

Abstract
Dystrophin deficiency leads to lethal dilated Duchenne cardiomyopathy. A promising therapy is to deliver a highly abbreviated microdystrophin gene to the heart using adeno-associated virus (AAV). Microdystrophin has been shown to mitigate dystrophin-deficient skeletal muscle disease. However, it is not clear whether microdystrophin is equally effective in treating Duchenne cardiomyopathy. To evaluate microdystrophin therapy in the heart, we injected 5 × 1012 viral genome particles/mouse of AAV-9 ΔR4-23/ΔC microdystrophin vector via tail vein to ~16–20-month-old (average 18.7–month-old) female mdx mice, a manifesting model of Duchenne cardiomyopathy. Cardiac transduction and heart function were examined at 2–8 months after gene transfer. We observed robust myocardial microdystrophin expression. Electrocardiography (ECG) and left ventricular catheter hemodynamic assays also revealed significant improvement. Furthermore, AAV-microdystrophin therapy prevented dobutamine-stress induced acute cardiac death. We demonstrate for the first time that AAV microdystrophin therapy significantly ameliorates functional deficiency in a phenotypic model of Duchenne cardiomyopathy. Our results support further exploration of microdystrophin therapy to treat Duchenne cardiomyopathy.
Introduction
Duchenne cardiomyopathy is a dystrophin-deficient heart disease. Dystrophin is a 427 kDa cytoskeletal protein essential for signaling and sarcolemmal integrity in normal muscle cells. In a dystrophin-deficient heart, cardiomyocytes undergo necrosis. Eventually, fibrotic tissues replace dead heart cells. Clinical features of Duchenne cardiomyopathy include abnormal electrocardiography (ECG), impaired hemodynamics, heart chamber dilation, and left ventricular pump failure. Current therapies are limited to pharmacological interventions.1 These treatments ameliorate symptoms but they cannot solve the central issue of dystrophin deficiency. Restoring dystrophin expression by gene therapy may lead to a fundamental breakthrough in Duchenne cardiomyopathy management.2,3
Many viral and nonviral vectors have been explored for heart gene transfer. So far, whole heart gene delivery has only been achieved with adeno-associated virus (AAV) in small mammals.4,5,6,7 AAV is a minute single-stranded DNA virus. Although AAV is exceptionally robust in delivering genes to the rodent heart, the application of AAV to dystrophin transfer is challenged by the 5 kb packaging limit of the virion. More than 60% of the 12 kb dystrophin coding sequence has to be removed for AAV packaging. Whereas in-frame deletion of ~50% dystrophin has been associated with very mild clinical disease in one patient,8 it is uncertain whether dystrophins with larger deletions are equally protective in general.9 A highly truncated, human dystrophin-derived ΔR4-23/ΔC microgene (3.8 kb) has been shown to effectively mitigate skeletal muscle disease in symptomatic dystrophic mice (Supplementary Figure S1).10,11,12 However, it is not completely clear whether the abbreviated gene can protect the heart in a phenotypic Duchenne cardiomyopathy model.13
Mdx mice are the most commonly used models of dystrophin deficiency. However, the hearts of young adult mdx mice are only mildly affected.14,15,16,17 Heart pathology becomes evident in 1-year-old mdx mice and it gets worse as mice age.17,18,19,20 We have recently shown that aged female mdx mice display similar dilated cardiomyopathy as seen in human patients.21,22 Here we tested the hypothesis that systemic delivery of the ΔR4-23/ΔC microdystrophin gene with AAV serotype-9 (AAV-9) vector can ameliorate heart disease in the aged mdx mouse, a manifesting model of Duchenne cardiomyopathy. 5 × 1012 viral genome particles/mouse of AAV-9 ΔR4-23/ΔC microdystrophin vectors were delivered to ~19-month-old (average: 18.7 ± 0.7; range: 16–20-month-old) female mdx mice via tail vein injection (Supplementary Table S1). Microgene expression, cardiac fibrosis, and left ventricular hemodynamics were examined ~4 months later (average: 4.1 ± 0.6; range, 2–8 months) (Supplementary Table S1). Pre- and post-AAV therapy ECG profiles were also compared in most experimental mice. We observed widespread microdystrophin expression, reduction of myocardial fibrosis, significant improvement in some ECG parameters and partial correction of the hemodynamic defects. Microgene therapy also greatly reduced dobutamine stress-induced acute cardiac death. Our results suggest that microdystrophin therapy may hold significant potential for treating symptomatic Duchenne cardiomyopathy.
Results
The myocardium of aged mdx mice was efficiently transduced by an AAV-9 microdystrophin vector
We have previously shown that systemically delivered AAV-9 effectively transduces the hearts of newborn and young adult mdx mice.23,24 Here, we examined AAV-9 transduction in ~19-month-old mdx mice (5 × 1012 viral genome particles/mouse). Despite the ongoing heart disease in aged female mdx mice,21,22 we observed robust widespread microdystrophin expression in the heart (Figure 1). A representative immunofluorescence staining is shown from an mdx mouse that was treated at the age of 20 months and harvested at the age of 22 months (Figure 1a). The human specific Dys-3 antibody revealed robust expression of human-derived microdystrophin (Figure 1a,b). The specificity of Dys-3 immunostaining was further confirmed using additional antibodies. The Mandys-8 antibody which recognizes an epitope deleted in the microgene showed negative staining (Figure 1b). Microdystrophin expression also correlated with a downregulation of utrophin, a dystrophin homolog (Figure 1b).21
Figure 1.

Adeno-associated virus serotype-9 (AAV-9) microdystrophin vector efficiently transduced the myocardium of aged mdx mice. (a) Representative Dys-3 immunofluorescence staining of whole mdx heart infected with AAV-9 microdystrophin vector. (b) Representative high-power images of immunofluorescence staining from untreated mdx and AAV-9 microdystrophin vector-treated mdx. Top panel: Dys-3 antibody immunostaining. Dys-3 is an antibody that specifically recognizes human dystrophin (Hum Dys). Dys-3 immunostaining reveals the expression of human-derived microdystrophin. Middle panel: Mandys-8 antibody (R11) immunostaining. Mandys-8 antibody specifically reacts with spectrin-like repeat 11, a repeat deleted in microdystrophin. Bottom panel: Utrophin immunostaining. (c) Representative western blot analysis of dystrophin expression in BL10, untreated mdx and AAV-infected mdx mice. Top panel: western blot with the DysB antibody; bottom panel: Rapid blue staining of a duplicated gel. µ-Dys, microdystrophin. Arrowhead, full-length mouse dystrophin; *Human ΔR4-23/ΔC microdystrophin.
To further validate immunostaining results (Figure 1a,b), we performed a western blot using the DysB antibody. This antibody reacts with an epitope presented in both mouse full-length dystrophin and human microdystrophin (Supplementary Figure S1). Consistent with a previous report,25 AAV gene transfer resulted in substantially higher microdystrophin expression than that of endogenous full-length mouse dystrophin expression in normal mice (Figure 1c).
Impact of AAV microdystrophin therapy on myocardial fibrosis and the anatomical properties of the mdx heart
Although there was minimal myocardial fibrosis in aged C57Bl/10 (BL10) mice, the aged mdx heart was highly fibrotic (Figure 2a,b).21 Interestingly, in two mdx mice that were treated at 16 months of age (the youngest starting age in our study), myocardial fibrosis was rarely detected (Figure 2c,d). In mice that received AAV therapy at later ages (≥17-month-old, most are treated at 20 months of age), fibrotic regions also appeared smaller than untreated mdx mice (Figure 2b,e,f). Quantification of the hydroxyproline content suggests that AAV microdystrophin therapy significantly reduced myocardial fibrosis (Figure 2g).
Figure 2.

Evaluation of cardiac fibrosis and cardiomyocyte size in adeno-associated virus serotype-9 (AAV-9) microdystrophin-treated mdx mice. Representative Masson trichrome staining photomicrographs from the hearts of BL10, untreated mdx, and AAV microdystrophin-treated mdx mice. Blue staining shows fibrosis. (a) Age-matched BL10 control. (b) Untreated mdx. (c) A 16-month-old mdx mouse treated for 4 months. (d) A 16-month-old mdx mouse treated for 8 months. (e) A 20-month-old mdx mouse treated for 3 months. (f) Another 20-month-old mdx mouse treated for 3 months. (g) Quantification of heart hydroxyproline content in untreated and AAV-treated mdx mice. *P ≤ 0.016. (h) Quantification of cardiomyocyte size in BL10, untreated mdx, and AAV microdystrophin-treated mdx mice. AU, arbitrary unit. *Significantly different from other groups. BL10, age and gender-matched control; mdx, untreated mdx; AAV, AAV microdystrophin-treated mdx.
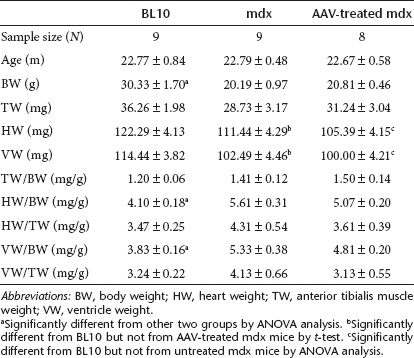
We have previously shown that the heart weight to body weight ratio and the ventricular weight to body weight ratio were significantly elevated in aged female mdx mice.21 AAV therapy did not alter these anatomic features (Table 1). We also quantified the cardiomyocyte size. Despite widespread microdystrophin expression in the heart of AAV-treated mdx mice (Figure 1a,b), we did not detect a significant change in the heart myofiber size between treated and untreated mdx mice (Figure 2h). In both cases, myofibers were significantly larger than those of control BL10 mice (Figure 2h).
Table 1. Weights and weight ratios.
AAV microdystrophin therapy resulted in beneficial ECG changes
In seven experimental mdx mice, ECG was recorded before AAV injection (pre-therapy ECG was not available in one treated mdx mouse). Paired pre- and post-therapy ECG comparison showed significant difference in several parameters (Table 2). Specifically, heart rate and QT interval were reduced, and PR interval prolonged after microdystrophin therapy. There was also a trend toward a smaller Q amplitude and a lower cardiomyopathy index although these changes did not reach statistical significance.
Table 2. ECG performance before and after AAV therapy (N = 7 pairs).

Compared to aged-matched BL10 and mdx mice, microgene-treated mdx mice showed significantly improved QT interval and Q amplitude (Figure 3). PR interval, QRS duration, and the cardiomyopathy index also leaned toward normal. Interestingly, microdystrophin therapy resulted in a heart rate significantly lower than that of normal control BL10 (Figure 3 and Supplementary Table S2).
Figure 3.

Adeno-associated virus serotype-9 (AAV-9) microdystrophin therapy partially improved the electrocardiography (ECG) profile. Quantitative evaluation of ECG in BL10, mdx, and AAV-treated mdx. HR, heart rate; bpm, beats/min; Q Amp, Q wave amplitude; C. Index, cardiomyopathy index. *Significantly different from the other two groups; cross, significantly different from BL10 only.
AAV microdystrophin therapy ameliorated left ventricular hemodynamic dysfunction and prevented dobutamine-induced acute cardiac death Left ventricular catheterization was performed at ~23 months of age (average: 22.7 ± 0.6; range: 20–24). A representative pressure–volume loop from an untreated female mdx mouse shows the rightward and downward shift characteristic of dilated cardiomyopathy (Figure 4a).21,22 Although each AAV microdystrophin-treated mdx mouse responded differently, they all showed some level of improvement in the pressure–volume loops (Figure 4a). The mouse that responded the best to therapy (treated for 4 months starting from the 16 months of age), exhibited a fully normalized pressure–volume loop (Figure 4a, top left panel). A 20-month-old mouse that was treated for 2 months also showed near-complete normalization (Figure 4a, second row right panel).
Figure 4.

Left ventricular hemodynamics was enhanced by adeno-associated virus serotype-9 (AAV-9) microdystrophin therapy. (a) Pressure–volume (PV) loops from eight AAV-treated mdx mice. The age of AAV injection and the duration of therapy are indicated at the bottom of each panel. Representative PV loops from BL10 and mdx mice are depicted in each panel for comparison. Vertical axis, pressure (mm Hg); horizontal axis, volume (µl). (b) Systolic parameters. (c) Diastolic parameters. (d) Overall cardiac function. *Significantly different from the other two groups; cross, significantly different from BL10 only. (e) Kaplan–Meier survival curve within 15 minutes of dobutamine administration. Neither BL10 nor AAV-treated mdx mice died during this period. The survival rate of untreated mdx is significantly lower than that of BL10 and AAV microdystrophin-treated mdx mice (P ≤ 0.0079). N = 7 for BL10, N = 7 for untreated mdx, N = 8 for AAV-treated mdx.
Collectively, AAV microdystrophin therapy appears to have completely rectified the systolic defects (Figure 4b). End-systolic volume, maximal pressure, and the maximal rate of left ventricular contraction (dP/dt max) were all within the normal range (Figure 4b). Diastolic function was also improved by AAV microdystrophin therapy (Figure 4c). End-diastolic volume and the minimal rate of left ventricular relaxation (dP/dt min) were significantly reduced in treated mdx mice. The time constant of heart relaxation (tau) was also reduced but the difference did not reach statistical significance. Compared with age and gender-matched untreated mdx mice and normal BL10 mice, mdx mice treated with AAV microdystrophin vector showed normal ejection fraction. The other indices of heart function (including stroke volume and cardiac output) were also significantly enhanced but these parameters were not restored to the wild-type levels (Figure 4d).
Administration of dobutamine resulted in acute cardiac death (death within 15 minutes of dobutamine injection) in 75% untreated mdx mice (Figure 4e). However, none of the AAV microdystrophin-treated mdx mice died within the same time frame (Figure 4e).
Discussion
The loss of dystrophin expression in the heart results in life threatening dilated cardiomyopathy. Expressing a functional dystrophin gene in cardiomyocytes may address the central issue of dystrophin deficiency. There are two key problems to overcome in gene therapy for Duchenne cardiomyopathy. First, we need a vector that can efficiently deliver the therapeutic gene to at least 50% cardiomyocytes in a diseased heart.14,21 Second, we need a functional dystrophin gene that can fulfill the needs of the heart.
Recent progress in gene delivery has begun to shed light on cardiac gene transfer.26,27 One of the most striking accomplishments is whole heart transduction with intravenous AAV injection.4,5,6,7 Among many different AAV serotypes, AAV-9 is particularly effective in reaching the entire myocardium in rodents.6,7 We have previously shown that dystrophin-deficiency may not constitute a barrier for AAV-9 mediated heart gene transfer in young mdx mice.23,24 However, dilated Duchenne cardiomyopathy has only been seen in aged female mdx mice.21,22 Here we tested whether systemic AAV-9 injection can achieve efficient myocardial transduction in a severely affected heart. Surprisingly, defective cardiac architecture and function in aged female mdx mice did not compromise AAV-9 transduction (Figure 1).11
Full-length dystrophin has four major structural domains including the N-terminal, rod, cysteine-rich and C-terminal domains (Supplementary Figure S1). The rod domain contains four hinges and 24 spectrin-like repeats. Recent studies suggest that different regions of dystrophin may contribute differently to the health and survival of cardiomyocytes.13,28,29 In the absence of a precise understanding of dystrophin structure–function relationship in the heart, delivery of a full-length dystrophin gene may offer the best protection to the heart. Unfortunately, the few vectors that are capable of carrying the full-length gene (such as gutted adenovirus and herpes virus) cannot effectively transduce the heart.30
Significant efforts have been invested over the past 10 years in developing the highly truncated microdystrophin genes. These microgenes carry ~30% of dystrophin coding sequence. They usually contain the N-terminal domain, a few selected hinges and spectrin-like repeats and the cysteine-rich domain (Supplementary Figure S1).31,32,33 Importantly, the synthetic microgenes can fit into a single AAV virion. The microgenes were initially designed to meet the functional needs of skeletal muscle. Independent studies from many investigators have now provided unequivocal evidence that AAV microdystrophin therapy can indeed ameliorate skeletal muscle dystrophy in dystrophin-deficient mice (reviewed in refs. 34,35). Several studies have examined the therapeutic potential of microdystrophin in the heart of newborn or young adult mdx mice.15,23,25 We first demonstrated that AAV-mediated ΔR4-23/ΔC microdystrophin expression strengthened the sarcolemmal integrity of cardiomyocytes in neonatal mdx mice.15 We later found that neonatal microdystrophin therapy also ameliorated ECG abnormality.23 Townsend et al. tested AAV ΔR4-23/ΔC microdystrophin therapy in 10-week-old mdx mice.25 Their results suggest that microdystrophin can prevent heart failure induced by a 30 minutes continuous dobutamine challenge.25 Nevertheless, since young adult mdx mice do not develop dystrophin deficient dilated cardiomyopathy,21,22 these promising reports still did not address the question of whether a microdystrophin gene designed for skeletal muscle can alleviate Duchenne cardiomyopathy.
The microdystrophin therapy is built on the findings that patients who carry certain rod-domain in-frame deletions show very mild skeletal muscle symptoms.8,36,37,38 Interestingly, some of these patients develop severe cardiomyopathy despite the absence of apparent muscle weakness.36,37,39,40,41,42 These observations suggest that there may exist one or multiple cardiac-specific regions in the rod domain of dystrophin.28,29 Lack of these cardiac-specific regions may not compromise skeletal muscle rescue but it may limit cardiac protection. The exact nature (or the presence) of the cardioprotective domain remains to be clarified. In this study, we used the ΔR4-23/ΔC microgene in which the vast majority of the dystrophin rod domain (including 20 spectrin-like repeats and one hinge) is deleted. We tested whether this highly abbreviated microgene can offer any help to the ongoing cardiomyopathy in a symptomatic model. We observed a significant improvement in ECG and left ventricular function. However, none of these were completely normalized (Figures 3 and 4 and Table 2).
We have previously shown that cardiac-specific expression of the 6 kb ΔH2-R19 minidystrophin gene significantly enhanced, but did not normalize, heart function in aged transgenic mdx mice (Supplementary Figure S1).13 Our results here suggest that systemic delivery of a much smaller microgene may also ameliorate Duchenne cardiomyopathy. Because systemic therapy also improved skeletal muscle morphology and function in 20-month-old mdx mice,11 it is possible that our observation may relate to global disease amelioration in aged mdx mice. In summary, our study represents a significant advancement for gene therapy of Duchenne cardiomyopathy. Our results strongly support further exploration of microdystrophin therapy in large animal models of Duchenne cardiomyopathy.
Materials and Methods
Experimental mice. Animal experiments were approved by the University of Missouri Animal Care and Use Committee in accordance with National Institutes of Health guidelines. Experimental BL10 and mdx mice were generated in a barrier facility using founders from The Jackson Laboratory (Bar Harbor, ME). In this study, we used female mdx mice that were at least 16-month-old (Supplementary Table S1). At this age, mdx mice show obvious cardiac pathology.17,18,19,20,21,22 We would like to point out that it is very challenging to obtain a large cohort of aged mdx mice. In the absence of external intervention, female mdx mice start to die at 14 months of age and ~30% die naturally by 20 months of age. The survival curve drops sharply thereafter.43,44 All mice were maintained in a specific-pathogen free animal care facility on a 12-hour light (25 lux):12-hour dark cycle with access to food and water ad libitum.
AAV-9 microdystrophin vector. The ΔR4-23/ΔC microdystrophin gene (a gift from Dr Jeffrey Chamberlain, University of Washington, Seattle, WA) is derived from the human dystrophin gene.31 Microdystrophin expression was regulated by the ubiquitous cytomegalovirus promoter. The microdystrophin expression cassette was packaged in AAV-9 and characterized according to our published protocols.7 AAV vector was delivered to conscious mice via tail vein injection. To evaluate microdystrophin therapy, eight 16–20-month-old (average 18.7 ± 0.6 months) female mdx mice were injected with a single dose of AAV microgene vector (5 × 1012 viral genome particles/mouse). Cardiac function was examined at 2–8 months (average 4.1 ± 0.6 months) after AAV therapy (Supplementary Table S1).
Morphological studies. Microdystrophin expression was evaluated by immunofluorescence staining using a human dystrophin-specific antibody (Dys-3, diluted 1:20, clone Dy10/12B2, IgG2a; Novocastra, Newcastle, UK). Additional immunofluorescence staining was performed using a dystrophin antibody specific to spectrin-like repeat 11 (Mandys-8, diluted 1:200; Sigma, St Louis, MO). This repeat is deleted in the ΔR4-23/ΔC microdystrophin construct. Utrophin expression was examined using a mouse monoclonal antibody against the utrophin N-terminal domain (VP-U579, diluted 1:20; clone DRP3/20C5, IgG1; Vector Laboratories, Burlingame, CA). Immunostaining was performed according to a previously published protocol.10,15 Slides were viewed at the identical exposure setting predetermined for each specific antibody. Masson trichrome staining was performed as we described before.13,21 To quantify cardiomyocyte size, mid-ventricular section of the heart was stained with hematoxylin and eosin. Digitized images (×200) were taken from endomyocardial (2 fields), interventricular (1 field), and epimyocardial (2 fields) areas. The minFeret diameter of 30–50 randomly picked cardiomyocytes was quantified with the National Institutes of Health ImageJ software (version 1.45h) in each photomicrograph. The minFeret diameter represents the shortest diameter across an ellipse. It is a commonly used morphometric parameter to measure muscle cell size.45
Western blot. The frozen heart was ground to fine powder in liquid nitrogen. Whole heart muscle lysate was prepared as we described before.13,21 Primary antibody of NCL-DysB (1:100, clone 34C5, IgG1; Novocastra) was applied at 4 °C overnight (Supplementary Figure S1).
Hydroxyproline quantification. The collagen content of the heart was determined by quantifying hydroxyproline level in previously frozen heart samples according to our published protocol.13,46
ECG and hemodynamic assay. A 12-lead ECG assay was performed using a system from AD Instruments (Colorado Springs, CO) according to our previously published protocol.13,21,22,23,47 The Q wave amplitude was determined using the lead I tracing. Other ECG parameters were analyzed using the lead II tracing. The cardiomyopathy index was calculated by dividing the QT interval by the PQ segment.48 Left ventricular hemodynamic assay was performed using a closed chest approach as we have previously described.13,21,22,47 The resulting pressure–volume loops were analyzed with the PVAN software (Millar Instruments, Houston, TX). After baseline determinations, 5 µg/g body weight of dobutamine (Sigma) was intraperitoneally injected. Mouse survival was monitored for 15 min after dobutamine administration. Cardiac death is diagnosed when heart rate was reduced to less than 250 b.p.m. and systolic pressure dropped to <30 mm Hg.13,21
Statistical analysis. Data are presented as mean ± s.e. of mean. The Prism 4 software (GraphPad, San Diego, CA) was used for statistical analysis in the survival study. The SPSS software (SPSS, Chicago, IL) was used for statistical analysis of other experiments. Student's t-test was used to compare hydroxyproline content between AAV microdystrophin-treated and -untreated mdx mice. One-way ANOVA analysis and Bonferroni post hoc analysis were used for multiple group comparisons. Dobutamine-stress survival was analyzed with the Kaplan–Meier method. The Mantel–Cox log-rank test was used for statistical analysis of survival with the Bonferroni corrected threshold used for determining significance. A P < 0.05 was considered as statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Schematic outlines of full-length dystrophin and abbreviated dystrophins that have been tested in the heart of mdx mice (not drawn to scale). Table S1. Overview of AAV-9ΔR4-23/ΔC-treated mdx mice. Table S2. P value for Figure 3.
Acknowledgments
This work was supported by grants from the National Institutes of Health (HL-91883, D.D.) and the Muscular Dystrophy Association (D.D.). We thank Keqing Zhang for excellent technical help. We thank Thomas McDonald and Alexandra Kellogg for the help with cardiomyocyte size quantification. We thank Duan Lab members for helpful discussion. The authors declared no conflict of interest.
Supplementary Material
Schematic outlines of full-length dystrophin and abbreviated dystrophins that have been tested in the heart of mdx mice (not drawn to scale).
Overview of AAV-9ΔR4-23/ΔC-treated mdx mice.
P value for Figure 3.
REFERENCES
- American Academy of Pediatrics, Section on Cardiology and Cardiac Surgery Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005;116:1569–1573. doi: 10.1542/peds.2005-2448. [DOI] [PubMed] [Google Scholar]
- Duan D. Challenges and opportunities in dystrophin-deficient cardiomyopathy gene therapy. Hum Mol Genet. 2006;15 Spec No 2:R253–R261. doi: 10.1093/hmg/ddl180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J-H, Bostick B, Yue Y., and, Duan D.2010Duchenne cardiomyopathy gene therapy Duan D.ed). Muscle Gene Therapy Springer Science + Business Media, LLC: New York; 141–162. [Google Scholar]
- Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG.et al. (2004Systemic delivery of genes to striated muscles using adeno-associated viral vectors Nat Med 10828–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J.et al. (2005Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart Nat Biotechnol 23321–328. [DOI] [PubMed] [Google Scholar]
- Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE.et al. (2006Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo Circ Res 99e3–e9. [DOI] [PubMed] [Google Scholar]
- Bostick B, Ghosh A, Yue Y, Long C., and, Duan D. Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther. 2007;14:1605–1609. doi: 10.1038/sj.gt.3303029. [DOI] [PubMed] [Google Scholar]
- England SB, Nicholson LV, Johnson MA, Forrest SM, Love DR, Zubrzycka-Gaarn EE.et al. (1990Very mild muscular dystrophy associated with the deletion of 46% of dystrophin Nature 343180–182. [DOI] [PubMed] [Google Scholar]
- Fanin M, Freda MP, Vitiello L, Danieli GA, Pegoraro E., and, Angelini C. Duchenne phenotype with in-frame deletion removing major portion of dystrophin rod: threshold effect for deletion size. Muscle Nerve. 1996;19:1154–1160. doi: 10.1002/mus.880190902. [DOI] [PubMed] [Google Scholar]
- Yue Y, Liu M., and, Duan D. C-terminal-truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther. 2006;14:79–87. doi: 10.1016/j.ymthe.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Blankinship MJ, Allen JM., and, Chamberlain JS. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther. 2008;16:657–664. doi: 10.1038/mt.2008.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L.et al. (2006rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice Nat Med 12787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick B, Yue Y, Long C, Marschalk N, Fine DM, Chen J.et al. (2009Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged Mdx mice Mol Ther 17253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Skimming JW, Liu M, Strawn T., and, Duan D. Full-length dystrophin expression in half of the heart cells ameliorates beta-isoproterenol-induced cardiomyopathy in mdx mice. Hum Mol Genet. 2004;13:1669–1675. doi: 10.1093/hmg/ddh174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS., and, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danialou G, Comtois AS, Dudley R, Karpati G, Vincent G, Des Rosiers C.et al. (2001Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury FASEB J 151655–1657. [DOI] [PubMed] [Google Scholar]
- Van Erp C, Loch D, Laws N, Trebbin A., and, Hoey AJ. Timeline of cardiac dystrophy in 3-18-month-old MDX mice. Muscle Nerve. 2010;42:504–513. doi: 10.1002/mus.21716. [DOI] [PubMed] [Google Scholar]
- Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS., and, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Lefaucheur JP, Pastoret C., and, Sebille A. Phenotype of dystrophinopathy in old mdx mice. Anat Rec. 1995;242:70–76. doi: 10.1002/ar.1092420109. [DOI] [PubMed] [Google Scholar]
- Wehling-Henricks M, Jordan MC, Roos KP, Deng B., and, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005;14:1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- Bostick B, Yue Y, Long C., and, Duan D. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res. 2008;102:121–130. doi: 10.1161/CIRCRESAHA.107.162982. [DOI] [PubMed] [Google Scholar]
- Bostick B, Yue Y., and, Duan D. Gender influences cardiac function in the mdx model of Duchenne cardiomyopathy. Muscle Nerve. 2010;42:600–603. doi: 10.1002/mus.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick B, Yue Y, Lai Y, Long C, Li D., and, Duan D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum Gene Ther. 2008;19:851–856. doi: 10.1089/hum.2008.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Yue Y, Shin JH., and, Duan D. Systemic Trans-splicing adeno-associated viral delivery efficiently transduces the heart of adult mdx mouse, a model for duchenne muscular dystrophy. Hum Gene Ther. 2009;20:1319–1328. doi: 10.1089/hum.2009.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D, Blankinship MJ, Allen JM, Gregorevic P, Chamberlain JS., and, Metzger JM. Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol Ther. 2007;15:1086–1092. doi: 10.1038/sj.mt.6300144. [DOI] [PubMed] [Google Scholar]
- Katz MG, Swain JD, White JD, Low D, Stedman H., and, Bridges CR. Cardiac gene therapy: optimization of gene delivery techniques in vivo. Hum Gene Ther. 2010;21:371–380. doi: 10.1089/hum.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly HQ, Kawase Y., and, Hajjar RJ. Advances in gene-based therapy for heart failure. J Cardiovasc Transl Res. 2008;1:127–136. doi: 10.1007/s12265-008-9022-4. [DOI] [PubMed] [Google Scholar]
- Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach s.d.et al. (2005Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy Circulation 1122799–2804. [DOI] [PubMed] [Google Scholar]
- Kaspar RW, Allen HD, Ray WC, Alvarez CE, Kissel JT, Pestronk A.et al. (2009Analysis of dystrophin deletion mutations predicts age of cardiomyopathy onset in becker muscular dystrophy Circ Cardiovasc Genet 2544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Yue Y, Bostick B., and, Duan D.2010Delivering large therapeutic genes for muscle gene therapy Duan D.ed). Muscle Gene Therapy Springer Science + Business Media, LLC: New York; 205–218. [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF.et al. (2002Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy Nat Med 8253–261. [DOI] [PubMed] [Google Scholar]
- Wang B, Li J., and, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C.et al. (2009Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy J Clin Invest 119624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trollet C, Athanasopoulos T, Popplewell L, Malerba A., and, Dickson G. Gene therapy for muscular dystrophy: current progress and future prospects. Expert Opin Biol Ther. 2009;9:849–866. doi: 10.1517/14712590903029164. [DOI] [PubMed] [Google Scholar]
- Odom GL, Gregorevic P., and, Chamberlain JS. Viral-mediated gene therapy for the muscular dystrophies: successes, limitations and recent advances. Biochim Biophys Acta. 2007;1772:243–262. doi: 10.1016/j.bbadis.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Di Lenarda A, Porcu M, Sinagra G, Mateddu A, Marrosu G.et al. (1997Dystrophin gene abnormalities in two patients with idiopathic dilated cardiomyopathy Heart 78608–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M.et al. (1996Myocardial involvement is very frequent among patients affected with subclinical Becker's muscular dystrophy Circulation 943168–3175. [DOI] [PubMed] [Google Scholar]
- Melis MA, Cau M, Muntoni F, Mateddu A, Galanello R, Boccone L.et al. (1998Elevation of serum creatine kinase as the only manifestation of an intragenic deletion of the dystrophin gene in three unrelated families Eur J Paediatr Neurol 2255–261. [DOI] [PubMed] [Google Scholar]
- Melis MA, Cau M., and, Deidda F. Mutation of dystrophin gene in two families with X-linked dilated cardiomyopathy. Neuromuscul Disord. 1998;8:244. [Google Scholar]
- Arbustini E, Diegoli M, Morbini P, Dal Bello B, Banchieri N, Pilotto A.et al. (2000Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy J Am Coll Cardiol 351760–1768. [DOI] [PubMed] [Google Scholar]
- Melacini P, Fanin M, Danieli GA, Fasoli G, Villanova C, Angelini C.et al. (1993Cardiac involvement in Becker muscular dystrophy J Am Coll Cardiol 221927–1934. [DOI] [PubMed] [Google Scholar]
- Nigro G, Politano L, Nigro V, Petretta VR., and, Comi LI. Mutation of dystrophin gene and cardiomyopathy. Neuromuscul Disord. 1994;4:371–379. doi: 10.1016/0960-8966(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Li D, Long C, Yue Y., and, Duan D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum Mol Genet. 2009;18:1209–1220. doi: 10.1093/hmg/ddp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain JS, Metzger J, Reyes M, Townsend D., and, Faulkner JA. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007;21:2195–2204. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- Lawlor MW, Read BP, Edelstein R, Yang N, Pierson CR, Stein MJ.et al. (2011Inhibition of activin receptor type IIB increases strength and lifespan in myotubularin-deficient mice Am J Pathol 178784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim CH, Grange RW., and, Duan D. The passive mechanical properties of the extensor digitorum longus muscle are compromised in 2- to 20-mo-old mdx mice. J Appl Physiol. 2011;110:1656–1663. doi: 10.1152/japplphysiol.01425.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostick B, Yue Y., and, Duan D. Phenotyping cardiac gene therapy in mice. Methods Mol Biol. 2011;709:91–104. doi: 10.1007/978-1-61737-982-6_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro G, Comi LI, Politano L., and, Nigro G.2004Cardiomyopathies associated with muscular dystrophies Engel A., and, Franzini-Armstrong C.eds). Myology: Basic and Clinical3rd edn., vol. 2. McGraw-Hill, Medical Pub. Division: New York; 1239–1256. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic outlines of full-length dystrophin and abbreviated dystrophins that have been tested in the heart of mdx mice (not drawn to scale).
Overview of AAV-9ΔR4-23/ΔC-treated mdx mice.
P value for Figure 3.