (average 3.7 × 109 plaque forming units (pfu)/g of tumor tissue) were still found from relapsed tumors when analyzed with a standard 50% tissue culture infectious dose (TCID50) assay (Figure 1b). Thus, the relapsed tumors were growing despite the presence of high amounts of functional virus. Underlining the resistant nature of relapsing tumors, susceptible tumors analyzed on day 4 after infection showed significantly lower virus titers (average 6.0 × 107 pfu/g) (Figure 1b).
Microarray analysis and virus-resistant genetic signature
In order to study mechanisms of relapse, five untreated and five recurrent xenografted SKOV3.ip1 tumors were surgically resected and RNA was extracted for microarray analysis. We used human-specific microarray chips and thus identified transcripts are from SKOV3.ip1 cells, not mouse stromal cells. The two groups of tumors were characterized by 241 genes differentially expressed of which 70 were upregulated and 171 downregulated in the resistant phenotype. Significantly upregulated genes are shown in Table 1 and downregulated genes in Supplementary Table S1 (q < 0.05 for all the listed genes). Other differentially expressed genes are shown in Supplementary Data.
Table 1. Genes upregulated in virus-resistant tumors in comparison to untreated tumors.
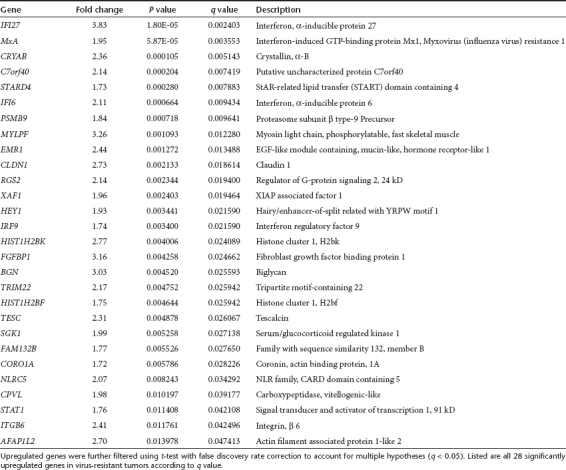
Data obtained with statistical tests were further analyzed using Ingenuity Pathways Analysis. We found that among the canonical pathways, the interferon pathway was one of the most differentially expressed (Figure 2a) and that these genes were frequently upregulated in the virotherapy resistant phenotype (Figure 2b). Graphical representation of interferon signaling pathways shows the molecular relationships between genes or gene products (Supplementary Figure S1a).
Figure 2.

Interferon pathway is significantly upregulated in virus-resistant tumors. Ingenuity Pathways Analysis of genes differentially expressed in virus-resistant versus untreated tumors. (a,b) The -log (P value) determines the probability that the association between the genes in the dataset and the canonical pathway is not explained by chance alone. Associations between the dataset and canonical pathways were measured in two ways: (a) The ratio of the number of genes from the dataset that map to the pathway divided by the total number of genes that map to the canonical pathway. (b) Percentage is showing the proportion of up- or downregulated genes over the total number of genes involved in the specific pathway (numbers at the end of the bars). Red is upregulated and green downregulated.
The crosstalk between the differentially expressed genes was further studied using pathway and protein–protein interactions stored in the Moksiskaan database22 which illustrated the connections between STAT1, IL-6, IRF9, and TNF (Supplementary Figure S1b). These genes are differentially expressed in response to virus infection according to Gene Ontology classification (Supplementary Table S2). Signaling Pathway Impact Analysis23 of the differentially expressed genes showed signs of inhibition of nucleotide-binding oligomerization domain-like receptor signaling,24 yet this regulation was nonsignificant with a false discovery rate corrected P value of 0.14 (Supplementary Data).
Quantitative reverse transcriptase-PCR confirmation of microarray results
To confirm the validity of the microarray analysis, changes in gene expression on mRNA level were analyzed in two separate experiments using both semiquantitative (Supplementary Materials and Methods and Figure S2) and fully quantitative reverse transcriptase-PCR with specific primers for some of the overexpressed genes (Figure 3a). The interferon pathway associated molecules MxA and interferon α-inducible protein 27 (IFI27) were confirmed to be upregulated in virus-resistant tumors (both P ≤ 0.001). In contrast, interferon α-inducible protein 16 (IFI16) was not significantly upregulated (P = 0.052), which was in accord with the microarray results (q = 0.055; Supplementary Data). Human β-actin was used as an internal control.
Figure 3.

Expression of Myxovirus resistance protein A (MxA) and interferon stimulated genes in tumors recurring after oncolytic adenoviral therapy. (a) RNA extracted from untreated control and Ad5/3-Δ24 treated tumors was reverse transcripted to cDNA and quantitative reverse transcriptase-PCR was performed to determine relative MxA, IFI27 and IFI16 mRNA levels (fold change). Data represent the mean ± SEM (n = 5). (b) Immunohistochemical MxA protein staining in untreated SKOV3.ip1 ovarian carcinoma cell line shows a small subpopulation of MxA positive cells. (c) Immunohistochemistry for MxA in treated tumors versus control tumors: higher MxA positive subpopulation was found in virus-resistant treated tumors. (d) MxA expression in SKOV3.ip1 cells after infection with adenovirus; To simulate the effect of stromal cells, SKOV3.ip1 cells were treated with recombinant universal type I interferon-α (IFN-α). The IFN-α treatment resulted in further upregulation of MxA expression 1 hour after infection (right panel).
MxA expression and distribution in naive SKOV3.ip1 cells
MxA is a key downstream protein in the interferon signaling cascade and reflects the virus-resistant phenotype as characterized by resistance to lysis by wild-type adenovirus.12 Monsurrò et al. recently published that up to 80% of cells in virus-resistant human pancreatic cancer cell lines are positive for MxA. In contrast, MxA expression was found only in 5% of the uninfected SKOV3.ip1 cells by immunocytochemistry (Figure 3b).
MxA upregulation in vivo
Based on the in vitro data, we hypothesized that killing of MxA negative (virus sensitive) cells would result in selection of MxA positive cells, which would then grow resulting in tumor relapse (Figure 4). This was corroborated by higher MxA staining in relapsed virus-resistant tumors compared to naive tumors (Figure 3c). As expected, staining was cytoplasmic. These experiments also corroborated the microarray data on a protein level.
Figure 4.

Virus-resistant population hypothesis. We hypothesized that already in the original cell line there is a subpopulation of cells resistant to adenovirus, and that these cells are selectively enriched under the pressure of viral oncolysis. Ultimately, emergence of this subpopulation causes recurrence of the tumor. Ad5, adenovirus serotype 5.
Induction of MxA in infected and interferon-α stimulated SKOV3.ip1 cells
To confirm that Ad5/3-δ24 infection results in MxA upregulation, SKOV3.ip1 cells were infected and then assessed for MxA (Figure 3d). Cytoplasmic MxA upregulation was seen already at 1 hour. To simulate the effect of stromal cells, IFN-α (recombinant universal A/D type I) was added and this further enhanced MxA expression (Figure 3d). MxA costained with adenovirus hexon suggested a link between infection, replication, and MxA expression (Supplementary Figure S3).
Possible new therapeutic targets for the reversion of the antiviral phenotype in tumors
The RNA expression profile of resistant cells was utilized to identify possible drug targets. We hypothesized that we could identify drugs that could be used to inhibit molecules upregulated during emergence of the virus-resistant phenotype.25 Such drugs might be useful in retaining sensitivity of tumors to virus. Alternatively, such targets might be useful druggable targets in tumors recurring after oncolytic virus treatment.
The Moksiskaan method maps drug data available in the Kyoto Encyclopedia of Genes and Genomes database26 to genes and their regulation information (Supplementary Materials and Methods). We did not find inhibitors for the upregulated genes, but instead we identified nuclear receptor subfamily 3 group C member 2 (NR3C2) as a gene that is downregulated in virus-resistant cells (Supplementary Figure S1c; Supplementary Table S1), and which could therefore be an upstream molecule responsible for the resistant phenotype. Importantly, Kyoto Encyclopedia of Genes and Genomes identified three agonists (desoxycortone, desoxycorticosterone pivalate, and desoxycorticosterone acetate) for the NR3C2 gene (Supplementary Figure S1c).27
In addition to this drug target, we identified several upregulated genes that code for membrane proteins (Table 2, Supplementary Data), which could be useful for identification of new drugs (e.g., monoclonal antibodies) for combination therapy with oncolytic adenoviruses.
Table 2. Genes upregulated in the plasma membrane of virus-resistant tumors in comparison to untreated tumors.

The adenovirus-resistant phenotype persists after transplantation
Severe combined immunodeficiency (SCID) mice (n = 10) were xenografted intraperitoneally with 5 × 106 luciferin expressing SKOV3-Luc cells and treated with phosphate-buffered saline (PBS) or Ad5/3-δ24 [total 3 × 109 viral particle (VP)] (Figure 5a). Tumor growth was monitored by in vivo bioluminescence imaging. After initially responding to the treatment, tumors started growing again suggesting acquired resistance against oncolytic adenovirus, as seen with the SKOV3.ip1 model (Figure 1). On day 27, both relapsed and untreated tumors were surgically removed and transplanted into new mice, which were then treated with either virus or PBS (Figure 5b). Adenovirus-resistant tumors could not be inhibited, while PBS treated tumors responded to treatment (P ≤ 0.001) (Figure 5b). TCID50 assay again demonstrated presence of functional virus in the resistant tumors (average 1.0 × 108 pfu/g).
Figure 5.

The adenovirus-resistant phenotype is sustained following transplantation into new mice. (a) Severe combined immunodeficiency (SCID) mice (n = 10) were xenografted intraperitoneally (i.p.) with 5 × 106 luciferase expressing SKOV3-Luc cells and were treated i.p. with total of 3 × 109 viral particle (VP) of Ad5/3-d24 or PBS (black arrows). Tumor growth was monitored by in vivo bioluminescence imaging. (a) On day 27 relapsed and untreated i.p. tumors were surgically removed and (b) reimplanted, with tumor stroma, into a second round of mice (n = 5–7). Following identical treatment (total 3 × 109 VP of Ad5/3-d24 or PBS; black arrows), virus-resistant tumors showed no inhibition of tumor growth, while susceptible tumors responded to treatment (P ≤ 0.001) (b). Each data point represents the mean ± SEM.
Tumor stroma was included in the transplants mentioned above. To evaluate the significance of the tumor microenvironment, tumor cells without stroma were obtained from ascites of both relapsing and untreated mice. Cells from the latter mice grew ex vivo on cell culture plates, while cells from the former did not grow (data not shown), suggesting that the tumor stroma plays a role in resistance to virus, perhaps again through interferons (Figure 3d, Supplementary Figure S3).
Discussion
Although much work remains in the evaluation of mechanisms and efficacy of adenoviral oncolytic therapy in humans, preclinical and clinical data have already suggested that initially sensitive tumors can eventually gain resistance to virus.11,28,29,30,31,32 This is not really surprising given the tremendous potential for selection and resistance exhibited by advanced tumors. Regarding resistance to chemotherapy, radiation, hormonal, and molecular therapies, many resistance mechanisms have been identified, which has then allowed development of new approaches for overcoming this resistance. For example, when the upregulation of the human epidermal growth factor receptor 2 and cyclooxygenase-2 were discovered important for multidrug resistance, their inhibitors could be developed which have subsequently been used with promising results.33,34 Key to these discoveries was availability of animal models that accurately capture the emergence of treatment resistance.
In this study, we developed a unique model where ovarian tumors recur after apparently curative oncolytic therapy. This allowed us to obtain information on possible escape mechanisms and to identify new therapeutic targets for overcoming resistance. In the initial microarray results comparing sensitive to resistant tumors, it was interesting to discover that the interferon pathways were highly upregulated in recurrent tumors. It seems that type I interferons play a critical role in innate immune response against adenoviral vectors.16 The fact that the functional virus is still present but ineffective in the recurring tumors is a further evidence that interferon signaling upregulated tumors, characterized by MxA expression, feature strong resistance to the lytic activity of the virus. In fact, up to 100-fold more virus was found in resistant tumors in comparison to naive tumors. Interestingly, an MxA related protein Mx235 was identified in a study where interferon signaling was implicated in resistance to vesicular stomatitis virus, another promising oncolytic virus.25 Furthermore, in that study, Mx2 could be downregulated by an interferon signaling inhibiting molecule, virus-sensitizers 1.25
To fully utilize the findings, we performed Moksiskaan alternative pathway analysis with cross-referencing to drug databases with the goal of identifying therapeutic targets to be used in a combination therapy with oncolytic adenovirus. In this regard, we identified NR3C2, which is downregulated in virus-resistant tumors and could be one of the upstream events possibly involved in upregulation of interferon signaling. This gene encodes for an aldosterone receptor with roles in the regulation of ion exchange as well as AP1 and NFkB-mediated fibrosis of cardiac tissues after injury.36 The therapeutic interest of this molecule resides in the fact that this protein can be upregulated by using common drugs including desoxycortone, desoxycorticosterone pivalate, and desoxycorticosterone acetate.27
Recent evidence suggests that the epithelial phenotype of ovarian cancer may represent a barrier to infection by oncolytic adenoviruses through impaired access to viral receptors.29 The resistant phenotype represented polarized expression of viral receptors and blockage of virion access by upregulated tight and adherens junctions.29 Interestingly, we identified several upregulated genes that code for membrane proteins. These could be useful for identification of new drugs for combination therapy with oncolytic adenovirus in order to achieve better accessibility to viral receptors. Particularly interesting could be the cluster of claudins (CLDN1, 10 and 16) that are probably upregulated in response to the viral stimulus since they were not overexpressed in uninfected pancreatic cancer xenografts positive for MxA.12
We were also able to confirm that in tumors with the antiviral phenotype there is overexpression of toll-like receptor 2 (TLR2) which was also observed in the pancreatic cancer xenograft model.12 TLR2 upregulation would fit well with enrichment of cells hyperactive in pathogen recognition and antiviral defenses. In fact, one possible mechanism for the induction of the antiviral interferon signature is chronic stimulus of TLR2.37 Emergence of the MxA positive phenotype and chronic stimulation of TLR2 by the virus may represent two facets of the same biological phenomenon and mechanistic links are likely. It will be interesting to evaluate whether TLR2 stimulation can lead to an increase in MxA positive cells.
To further evaluate emergence of resistance, we developed another in vivo model, this time stably expressing firefly luciferase so that relapse could be detected without killing of the animals. Upon transplantation of tumors into new mice, resistance to adenovirus was retained. This suggested a tumor-specific effect as opposed to a systemic effect mediated e.g., by the immune system, keeping in mind that these were SCID mice.
The tumor microenvironment has been suggested as an important resistance mediator against viruses. Stromal cells have been shown to contribute to immunity by secreting antiviral cytokines, restricting production and spread of viral progeny, and modulating the recruitment and maturation of invading immune cells.38 Our results support the importance of the stroma in the antiviral phenotype, since without it adenovirus-resistant cells did not grow ex vivo, while nonresistant cells did. One possible contribution of the stroma could be production of interferons which then triggers interferon pathways in tumor cells resulting in the antiviral phenotype. The stroma could be particularly important in the context of deficient interferon production by the tumor cells themselves, which is known to be a classic tumor associated defect. Therefore, the tumor cells per se were perhaps not completely resistant to the virus in them, when stromal factors (e.g., interferon) were removed.
Further approaches for studying the relationship between interferon signaling and resistance to adenovirus could include downregulation of interferon pathways e.g., through small hairpin RNA-mediated knockdown of STAT1, to test whether this prevents tumor cells from relapsing after adenovirus treatment. However, complete abrogation of a target gene is difficult to achieve and since STAT1-negative cells would be rapidly killed by the virus, unsuccessfully knocked-down (STAT1-positive) cells could over-grow ultimately causing relapse. Also, given the complexity of interferon signaling, there are other mediators, such as the ubiquitously expressed interferon regulatory factor 3, which can cause STAT1-independent upregulation of interferon stimulated genes.39
In summary, we: (i) established two in vivo models for studying mechanisms of resistance to oncolytic adenoviral therapies; (ii) identified a virus-resistant molecular phenotype of ovarian cancer, characterized by upregulation of interferon-related gene expression, easily identified by MxA protein expression; (iii) provided data suggesting that the antiviral phenotype can lead to failure of oncolytic therapy in vivo; (iv) proposed an escape mechanism based on progressive in vivo enrichment of antiviral phenotype containing cells under adenoviral pressure; (v) suggested possible molecular targets to overcome or prevent virus resistance; (vi) obtained preliminary evidence suggesting that the tumor stroma contributes to the antiviral phenotype.
We surmise that the cells with the potential for resistance are present already beforehand. Virus treatment then applies selective pressure leading to enrichment of those cells. This is in accord with proposed resistance mechanism against many chemotherapeutics, as e.g., multidrug resistant cells have been proposed being present even in early tumors, but are enriched under selective pressure mediated by treatment.40,41 Alternatively, adenovirus treatment might also induce resistance in initially susceptible cells which then cause relapse. This would be supported by observation that adenovirus infection itself can rapidly upregulate MxA expression in naive SKOV3.ip1 cells. These two possibilities are not mutually exclusive. If the MxA positive subpopulation could be depleted prior to transplantation of resistant cells, this might help differentiate selection versus induced resistance.
Given that the cellular defenses against many different oncolytic viruses are similar, it will be interesting to study whether these findings apply to other viruses. This possibility seems to be supported by a recent report where vesicular stomatitis virus replication could be enhanced with a molecule with anti-interferon activity.25 Oncolytic viruses are rapidly entering the clinical arena and therefore it is relevant to understand resistance against them as a first step for counteracting it and for identifying rational combination treatments. Eventually these developments can contribute to the utility of oncolytic viruses in the treatment of cancer in a process toward personalized combinatory therapies.
Materials and Methods
Cells and viruses. Human ovarian adenocarcinoma cell line SKOV3.ip1 was provided by Dr Price (M. D. Anderson Cancer Center, Houston, TX). Firefly luciferase expressing ovarian adenocarcinoma cell line SKOV3-Luc was kindly provided by Dr Negrin (Stanford Medical School, Stanford, CA). Human lung adenocarcinoma cell line A549 was purchased from American Type Culture Collection (Manassas, VA) and human E1-transformed embryonal kidney cell line 293 was obtained from Microbix (Toronto, Ontario, Canada). Cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and maintained in a humidified atmosphere at 37 °C and 5% CO2. Oncolytic adenovirus Ad5/3-δ2421 was amplified on A549 cells and purified on double cesium chloride gradients. The VP concentration was measured spectrophotometrically and the amount of infectious particles was determined by a standard 50% TCID50 assay on 293 cells. The ratio of VP/infectious units was 6.
Animal experiments. Three- to four-week-old female C.B-17 SCID mice were purchased from Taconic (Ejby, Denmark) and quarantined for 2 weeks. Survival experiment: Ten million SKOV3.ip1 ovarian cancer cells were injected intraperitoneally and 10 days later mice received intraperitoneally either 500 µl of modified Eagle's medium (n = 5) or 3 × 107 VP of Ad5/3-Δ24 (n = 5) in equal volume. Tumor growth follow-up experiment: 5 × 106 SKOV3-Luc cells were injected intraperitoneally in 300 µl of Dulbecco's modified Eagle's medium, and on days 3, 7, and 10 mice were treated intraperitoneally with either 300 µl of PBS (n = 10) or 1 × 109 VP of Ad5/3-Δ24 (n = 10) in equal volume. Intraperitoneal tumor growth was monitored noninvasively by bioluminescence imaging as described:42 Briefly, 150 mg/kg D-luciferin (Promega, Madison, WI) was injected intraperitoneally and imaged by IVIS 100 (Xenogen, Alameda, CA). On day 27 all mice were killed, tumors were surgically removed, and 2 × 0.3 cm3 of tumor material—including stroma—was freshly transplanted in laparotomy into new SCID mice (n = 7). To assess virus in susceptible tumors, 5 × 106 SKOV3-Luc cells were injected intraperitoneally into SCID mice (n = 5). Then the mice were treated intraperitoneally with PBS or virus (total 3 × 109 VP of Ad5/3-Δ24) on days 5, 8, and 11 postimplantation. The health of the mice was followed daily and mice were killed according to humane end-point guidelines and tumors were collected. All animal experiments were approved by the Experimental Animal Committee of the University of Helsinki and the Provincial Government of Southern Finland.
In vivo statistics. Survival data was plotted into a Kaplan–Meier curve and groups were compared pair-wise with log-rank test (SPSS 11.5; SPSS, Chicago, IL). Statistical analyses for in vivo tumor growth follow-up were performed using two-tailed Student's t-test and the nonparametric Mann–Whitney U-test (SPSS 15.0; SPSS). For all analyses P value of <0.05 was deemed statistically significant.
TCID50 for tumor tissue. To determine the number of functional adenoviral particles in tumor tissue, control and virus-treated ovarian cancer tumors were mechanically homogenized followed by three freeze/thaw cycles. Cell remnants were centrifuged and collected supernatants were used for TCID50 assay. Data are expressed as plaque forming unit (pfu)/g of tumor tissue.
RNA extraction and microarray analysis. Five untreated and five recurrent xenografted tumors were surgically resected and the RNA was extracted for microarray analysis. The analysis of the gene expression data obtained with Affymetrix GeneChip HG-U133A (Affymetrix, Santa Clara, CA) was conducted using Bioconductor in R.43 We used Hs133P_Hs_ENSG annotation library (version 9) to bind the probes in the array to transcripts. Normalization was done using robust multiarray average normalization.44 The raw data were then analyzed with statistical tests in order to identify differentially expressed genes. A gene was considered differentially expressed if the ratio of medians of normalized intensities of cases and controls was either less than 1/1.7 or greater than 1.7 and the SD between all samples was at least 0.4.
Canonical pathway analysis. Data were further analyzed using the Ingenuity Pathways Analysis (Ingenuity Systems, Mountain View, CA; http://www.ingenuity.com/): Canonical pathway analysis identified the most significant pathways of the data set according to the Ingenuity Pathways Analysis library. Genes from the dataset that met the selected cutoff and were associated with a canonical pathway in the Ingenuity Pathways Knowledge Base were considered for the analysis. The significance of the association between the data set and the canonical pathway was measured in two ways: (i) A ratio of the number of genes from the data set that map to the pathway divided by the total number of genes that map to the canonical pathway. (ii) Fisher's exact test was used to calculate a P value determining the probability that the association between the genes in the dataset and the canonical pathway is not explained by chance alone. The threshold P value was set to 0.05 based on statistical hypothesis testing.
Quantitative reverse transcriptase-PCR. Total cellular RNA of SKOV3.ip1 xenografts was reverse-transcribed into cDNA using Qiagen QuantiTect Reverse Transcription Kit (205311; Hilden, Germany), which included genomic DNA wipeout, and concentrations of the cDNAs were balanced to 100 ng/µl using NanoDrop 2000 Spectrophotometer (Thermo Scientific, Waltham, WA). cDNA samples were amplified using LightCycler480 SYBR Green I Master mix (Roche, Mannheim, Germany) and specific primers to analyze mRNA levels for human MxA (5′-ACCTACAGCTGGCTCCTGAA-3′ and 5′-CGGCTAACGGATAAGCAGAG-3′), IFI27 (5′-ACCTCATCAGCAGTGACCAGT-3′ and 5′-ACATCATCTTGGCTGCTATGG-3′), IFI16 (5-ACTGAGTACAACAAAGCCATTTGA-3 and 5′-TTGTGACATTGTCCTGTCCCCAC-3′). Human β-actin primers (5′-TCACCCACACTGTGCCCATCT-3′ and 5′-GTGAGGATCTTCATGAGGTAGTCAGTC-3′) were used for normalization to human genomic mRNA. The relative amounts of cytokine mRNA were calculated with the δδcomparative threshold method. The expression levels of each gene were expressed as fold increase in five treated tumors compared to five nontreated tumors, and analyzed using two-tailed Student's t-test (SPSS 15.0; SPSS).
Immunohistochemistry analysis. Normal SKOV3.ip1 cell line, frozen tissues of five SKOV3.ip1 untreated xenografts and five SKOV3.ip1 adenovirus-resistant xenografts were stained with MxA antibody (sc-50509; Santa Cruz Biotechnology, Santa Cruz, CA). Sections were boiled for 30 minutes at 98 °C in 10 mmol/l citrate buffer pH 6, treated with 3% hydrogen peroxide 10 minutes and then with Protein Blocking Agent (Novocastra Laboratories, Newcastle, UK) for 10 minutes. MxA antibody (diluted 1:1000 in PBS) was applied for 60 minutes at room temperature. Sections were washed and treated with NovoLink Polymer Detection System according to manufacturer's instructions (Novocastra Laboratories) for immunohistochemistry. Assays were performed in triplicates.
Immunofluorescence and microscopy. Cells were plated on coverslips and 16 hours later infected on ice for 30 minutes with adenovirus (100 VP/cell). The cells were washed with PBS and medium replaced with growth medium containing 10% fetal calf serum. For exogenous interferon induction cells were pretreated with IFN-αA/D recombinant universal type I interferon 100 IU/ml (Sigma-Aldrich, St Louis, MO) 16 hours prior to infection and during infection. After 0 minute, 30 minutes, 1 hour or 2 hours chase cells were washed with PBS and fixed with 4% paraformaldehyde 10 minutes at room temperature and stored at +4 °C in PBS. Cells were stained with rabbit polyclonal anti-MxA (1:100, H-285; Santa Cruz Biotechnology) and goat polyclonal anti-adenovirus hexon Virostat #1401 (1:50, Portland, ME) primary antibodies, and then Alexa405 and Alexa594 conjugated secondary antibodies (Molecular Probes/Invitrogen, Carlsbad, CA), respectively. Cells were mounted with Vectashield (Vectorlabs, Peterborough, UK). Cells were visualized using Zeiss LSM 5 Duo laser scanning confocal microscope (Jena, Germany). Images were processed with Adobe Photoshop CS3 and Illustrator CS3 software (Adobe Systems, San Jose, CA).
SUPPLEMENTARY MATERIAL Figure S1. Relationships between differentially expressed interferon pathway genes and a possible drug target for the reversion of the resistant phenotype. Graphical representation of interferon signaling pathways by Ingenuity Pathways Analysis (a): Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge. Upregulated genes of the dataset are highlighted. Moksiskaan candidate pathway of the genes differentially expressed between untreated and virus resistant tumors (b). STAT1 is linked to IL6, IRF9, CXCR4, and TNF and possibly responsible for the downstream effect of chemokine ligands. Green and blue borders refer to up- and downregulated genes, respectively. Known regulations between differentially expressed genes are shown with bold borders whereas the predictions (CXCL9-11) are kept thin and the nodes are grey. Drugs that can influence the downregulated gene NR3C2 as identified by Moksiskaan analysis (c). Figure S2. Expression of MxA and interferon stimulated genes in recurring versus control tumors. RNA extracted from untreated control and Ad5/3-?24 treated tumors was reverse transcripted to cDNA and analyzed with semi-quantitative RT-PCR using specific primers for human Myxovirus resistance protein A (MxA), interferon alpha-inducible protein 27 (IFI27) and interferon alpha-inducible protein 16 (IFI16). Human β-actin primers were used as controls. Figure S3. Expression of MxA in infected SKOV3.ip1 cells after interferon-α treatment. MxA expression and adenovirus hexon staining in SKOV3.ip1 cells after infection with adenovirus (a). To simulate the effect of stromal cells the SKOV3.ip1 cells were treated with recombinant universal type I interferon-α prior to infection and during infection (b). The interferon-α treatment resulted in further upregulation of MxA expression 1h after infection. IFN, interferon; Ad, adenovirus. Table S1. Genes downregulated in virus-resistant tumors in comparison to untreated tumors. Downregulated genes were further filtered using t-test with false discovery rate correction to account for multiple hypotheses (q<0.05). Listed are all 113 significantly downregulated genes in virus resistant tumors. Table S2. Genes differentially expressed in response to virus infection in recurrent versus untreated tumors. Enriched Gene Ontology (GO) terms: response to virus. GO enrichment analysis was done using Fisher's exact test, which compares the observed frequency of each present GO term to the frequency in a reference gene set. A GO term is present if some input gene is annotated with the GO term or its descendants. Materials and Methods. Data.
Acknowledgments
We thank Eerika Karli, Aila Karioja-Kallio and Kikka Holm for expert assistance (Cancer Gene Therapy Group, University of Helsinki, Helsinki, Finland). We thank Giulio Innamorati (Department of Pathology, University of Verona, Verona, Italy) for critical discussions and continuous support, Luciano Castiello for the Ingenuity Pathways analysis assistance and Stefano Eleuteri for technical support in the immunohistochemical experiments (University of Verona). We would like to thank Biomedicum Genomics facility for carrying out the microarray hybridizations, and the Biomedicum SysBio core unit for preprocessing and fold-change calculations of the microarray data (University of Helsinki). We thank the Molecular Imaging Unit (MIU, Biomedicum Helsinki, Finland) for providing the facilities and software for in vitro imaging. This work was supported by: European Research Council, American Society of Clinical Oncology (ASCO) Foundation, Helsinki University Central Hospital Research Funds (EVO), Helsinki Biomedical Graduate School (HBGS), Sigrid Juselius Foundation, Academy of Finland, Biocentrum Helsinki, Biocenter Finland, University of Helsinki; Associazione Italiana Ricerca Cancro (AIRC), Milan, Italy; Ministero della Salute, Rome, Italy. V.M. was supported by Azienda Ospedaliera Universitaria di Verona. A.H. is K. Albin Johansson Research Professor of the Foundation for the Finnish Cancer Institute. A.H. and O.H. are shareholders in Oncos Therapeutics, Ltd.
Supplementary Material
Relationships between differentially expressed interferon pathway genes and a possible drug target for the reversion of the resistant phenotype. Graphical representation of interferon signaling pathways by Ingenuity Pathways Analysis (a): Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge. Upregulated genes of the dataset are highlighted. Moksiskaan candidate pathway of the genes differentially expressed between untreated and virus resistant tumors (b). STAT1 is linked to IL6, IRF9, CXCR4, and TNF and possibly responsible for the downstream effect of chemokine ligands. Green and blue borders refer to up- and downregulated genes, respectively. Known regulations between differentially expressed genes are shown with bold borders whereas the predictions (CXCL9-11) are kept thin and the nodes are grey. Drugs that can influence the downregulated gene NR3C2 as identified by Moksiskaan analysis (c).
Expression of MxA and interferon stimulated genes in recurring versus control tumors. RNA extracted from untreated control and Ad5/3-?24 treated tumors was reverse transcripted to cDNA and analyzed with semi-quantitative RT-PCR using specific primers for human Myxovirus resistance protein A (MxA), interferon alpha-inducible protein 27 (IFI27) and interferon alpha-inducible protein 16 (IFI16). Human β-actin primers were used as controls.
Expression of MxA in infected SKOV3.ip1 cells after interferon-α treatment. MxA expression and adenovirus hexon staining in SKOV3.ip1 cells after infection with adenovirus (a). To simulate the effect of stromal cells the SKOV3.ip1 cells were treated with recombinant universal type I interferon-α prior to infection and during infection (b). The interferon-α treatment resulted in further upregulation of MxA expression 1h after infection. IFN, interferon; Ad, adenovirus.
Genes downregulated in virus-resistant tumors in comparison to untreated tumors. Downregulated genes were further filtered using t-test with false discovery rate correction to account for multiple hypotheses (q<0.05). Listed are all 113 significantly downregulated genes in virus resistant tumors.
Genes differentially expressed in response to virus infection in recurrent versus untreated tumors. Enriched Gene Ontology (GO) terms: response to virus. GO enrichment analysis was done using Fisher's exact test, which compares the observed frequency of each present GO term to the frequency in a reference gene set. A GO term is present if some input gene is annotated with the GO term or its descendants.
REFERENCES
- Pesonen S, Kangasniemi L., and, Hemminki A. Oncolytic adenoviruses for the treatment of human cancer: focus on translational and clinical data. Mol Pharm. 2011;8:12–28. doi: 10.1021/mp100219n. [DOI] [PubMed] [Google Scholar]
- Bachtarzi H, Stevenson M., and, Fisher K. Cancer gene therapy with targeted adenoviruses. Expert Opin Drug Deliv. 2008;5:1231–1240. doi: 10.1517/17425240802507636. [DOI] [PubMed] [Google Scholar]
- Shirakawa T. The current status of adenovirus-based cancer gene therapy. Mol Cells. 2008;25:462–466. [PubMed] [Google Scholar]
- Guse K., and, Hemminki A. Cancer gene therapy with oncolytic adenoviruses. J BUON. 2009;14 Suppl 1:S7–15. [PubMed] [Google Scholar]
- Immonen A, Vapalahti M, Tyynelä K, Hurskainen H, Sandmair A, Vanninen R.et al. (2004AdvHSV-tk gene therapy with intravenous ganciclovir improves survival in human malignant glioma: a randomised, controlled study Mol Ther 10967–972. [DOI] [PubMed] [Google Scholar]
- Pearson S, Jia H., and, Kandachi K. China approves first gene therapy. Nat Biotechnol. 2004;22:3–4. doi: 10.1038/nbt0104-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W., and, Fang H. Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets. 2007;7:141–148. doi: 10.2174/156800907780058817. [DOI] [PubMed] [Google Scholar]
- Nayak S., and, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010;17:295–304. doi: 10.1038/gt.2009.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Davis JJ, Zhu H, Dong F, Guo W, Ang J.et al. (2007Redirecting adaptive immunity against foreign antigens to tumors for cancer therapy Cancer Biol Ther 61773–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuve S, Liu Y, Tragoolpua K, Jacobs JD, Yumul RC, Li ZY.et al. (2009In situ adenovirus vaccination engages T effector cells against cancer Vaccine 274225–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, Ugolini M.et al. (2010Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients Cancer Res 704297–4309. [DOI] [PubMed] [Google Scholar]
- Monsurrò V, Beghelli S, Wang R, Barbi S, Coin S, Di Pasquale G.et al. (2010Anti-viral state segregates two molecular phenotypes of pancreatic adenocarcinoma: potential relevance for adenoviral gene therapy J Transl Med 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N.et al. (2008An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer Proc Natl Acad Sci USA 10518490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MH, Cook JA, Chandramouli GV, DeGraff W, Yan H, Zhao S.et al. (2007Gene expression profiling of breast, prostate, and glioma cells following single versus fractionated doses of radiation Cancer Res 673845–3852. [DOI] [PubMed] [Google Scholar]
- Sadler AJ., and, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Huang X., and, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J Virol. 2007;81:3170–3180. doi: 10.1128/JVI.02192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nociari M, Ocheretina O, Schoggins JW., and, Falck-Pedersen E. Sensing infection by adenovirus: Toll-like receptor-independent viral DNA recognition signals activation of the interferon regulatory factor 3 master regulator. J Virol. 2007;81:4145–4157. doi: 10.1128/JVI.02685-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung RS, Qin L., and, Bromberg JS. TNFalpha and IFNgamma induced by innate anti-adenoviral immune responses inhibit adenovirus-mediated transgene expression. Mol Ther. 2001;3 5 Pt 1:757–767. doi: 10.1006/mthe.2001.0318. [DOI] [PubMed] [Google Scholar]
- Randall RE., and, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89 Pt 1:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Staeheli P., and, Pavlovic J. Inhibition of vesicular stomatitis virus mRNA synthesis by human MxA protein. J Virol. 1991;65:4498–4501. doi: 10.1128/jvi.65.8.4498-4501.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanerva A, Zinn KR, Chaudhuri TR, Lam JT, Suzuki K, Uil TG.et al. (2003Enhanced therapeutic efficacy for ovarian cancer with a serotype 3 receptor-targeted oncolytic adenovirus Mol Ther 8449–458. [DOI] [PubMed] [Google Scholar]
- Laakso M., and, Hautaniemi S. Integrative platform to translate gene sets to networks. Bioinformatics. 2010;26:1802–1803. doi: 10.1093/bioinformatics/btq277. [DOI] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS.et al. (2009A novel signaling pathway impact analysis Bioinformatics 2575–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, McDonald C, Kanneganti TD, Amer A., and, Núñez G. Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol. 2006;177:3507–3513. doi: 10.4049/jimmunol.177.6.3507. [DOI] [PubMed] [Google Scholar]
- Diallo JS, Le Boeuf F, Lai F, Cox J, Vaha-Koskela M, Abdelbary H.et al. (2010A high-throughput pharmacoviral approach identifies novel oncolytic virus sensitizers Mol Ther 181123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M., and, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38 Database issue:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling M, Eisen C., and, Christ M. Aldosterone-specific membrane receptors and rapid non-genomic actions of mineralocorticoids. Mol Cell Endocrinol. 1992;90:C5–C9. doi: 10.1016/0303-7207(92)90092-k. [DOI] [PubMed] [Google Scholar]
- Strauss R, Sova P, Liu Y, Li ZY, Urban N,, Drescher C.et al. (2006Resistance of primary ovarian cancer cells to viral oncolysis Mol Ther 13S18–S18. [Google Scholar]
- Strauss R, Sova P, Liu Y, Li ZY, Tuve S, Pritchard D.et al. (2009Epithelial phenotype confers resistance of ovarian cancer cells to oncolytic adenoviruses Cancer Res 695115–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I.et al. (2010Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF Mol Ther 181874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesonen S, Nokisalmi P, Escutenaire S, Särkioja M, Raki M, Cerullo V.et al. (2010Prolonged systemic circulation of chimeric oncolytic adenovirus Ad5/3-Cox2L-D24 in patients with metastatic and refractory solid tumors Gene Ther 17892–904. [DOI] [PubMed] [Google Scholar]
- Pesonen S, Diaconu I, Cerullo V, Escutenaire S, Raki M, Kangasniemi L.et al. (2011Integrin targeted oncolytic adenoviruses Ad5-D24-RGD and Ad5-RGD-D24-GMCSF for treatment of patients with advanced chemotherapy refractory solid tumors Int J Cancerepub ahead of print). [DOI] [PubMed]
- Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM., and, Hortobagyi GN. The HER-2 receptor and breast cancer: ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist. 2009;14:320–368. doi: 10.1634/theoncologist.2008-0230. [DOI] [PubMed] [Google Scholar]
- Menter DG, Schilsky RL., and, DuBois RN. Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward. Clin Cancer Res. 2010;16:1384–1390. doi: 10.1158/1078-0432.CCR-09-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zürcher T, Pavlovic J., and, Staeheli P. Mouse Mx2 protein inhibits vesicular stomatitis virus but not influenza virus. Virology. 1992;187:796–800. doi: 10.1016/0042-6822(92)90481-4. [DOI] [PubMed] [Google Scholar]
- Fiebeler A, Schmidt F, Müller DN, Park JK, Dechend R, Bieringer M.et al. (2001Mineralocorticoid receptor affects AP-1 and nuclear factor-kappab activation in angiotensin II-induced cardiac injury Hypertension 372 Part 2787–793. [DOI] [PubMed] [Google Scholar]
- Appledorn DM, Patial S, McBride A, Godbehere S, Van Rooijen N, Parameswaran N.et al. (2008Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo J Immunol 1812134–2144. [DOI] [PubMed] [Google Scholar]
- Wojton J., and, Kaur B. Impact of tumor microenvironment on oncolytic viral therapy. Cytokine Growth Factor Rev. 2010;21:127–134. doi: 10.1016/j.cytogfr.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN.et al. (2002Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes J Virol 765532–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagno AM, Salcido CD, Gillet JP, Wu CP, Fostel JM, Mumau MD.et al. (2010Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics J Natl Cancer Inst 1021637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okugawa K, Kobayashi H, Hirakawa T, Sonoda T, Ogura T., and, Nakano H. In vivo establishment and characterization of a paclitaxel-resistant human ovarian cancer cell line showing enhanced growth properties and drug-resistance only in vivo. J Cancer Res Clin Oncol. 2004;130:178–186. doi: 10.1007/s00432-003-0516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki O, Bauerschmitz G, Hemmi S, Lavilla-Alonso S, Diaconu I, Guse K.et al. (2011Oncolytic adenovirus based on serotype 3 Cancer Gene Ther 18288–296. [DOI] [PubMed] [Google Scholar]
- Durinck S. Pre-processing of microarray data and analysis of differential expression. Methods Mol Biol. 2008;452:89–110. doi: 10.1007/978-1-60327-159-2_4. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B., and, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relationships between differentially expressed interferon pathway genes and a possible drug target for the reversion of the resistant phenotype. Graphical representation of interferon signaling pathways by Ingenuity Pathways Analysis (a): Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge. Upregulated genes of the dataset are highlighted. Moksiskaan candidate pathway of the genes differentially expressed between untreated and virus resistant tumors (b). STAT1 is linked to IL6, IRF9, CXCR4, and TNF and possibly responsible for the downstream effect of chemokine ligands. Green and blue borders refer to up- and downregulated genes, respectively. Known regulations between differentially expressed genes are shown with bold borders whereas the predictions (CXCL9-11) are kept thin and the nodes are grey. Drugs that can influence the downregulated gene NR3C2 as identified by Moksiskaan analysis (c).
Expression of MxA and interferon stimulated genes in recurring versus control tumors. RNA extracted from untreated control and Ad5/3-?24 treated tumors was reverse transcripted to cDNA and analyzed with semi-quantitative RT-PCR using specific primers for human Myxovirus resistance protein A (MxA), interferon alpha-inducible protein 27 (IFI27) and interferon alpha-inducible protein 16 (IFI16). Human β-actin primers were used as controls.
Expression of MxA in infected SKOV3.ip1 cells after interferon-α treatment. MxA expression and adenovirus hexon staining in SKOV3.ip1 cells after infection with adenovirus (a). To simulate the effect of stromal cells the SKOV3.ip1 cells were treated with recombinant universal type I interferon-α prior to infection and during infection (b). The interferon-α treatment resulted in further upregulation of MxA expression 1h after infection. IFN, interferon; Ad, adenovirus.
Genes downregulated in virus-resistant tumors in comparison to untreated tumors. Downregulated genes were further filtered using t-test with false discovery rate correction to account for multiple hypotheses (q<0.05). Listed are all 113 significantly downregulated genes in virus resistant tumors.
Genes differentially expressed in response to virus infection in recurrent versus untreated tumors. Enriched Gene Ontology (GO) terms: response to virus. GO enrichment analysis was done using Fisher's exact test, which compares the observed frequency of each present GO term to the frequency in a reference gene set. A GO term is present if some input gene is annotated with the GO term or its descendants.