

Abstract
Genomic disorders constitute a class of diseases that are associated with DNA rearrangements resulting from region-specific genome instability, that is, genome architecture incites genome instability. Nonallelic homologous recombination (NAHR) or crossing-over in meiosis between sequences that are not in allelic positions (i.e., paralogous sequences) can result in recurrent deletions or duplications causing genomic disorders. Previous studies of NAHR have focused on description of the phenomenon, but it remains unclear how NAHR occurs during meiosis and what factors determine its frequency. Here we assembled two patient cohorts with reciprocal genomic disorders; deletion associated Smith-Magenis syndrome and duplication associated Potocki-Lupski syndrome. By assessing the full spectrum of rearrangement types from the two cohorts, we find that complex rearrangements (those with more than one breakpoint) are more prevalent in copy-number gains (17.7%) than in copy-number losses (2.3%); an observation that supports a role for replicative mechanisms in complex rearrangement formation. Interestingly, for NAHR-mediated recurrent rearrangements, we show that crossover frequency is positively associated with the flanking low-copy repeat (LCR) length and inversely influenced by the inter-LCR distance. To explain this, we propose that the probability of ectopic chromosome synapsis increases with increased LCR length, and that ectopic synapsis is a necessary precursor to ectopic crossing-over.
Main Text
During the last two decades, studies of genomic disorders have uncovered mechanisms for generating human genomic rearrangements.1,2 One prevalent mechanism, nonallelic homologous recombination (NAHR), utilizes directly oriented paralogous low-copy repeat (LCR) substrates to produce recurrent reciprocal deletions and duplications by an ectopic crossover.3 Some evidence has suggested that genome-wide NAHR frequency might be proportional to the flanking LCR length but inversely proportional to the distance between the LCRs.1 Sperm PCR analyses of de novo germline rearrangement rates show that NAHR-generated deletions occur approximately twice as frequently as duplications.4 Nonrecurrent rearrangements, occurring where there is insufficient ectopic homology to allow NAHR, might happen by nonhomologous end joining (NHEJ) or by replicative mechanisms (e.g., fork stalling and template switching [FoSTeS] or microhomology-mediated break-induced replication [MMBIR]).5–7
One of the most extensively studied genomic disorders is Smith-Magenis syndrome (SMS [MIM 182290]). Early efforts investigating microdeletions causing SMS revealed NAHR as the major underlying rearrangement mechanism.8 The NAHR model predicts that the reciprocal duplication will also occur, and it was reported later and described as causative for another genomic disorder, Potocki-Lupski syndrome (PTLS [MIM 610883]).9,10 As human genome analyses and copy-number variation (CNV) detection methods improved in resolution and robustness, it was also shown that some PTLS duplications are nonrecurrent, and these duplications are generated by different mechanisms distinct from NAHR.11,12 However, there has been no comprehensive characterization of the molecular hallmarks of different SMS deletions.
In this report of 131 families with a child diagnosed with SMS, we investigated 131 de novo deletions at this chromosomal 17p11.2 region and compared their mechanisms to those from a cohort carrying the reciprocal duplications of the locus. The distribution and relative frequencies of various mechanisms contributing to deletions and duplications at the same locus provide insights into the nature of these rearrangement mechanisms. In addition to the two previously documented types of recurrent SMS and PTLS rearrangements,8,9,12,13 we now report a third type of recurrent SMS deletion and a PTLS duplication reciprocal to this deletion that occurs by utilizing yet a third set of paralogous LCRs as NAHR substrates. Analysis of the NAHR rates responsible for different recurrent rearrangements reveals a positive correlation with the flanking LCR length and suggests an inverse influence of the distance between LCRs.
A total of 131 patients who have a deletion involving the retinoic acid-induced gene 1 (RAI1 [MIM 607642]) were recruited after informed consent was procured; the study was approved by the institutional review board of Baylor College of Medicine. These samples were initially analyzed by pulsed-field gel electrophoresis,8 fluorescence in situ hybridization,14 bacterial artificial chromosome (BAC)-array comparative genomic hybridization (aCGH),15 or clinical oligonucleotide aCGH. The results indicate that 96 of them represented common recurrent (CR) deletions; characteristic features include deletion of a 3.6 Mb genomic interval flanked by the distal and proximal ∼170 kb, >97% sequence identical SMS-REP LCRs.16 Of the 35 remaining deletions, the breakpoint junctions have been previously sequenced in ten subjects; six subjects had uncommon recurrent deletions (or uncommon recurrent type 1 [UR1]), and four subjects had nonrecurrent deletions.13,17
We mapped the remaining 25 deletions by using Agilent targeted oligonucleotide based aCGH (Figure 1A). The array designs are in either a 4 × 44K format or a 4 × 180K format, interrogating chromosome 17p at a resolution of ∼500 bp or ∼200 bp. Three patients have array results consistent with UR1 deletions. Subjects BAB1190 and BAB1456 have apparently identical deletions, which probably represent recurrent deletions (uncommon recurrent type 2 [UR2]) because they are flanked by directly oriented paralogous LCRs. Simple nonrecurrent deletions ranging from 1.4 Mb to 8.4 Mb were identified in 15 subjects. Although subjects BAB540 and BAB2245 seem to have nearly identical losses by aCGH, they are still considered as nonrecurrent deletions because their deletion boundaries fall into two regions that cannot be interrogated by unique sequence probes in aCGH in which no apparent direct LCR pairs are located in the reference haploid human genome (hg18). However, we cannot rule out the possibility that the parents of these two subjects could carry direct LCRs flanking the deletions specific to their own personal genomes. Complex rearrangements, having more than one breakpoint, are observed in three subjects, BAB1221,18 1931, and 649. Breakpoint junctions of all nonrecurrent or complex deletions whose aCGH results show deletion boundaries mapping to regions that do not include large LCRs were amplified by PCR and sequenced (Figures 1B and 1C; Table 1). The breakpoint sequences represented the products of recombination whose features enable us to surmise the possible mechanisms that produced such deletions and also to categorize these deletions as being either complex or simple.
Figure 1.
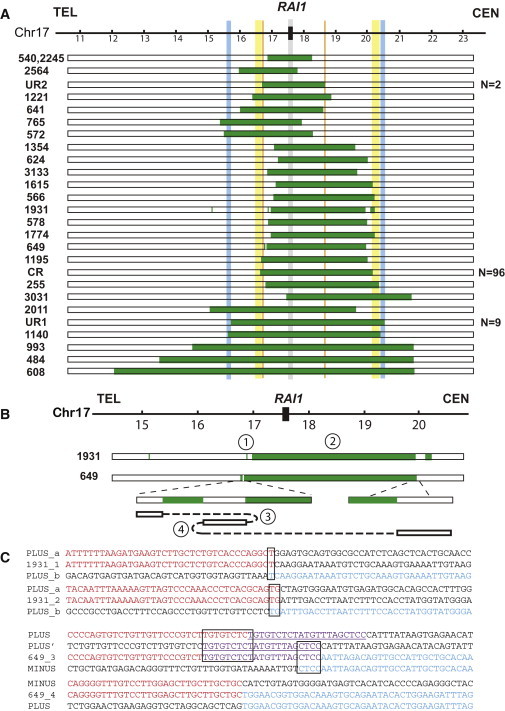
Summary of Rearrangement Results for 131 SMS Deletions
(A) A schematic representation of part of the human chromosome 17p (hg18) is illustrated as a horizontal line on the top of the figure with megabase pair genomic coordinates below. The critical region of the SMS deletion where the predominant dosage-sensitive critical gene RAI1 maps is indicated as a black rectangle and a vertical gray shadow. The vertical yellow, blue and orange shadow areas represent LCRs that mediate the CR, UR1, and UR2 SMS deletions. Below, horizontal bars depict the involved genomic intervals for each subject (BAB identification number on the left) from the interpretations of aCGH results: green or white bars represent deletion or normal copy, respectively. The recurrent deletions, flanked by directly oriented LCRs, are most likely produced by NAHR. The simple nonrecurrent or complex deletions have breakpoints not located in LCRs; they were most likely produced by mechanisms other than NAHR, such as NHEJ and/or replicative mechanisms such as FoSTeS or MMBIR. The totals for numbers of subjects with recurrent deletions are shown to the right of the aCGH interpretations.
(B) Breakpoint junctions and rearrangement structure in complex deletions BAB1931 and BAB649. The copy-number interpretations from aCGH results for both cases are shown at the top of this panel. For BAB649, the structure of the rearranged product is displayed under the aCGH interpretation. The normal copy segment between the two deletions is inverted. The breakpoint junctions are numbered corresponding to the breakpoint sequences listed below.
(C) Junction sequences are aligned to the reference sequence, and the transition between DNA sequences with different colors indicates the breakpoint interval. The black boxes outline microhomologies identified at the breakpoint junctions. The underlined purple sequences are the segments involved in one additional rearrangement step.
Table 1.
Breakpoint Features of Simple Nonrecurrent and Complex SMS Deletions
| BAB Number | Rearrangement Type | Breakpoint Feature |
|---|---|---|
| 624 | simple nonrecurrent | 3 bp microhomology |
| 2011 | simple nonrecurrent | 1 bp microhomology |
| 2564 | simple nonrecurrent | 4 bp microhomology |
| 3031 | simple nonrecurrent | 3 bp microhomology |
| 1774a | simple nonrecurrent | 4 bp microhomology |
| 765a | simple nonrecurrent | AluY-AluSg |
| 1354a | simple nonrecurrent | AluY-AluSc |
| 578 | simple nonrecurrent | No homology |
| 566a | simple nonrecurrent | 1 bp insertion |
| 649 | complex (three breakpoints sequenced) | 9 bp and 4 bp microhomologies; no homology |
| 1931 | complex (two breakpoints sequenced) | 1 bp and 2 bp microhomologies |
Breakpoint sequence reported in Shaw et al.17
Since the discovery of common recurrent SMS deletions and PTLS duplications, there have been on-going efforts to find additional types of recurrent rearrangements at this locus. Shaw et al.13 and Zhang et al.12 reported that an alternative pair of LCRs, consisting of 112 kb of ∼98% identity and termed 17pA/D,19 can act as substrates to generate recurrent reciprocal deletions and duplications, the UR1 deletion. In this study, we have identified yet a third type of recurrent deletion (found in BAB1190 and BAB1456) based on aCGH results, which we term the UR2 deletion (Figure 2A). The duplication reciprocal to this deletion was also identified in one subject (BAB3142) when we performed high-resolution genome analysis on more PTLS patients (Figure 2A). The LCRs flanking these rearrangements are ∼24 kb in length, share ∼98.6% identity, and are oriented in the same direction, thus representing directly oriented paralogous segments fulfilling criteria for NAHR substrate pairs. Three additional copies of these 24 kb paralogous LCRs exist in 17p11.2. Allele-specific PCR enabled us to determine the precise crossover interval in BAB1456. The crossover occurred at genome position chr17:16,541,605-16,541,718 (human genome assembly hg18), approximately 1.3 kb and 1.2 kb proximal to two homologous recombination (HR) hotspot motifs and representing potential binding sites for a PR domain containing protein 9 (PRDM9 [MIM 609760])20–23 (Figure 2A). Allele-specific PCR did not successfully map the breakpoint regions in BAB1190 and 3142 probably because of polymorphisms within the 24 kb LCR in these two samples.
Figure 2.
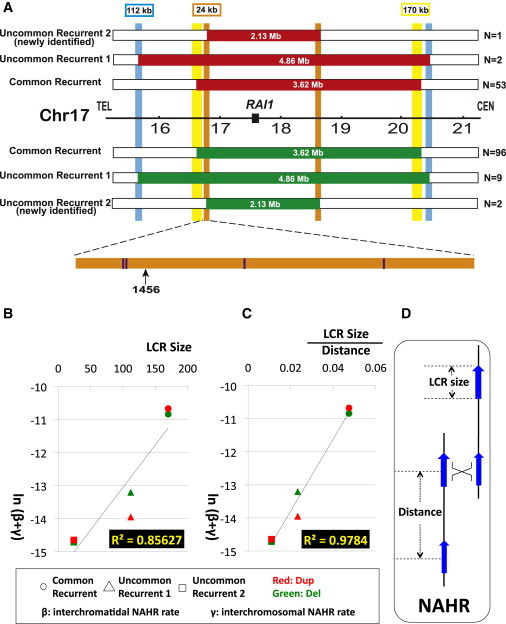
The NAHR Frequency Varies as the LCR Length and Is Inversely Influenced by Inter-LCR Distance
(A) Identification of the type 2 uncommon recurrent SMS deletion and PTLS duplication. The schematic graph of the newly identified uncommon recurrent type 2 (UR2) deletion (green) and duplication (red) are compared to the common recurrent (CR) and uncommon recurrent type 1 (UR1) rearrangements. The lengths of the flanking LCRs are marked above the LCRs. The inter-LCR distances are listed inside the rearranged bars. One copy of the 24 kb LCRs is expanded to show the exact crossover regions. The black arrow indicates where the crossover occurs in BAB1456. The positions of the 13-mer HR hotspot motif (CCNCCNTNNCCNC)20 are highlighted as purple vertical bars.
(B–C) The natural logarithm of calculated intermolecular NAHR rates in male meiosis as a function of (B) LCR length or (C) LCR length divided by inter-LCR distance. Interchromatidal NAHR (β) is much less frequent than interchromosomal NAHR (γ) in male meiosis.4 In other words, intermolecular NAHR events (β + γ) occur predominantly by interchromosomal recombinations (γ), and their frequency probably reflects interactions between homologous chromosomes that pair at synapsis. The strongest correlation for the data appears to occur when both LCR length and distance between LCRs are taken into account; i.e., when LCR length divided by inter-LCR distance is the variable. Coordinates for each dot were calculated with data from Tables 2 and 3.
(D) Definition of LCR length and distance. The inter-LCR distance is length of the segment in between LCRs plus the length of one LCR.
The relative contributions of deletions and duplications to genomic disorders vary with the nature of the process that produces them. With rearrangement data from the SMS cohort in this study and the PTLS duplication cohort of 79 index patients (74 previously reported patients12 and five new patients, Figure S1, available online), we are able to examine the ratios of de novo deletions versus duplications occurring by diverse mechanisms (Table 2). Both the 17p11.2 deletions and duplications cause fully penetrant genomic disorders, SMS and PTLS, respectively. There are no data suggesting any strong ascertainment bias between deletions (i.e., SMS) versus duplications (i.e., PTLS).
Table 2.
Distribution of Recurrent, Simple Nonrecurrent, and Complex Rearrangements in the Deletion and Duplication Cohorts
|
Observed Types |
Recurrent |
Simple Nonrecurrent |
Complex |
Total |
|---|---|---|---|---|
| Mechanisms | NAHR | FoSTeS, MMBIR, or NHEJ | FoSTeS, MMBIR, or multiple NHEJ | |
| Deletions | 107 (81.7%) | 21 (16.0%) | 3 (2.3%) | 131 |
| Duplications | 56 (70.9%) | 9 (11.4%) | 14 (17.7%) | 79 |
The generally accepted model for the mechanism of formation of recurrent rearrangements, that is deletions or duplications flanked by paralogous LCRs, is NAHR. It is possible that break-induced replication (BIR)24 can contribute to recurrent rearrangements. However, if these rearrangements occur in meiosis, homologous recombination repair of two-ended double-strand breaks is more likely than BIR (i.e., repair of one-ended breaks). According to the NAHR model, deletions are expected to occur de novo more frequently than duplications because intrachromatidal NAHR can produce only deletions, whereas interchromatidal and interchromosomal NAHR can mediate both deletions and duplications.3 Sperm PCR analysis assaying for de novo mutation rates at three different autosomal loci, including the crossovers responsible for UR1, showed that NAHR-driven deletions occur approximately twice as frequently as the reciprocal duplications.4 In this current report, there are 107 deletions and 56 duplications that clearly occurred by the NAHR mechanism. The proportion of the numbers of patients with deletions versus duplications is likely to reflect the relative occurrence, because our SMS and PTLS populations were ascertained from similar referral populations and we assume no disease-specific ascertainment or selection biases. The deletion to duplication ratio observed in the patient population is approximately 1.9:1; similar to the 2.14:1 observation for the de novo NAHR events at the UR1 locus from sperm PCR analysis (two-tailed exact binominal test of goodness-of-fit, p = 0.502). The ratios of deletion to duplication for CR1 was 96:53 or 1.8:1 (p = 0.334); for UR1 it was 9:2 or 4.5:1 (p = 0.52) and that for UR2 was 2:1 (p = 1). Hence, our analysis with data from SMS and PTLS patients supports the relative ratios for NAHR-derived deletion versus duplication rearrangements provided by observation of de novo events in normal males from the sperm PCR data. Furthermore, our patient population data suggest that disease prevalence for these sporadic genomic disorders reflects the rate of new mutation for deletion relative to duplication (i.e., ∼2:1), suggesting that the pathogenic SMS deletions and PTLS duplications undergo similar selection pressures after the mutations are formed during spermatogenesis.
Complex rearrangements are those that include more than one breakpoint or novel junction. FoSTeS, MMBIR and other replication-based mechanisms have been proposed to explain complex rearrangements.25 However, the proportions of the complex rearrangements these replication-based mechanisms can account for is unknown. Other potential mechanisms, such as multiple NHEJ events, can potentially explain features observed with complex rearrangements. Strikingly, when comparing the prevalence of complex rearrangements between the deletion and duplication cohorts (Table 2), the frequency in duplications (14/79, 17.7%) is significantly higher than that in deletions (3/131, 2.3%) (two-tailed Fisher's exact test, p = 1.2 × 10−4). When using the number of nonrecurrent rearrangements instead of all the rearrangements as the denominator (i.e., excluding NAHR-mediated recurrent rearrangements), the prevalence of complex rearrangements is still significantly higher in duplications (14/23) than in deletions (3/24) (p = 7.8 × 10−4). One might argue that more complex deletions are mechanistically possible but that larger deletions are not observed because of lethality. To account for this theoretical possibility, we excluded the duplications (three simple and six complex duplications) that have copy-number gains extending beyond the largest deletion, in BAB608, and repeated the statistical analysis. The difference (8/14 for duplications versus 3/24 for deletions) remains significant (p = 7.6 × 10−3). Thus, complex rearrangements are observed more with gains than with losses.
This propensity for copy-number gains versus losses in complex rearrangements is consistent with a characteristic attribute of the replication-based rearrangement mechanism. The replicative mechanism is an additive process that introduces genomic copy-number changes either by failing to copy or over-copying lengths of sequence, that is a loss or deletion represents a forward template switch that omits a length of sequence template, whereas duplication, triplication, and amplification represent an iterative process of copying the same genomic interval more than once.7 The template switch mechanism cannot delete segments from the replication product once they are formed. If a deletion is produced early in fork progression from an origin of replication, it can be recovered by template switching to upstream of the deletion, and it can be further converted into a duplication by additional backward template switches; in contrast, a duplication or a higher copy-number gain cannot be erased by a replication-based mechanism during the round of replication in which it was generated because it is now in cis with the DNA end that is switching templates.
Other mechanisms that might generate complex rearrangement do not have properties that favor gains over losses. For example, NAHR has the preference to generate deletions, as discussed in a previous section. In principle, NHEJ can generate a duplication by using a genomic fragment from a homolog or sister, but NHEJ cannot readily explain triplication or any other iterative process resulting in amplification. Although there are no experimental data on the frequency of products generated by multiple NHEJ events, it is conceivable that NHEJ contributes to a simple deletion-generating mechanism but also distinctly plausible that such events represent products of a replicative mechanism utilizing a single template switch.11,26,27
Benefitting from access to a large collection of de novo rearrangements at a single locus, we can investigate relative contributions of distinct paralogous substrates to NAHR events by using a statistical approach. With the newly discovered UR2 deletions and duplication in this report, we now have documented a total of six types of recurrent rearrangements at the human 17p11.2 locus associated with either SMS or PTLS, both deletions and duplications of CR, UR1, and UR2. We assume that their relative prevalence in our patient cohort is in proportion to the corresponding NAHR frequency in spermatogenesis, that is the selection pressures for CR, UR1, and UR2 deletions and duplications are comparable, and NAHR rates for deletions versus duplications at this locus in sperm are representative of de novo germline events. The empirical data for the UR1 duplication frequency in sperm4 are used to estimate male germline NAHR rates for the other five recurrent rearrangements (Table 3). Note that individual types of recurrent rearrangements exhibit divergent frequencies of occurrence.
Table 3.
LCR Length, Distance, and Estimated NAHR Rates in Male Meiosis for the Recurrent Rearrangements
| Rearrangement Type | LCR Length (kb) | Distance (kb) | Number of Patients Observed | Calculated NAHR Rate (α + β + γ) | Calculated NAHR Rate (β + γ) | |
|---|---|---|---|---|---|---|
| Common recurrent | deletion | 170 | 3586 | 96 | 4.20 × 10−5 | 1.96 × 10−5 |
| Common recurrent | duplication | 170 | 3586 | 53 | 2.32 × 10−5 | 2.32 × 10−5 |
| Uncommon recurrent 1 | deletion | 112 | 4802 | 9 | 3.93 × 10−6 | 1.84 × 10−6 |
| Uncommon recurrent 1 | duplication | 112 | 4802 | 2 | 8.74 × 10−7 a | 8.74 × 10−7 |
| Uncommon recurrent 2 | deletion | 24 | 2179 | 2 | 8.74 × 10−7 | 4.08 × 10−7 |
| Uncommon recurrent 2 | duplication | 24 | 2179 | 1 | 4.37 × 10−7 | 4.37 × 10−7 |
See Table S1 for details of LCR length calculation. a: This frequency is obtained from empirical data from sperm PCR analysis.4 The other frequencies in the same column are calculated based on the ratio of the observed prevalence (number of patients) relative to the prevalence of UR1 duplication. The following abbreviations are used: α, intrachromatidal NAHR rate; β, interchromatidal NAHR rate; γ, interchromosomal NAHR rate.
We next explored the relationship between the frequency of an NAHR rearrangement and the sizes of flanking LCRs and/or inter-LCR distance. The NAHR deletion rates are not directly comparable to those of duplications in that crossovers leading to deletions consist of intrachromatidal (α), interchromatidal (β), and interchromosomal (γ) rearrangements, whereas intrachromatidal crossovers (α) do not contribute to duplications.4 To reconcile this difference, the intermolecular NAHR rates (β + γ) were calculated for the deletions. The calculations are based on the hypothesis that CR and UR2 have similar (α + β + γ):(β + γ) ratios to UR1, which has been estimated as 2.14:1 for this region.4
We first calculated NAHR (β + γ) rates versus LCR length and observe a significant correlation (Figure 2B, correlation coefficient = 0.85627). No significant correlation is observed for rates versus distance between LCR pairs. However, of even greater interest, when we plotted NAHR (β + γ) rates against LCR lengths and distances by using various functions such as linear, power, exponential relationships or a combination thereof, we observe even greater correlations. Our data show the strongest correlation (Figure 2C, correlation coefficient = 0.9784) when LCR length is divided by inter-LCR distance and plotted against the logarithm of the estimated frequencies of intermolecular NAHR rate (β + γ).
It is not immediately apparent how the observed frequency of a HR mechanism might depend on substrate lengths beyond the minimal efficient processing segment (MEPS)28–30 required for HR. Nor is it obvious why linear distance between substrates might influence the event frequencies. If these crossovers occur as HR events during meiosis, our results suggest that the relationship might be explained by considering ectopic synapsis as a precursor to ectopic crossing-over. We suggest that the dependence on inter-LCR distance could reflect the declining probability of a successful 3D search of two ended double-strand breaks (DSB) generated prior to synaptonemal complex formation.31 The probability of synapsis, we propose, might depend on the length of LCR because the probability of presynaptic contacts will be increased. Thus, one interpretation of our observed LCR length dependency for NAHR frequency and inverse influence of inter-LCR distance is that they might reflect the probability of ectopic synapsis formation. Ectopic synapsis provides the necessary components for ectopic crossing-over,32 and the probability of a crossover might be further enhanced by the presence of PRDM9-mediated hotspots.21–23,33 In support of the potential role of PRDM9, we identified two PRDM9 binding motifs (i.e., the 13-mer HR hotspot motif) ∼1 kb away from the 114 bp crossover interval of UR2 in this study (Figure 2A); UR1 and CR have also been shown to have PRDM9 binding motifs within the empirically defined hotspot for crossing-over.12,34 Although it is possible that these recombination events can occur premeiotically, the current experimental evidence, including segregation of marker genotypes,35 measurements by pooled sperm PCR4 and the presence of PRDM9 binding site motif(s) in the NAHR hotspot interval,12 favors most of these recurrent 17p11.2 rearrangements being meiotic. Because these crossovers are flanked by repeats and are therefore probably produced by homology driven mechanisms, they are more likely to occur in meiosis when homologous recombination and crossing-over occur at high levels.
Interestingly, similar correlation patterns have been observed in rearrangements on the Y chromosome.36 Different LCR pairs, such as b2/b4, b2/b3, and gr/gr, can be used as NAHR substrates to produce these rearrangements. Individuals carrying a haplotype with a polymorphic CNV deletion that results in a reduced b2-b4 inter-LCR distance present increased frequency of b2/b4 deletion, the latter associated with male infertility.37 Comparing NAHR rates with alternative LCR pairs as substrates, b2/b3 versus gr/gr, supports the contention that the rearrangement formation frequency is positively associated with LCR length.38 Further insights into how either LCR length or inter-LCR distance might influence crossovers at allelic versus ectopic positions could be provided from studies at other loci. It should be noted that the degree of homology between LCR pairs might also play a role in determining NAHR rates. In our case, the percent identities of the LCR substrate pairs are similar (∼98%) for the NAHR-generated CR, UR1, and UR2 rearrangements (Table S1).
These data encourage a model of meiotic recombination (Figure 3) that offers a solution to the long-standing problem of why crossing-over can occur ectopically in meiosis.39 We propose that numerous presynaptic contacts are made by processed ends from programmed double-strand breaks.40 These ends search through neighboring space and pair with regions of limited homology in ectopic as well as allelic positions.41,42 Most ectopic interactions are dispersed by becoming noncrossover events, possibly by the mechanism of synthesis-dependent strand-annealing,31,32,43 which leads only to noncrossovers.44 We postulate that a certain threshold number or density of nearby presynaptic contacts is required to establish synapsis by the synaptonemal complex.31 Synaptonemal complex formation proceeds from subtelomeric regions toward the centromere, with the centromere acting as a barrier.45 This barrier might perhaps facilitate ectopic synapsis in pericentromeric regions, such as 17p11.2. Synapsis allows crossing-over,31,32,41,46 which is thus regularized to occur between tracts of extensive homology, usually homology in allelic positions, thus allowing regularized pairing and segregation of homologous bivalent chromosomes in the first meiotic division. Ectopic synapsis for linked loci could potentially be influenced by inter-LCR distance; however, we see no correlation between crossover frequency and inter-LCR distance alone.
Figure 3.
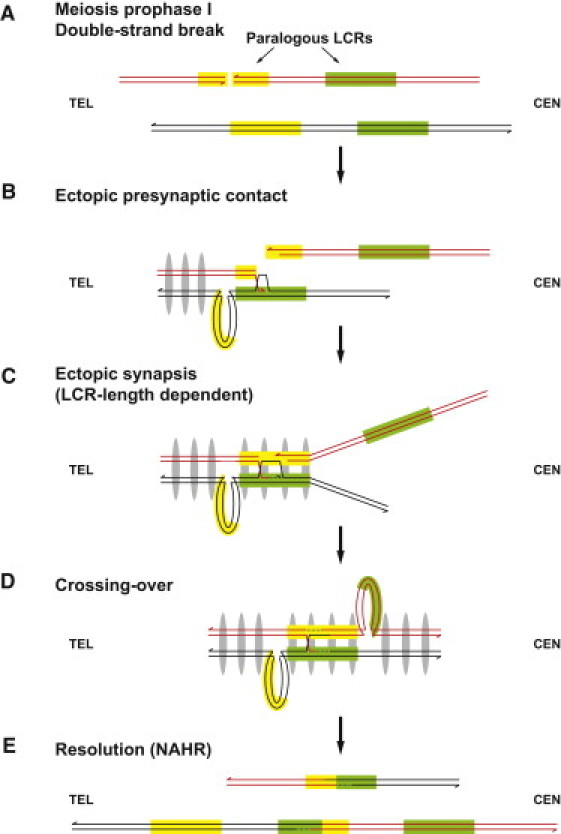
Ectopic Synapsis Model
(A) In meiosis prophase I, a double-strand break (DSB) occurs on one LCR. The yellow and green rectangles indicate paralogous LCRs.
(B) The processed double-strand end searches the neighboring regions, making ectopic presynaptic contacts. The presence of gray ellipses indicates establishment of synapsis, which proceeds from subtelomeric regions toward centromeres.
(C) Ectopic synapsis is formed after a certain number or density of presynaptic contacts is made. The successful establishment of ectopic synapsis is dependent upon the length of ectopic homology.
(D) After ectopic synapsis is set up, crossing-over occurs between nonallelic LCRs.
(E) Resolution can lead to NAHR.
This model predicts that, because the probability of achieving a sufficient number of presynaptic contacts will vary as the length of homology, establishing an ectopic crossover will also vary directly as the length of homologous sequence. Once synapsis is established, ectopic crossing-over can occur by the same mechanism as AHR, a contention supported by the observation that AHR and NAHR share common features,47 including association with identical hotspot motifs.48 In summary, recurrent duplications and deletions are formed during meiosis because ectopic homology allows ectopic presynaptic contacts; extensive ectopic homology in the case of a large LCR allows a sufficient number of presynaptic contacts to allow synaptonemal complex formation, which, in turn, allows crossovers to form.
In conclusion, a variety of mechanisms can generate copy-number gains and losses at a given locus. NAHR can be the predominant mechanism when there are nearby LCRs, and different pairs can be utilized. Nevertheless, the probability of which LCR pair is used correlates positively with LCR length and might be inversely influenced by the distance between repeats. Copy-number gains show greater complexity than losses, consistent with a major contribution of replicative mechanisms to the formation of copy-number change.
Acknowledgments
We thank all the participating subjects and families for their time, efforts, and collaboration; the referring physicians and genetic counselors for sample acquisition; and Weimin Bi, Wenli Gu, and Pawel Stankiewicz for comments. This work was supported in part by the SMS Research Foundation and the National Institutes of Health, National Institute of Neurological Disorders and Stroke grant R01NS058529 to J.R.L., the National Institutes of General Medical Science grant R01GM064022 to P.J.H., Texas Children's Hospital General Clinical Research Center grant M01RR00188, and the Intellectual and Developmental Disabilities Research Centers grant P30HD024064. J.R.L. is a consultant for Athena Diagnostics and Ion Torrent Systems, holds stock ownership of 23andMe, and is a coinventor on multiple US and European patents for DNA diagnostics. The Baylor College of Medicine and Department of Molecular and Human Genetics derive revenue from diagnostic testing provided by the Medical Genetics Laboratories.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Medical Genetics Laboratories, Baylor College of Medicine, https://www.bcm.edu/geneticlabs
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Lupski J.R. Genomic disorders: Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 2.Lupski J.R. Genomic disorders ten years on. Genome Med. 2009;1:42. doi: 10.1186/gm42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stankiewicz P., Lupski J.R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 4.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slack A., Thornton P.C., Magner D.B., Rosenberg S.M., Hastings P.J. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006;2:e48. doi: 10.1371/journal.pgen.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 7.Hastings P.J., Ira G., Lupski J.R. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen K.S., Manian P., Koeuth T., Potocki L., Zhao Q., Chinault A.C., Lee C.C., Lupski J.R. Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat. Genet. 1997;17:154–163. doi: 10.1038/ng1097-154. [DOI] [PubMed] [Google Scholar]
- 9.Potocki L., Chen K.S., Park S.S., Osterholm D.E., Withers M.A., Kimonis V., Summers A.M., Meschino W.S., Anyane-Yeboa K., Kashork C.D. Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat. Genet. 2000;24:84–87. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- 10.Potocki L., Bi W., Treadwell-Deering D., Carvalho C.M., Eifert A., Friedman E.M., Glaze D., Krull K., Lee J.A., Lewis R.A. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang F., Potocki L., Sampson J.B., Liu P., Sanchez-Valle A., Robbins-Furman P., Navarro A.D., Wheeler P.G., Spence J.E., Brasington C.K. Identification of uncommon recurrent Potocki-Lupski syndrome-associated duplications and the distribution of rearrangement types and mechanisms in PTLS. Am. J. Hum. Genet. 2010;86:462–470. doi: 10.1016/j.ajhg.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw C.J., Withers M.A., Lupski J.R. Uncommon deletions of the Smith-Magenis syndrome region can be recurrent when alternate low-copy repeats act as homologous recombination substrates. Am. J. Hum. Genet. 2004;75:75–81. doi: 10.1086/422016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stankiewicz P., Shaw C.J., Dapper J.D., Wakui K., Shaffer L.G., Withers M., Elizondo L., Park S.S., Lupski J.R. Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am. J. Hum. Genet. 2003;72:1101–1116. doi: 10.1086/374385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw C.J., Shaw C.A., Yu W., Stankiewicz P., White L.D., Beaudet A.L., Lupski J.R. Comparative genomic hybridisation using a proximal 17p BAC/PAC array detects rearrangements responsible for four genomic disorders. J. Med. Genet. 2004;41:113–119. doi: 10.1136/jmg.2003.012831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park S.S., Stankiewicz P., Bi W., Shaw C., Lehoczky J., Dewar K., Birren B., Lupski J.R. Structure and evolution of the Smith-Magenis syndrome repeat gene clusters, SMS-REPs. Genome Res. 2002;12:729–738. doi: 10.1101/gr.82802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw C.J., Lupski J.R. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum. Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- 18.Park J.P., Moeschler J.B., Davies W.S., Patel P.I., Mohandas T.K. Smith-Magenis syndrome resulting from a de novo direct insertion of proximal 17q into 17p11.2. Am. J. Med. Genet. 1998;77:23–27. [PubMed] [Google Scholar]
- 19.Stankiewicz P., Shaw C.J., Withers M., Inoue K., Lupski J.R. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004;14:2209–2220. doi: 10.1101/gr.2746604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers S., Freeman C., Auton A., Donnelly P., McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 21.Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers S., Bowden R., Tumian A., Bontrop R.E., Freeman C., MacFie T.S., McVean G., Donnelly P. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science. 2010;327:876–879. doi: 10.1126/science.1182363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parvanov E.D., Petkov P.M., Paigen K. Prdm9 controls activation of mammalian recombination hotspots. Science. 2010;327:835. doi: 10.1126/science.1181495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrow D.M., Connelly C., Hieter P. “Break copy” duplication: A model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics. 1997;147:371–382. doi: 10.1093/genetics/147.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang F., Carvalho C.M., Lupski J.R. Complex human chromosomal and genomic rearrangements. Trends Genet. 2009;25:298–307. doi: 10.1016/j.tig.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bi W., Sapir T., Shchelochkov O.A., Zhang F., Withers M.A., Hunter J.V., Levy T., Shinder V., Peiffer D.A., Gunderson K.L. Increased LIS1 expression affects human and mouse brain development. Nat. Genet. 2009;41:168–177. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boone P.M., Liu P., Zhang F., Carvalho C.M., Towne C.F., Batish S.D., Lupski J.R. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet. Med. 2011;13:582–592. doi: 10.1097/GIM.0b013e3182106775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jinks-Robertson S., Michelitch M., Ramcharan S. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1993;13:3937–3950. doi: 10.1128/mcb.13.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liskay R.M., Letsou A., Stachelek J.L. Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics. 1987;115:161–167. doi: 10.1093/genetics/115.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiter L.T., Hastings P.J., Nelis E., De Jonghe P., Van Broeckhoven C., Lupski J.R. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 1998;62:1023–1033. doi: 10.1086/301827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleckner N. Chiasma formation: Chromatin/axis interplay and the role(s) of the synaptonemal complex. Chromosoma. 2006;115:175–194. doi: 10.1007/s00412-006-0055-7. [DOI] [PubMed] [Google Scholar]
- 32.Hastings P.J. Mechanisms of Ectopic Gene Conversion. Genes. 2010;1:427–439. doi: 10.3390/genes1030427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berg I.L., Neumann R., Lam K.W., Sarbajna S., Odenthal-Hesse L., May C.A., Jeffreys A.J. PRDM9 variation strongly influences recombination hot-spot activity and meiotic instability in humans. Nat. Genet. 2010;42:859–863. doi: 10.1038/ng.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bi W., Park S.S., Shaw C.J., Withers M.A., Patel P.I., Lupski J.R. Reciprocal crossovers and a positional preference for strand exchange in recombination events resulting in deletion or duplication of chromosome 17p11.2. Am. J. Hum. Genet. 2003;73:1302–1315. doi: 10.1086/379979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw C.J., Bi W., Lupski J.R. Genetic proof of unequal meiotic crossovers in reciprocal deletion and duplication of 17p11.2. Am. J. Hum. Genet. 2002;71:1072–1081. doi: 10.1086/344346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carvalho C.M., Zhang F., Lupski J.R. Structural variation of the human genome: Mechanisms, assays, and role in male infertility. Syst Biol Reprod Med. 2011;57:3–16. doi: 10.3109/19396368.2010.527427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang F., Lu C., Li Z., Xie P., Xia Y., Zhu X., Wu B., Cai X., Wang X., Qian J. Partial deletions are associated with an increased risk of complete deletion in AZFc: A new insight into the role of partial AZFc deletions in male infertility. J. Med. Genet. 2007;44:437–444. doi: 10.1136/jmg.2007.049056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu C., Zhang J., Li Y., Xia Y., Zhang F., Wu B., Wu W., Ji G., Gu A., Wang S. The b2/b3 subdeletion shows higher risk of spermatogenic failure and higher frequency of complete AZFc deletion than the gr/gr subdeletion in a Chinese population. Hum. Mol. Genet. 2009;18:1122–1130. doi: 10.1093/hmg/ddn427. [DOI] [PubMed] [Google Scholar]
- 39.Lichten M., Borts R.H., Haber J.E. Meiotic gene conversion and crossing over between dispersed homologous sequences occurs frequently in Saccharomyces cerevisiae. Genetics. 1987;115:233–246. doi: 10.1093/genetics/115.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keeney S., Neale M.J. Initiation of meiotic recombination by formation of DNA double-strand breaks: Mechanism and regulation. Biochem. Soc. Trans. 2006;34:523–525. doi: 10.1042/BST0340523. [DOI] [PubMed] [Google Scholar]
- 41.Haber J.E., Leung W.Y., Borts R.H., Lichten M. The frequency of meiotic recombination in yeast is independent of the number and position of homologous donor sequences: Implications for chromosome pairing. Proc. Natl. Acad. Sci. USA. 1991;88:1120–1124. doi: 10.1073/pnas.88.4.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldman A.S., Lichten M. The efficiency of meiotic recombination between dispersed sequences in Saccharomyces cerevisiae depends upon their chromosomal location. Genetics. 1996;144:43–55. doi: 10.1093/genetics/144.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hastings P.J., Lupski J.R., Rosenberg S.M., Ira G. Mechanisms of change in gene copy number. Nat. Rev. Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pâques F., Haber J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown P.W., Judis L., Chan E.R., Schwartz S., Seftel A., Thomas A., Hassold T.J. Meiotic synapsis proceeds from a limited number of subtelomeric sites in the human male. Am. J. Hum. Genet. 2005;77:556–566. doi: 10.1086/468188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zickler D., Kleckner N. Meiotic chromosomes: Integrating structure and function. Annu. Rev. Genet. 1999;33:603–754. doi: 10.1146/annurev.genet.33.1.603. [DOI] [PubMed] [Google Scholar]
- 47.Lupski J.R. Hotspots of homologous recombination in the human genome: Not all homologous sequences are equal. Genome Biol. 2004;5:242. doi: 10.1186/gb-2004-5-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Myers S.R., McCarroll S.A. New insights into the biological basis of genomic disorders. Nat. Genet. 2006;38:1363–1364. doi: 10.1038/ng1206-1363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.