

Abstract
Severe combined deficiency of the 2-oxoacid dehydrogenases, associated with a defect in lipoate synthesis and accompanied by defects in complexes I, II, and III of the mitochondrial respiratory chain, is a rare autosomal recessive syndrome with no obvious causative gene defect. A candidate locus for this syndrome was mapped to chromosomal region 2p14 by microcell-mediated chromosome transfer in two unrelated families. Unexpectedly, analysis of genes in this area identified mutations in two different genes, both of which are involved in [Fe-S] cluster biogenesis. A homozygous missense mutation, c.545G>A, near the splice donor of exon 6 in NFU1 predicting a p.Arg182Gln substitution was found in one of the families. The mutation results in abnormal mRNA splicing of exon 6, and no mature protein could be detected in fibroblast mitochondria. A single base-pair duplication c.123dupA was identified in BOLA3 in the second family, causing a frame shift that produces a premature stop codon (p.Glu42Argfs∗13). Transduction of fibroblast lines with retroviral vectors expressing the mitochondrial, but not the cytosolic isoform of NFU1 and with isoform 1, but not isoform 2 of BOLA3 restored both respiratory chain function and oxoacid dehydrogenase complexes. NFU1 was previously proposed to be an alternative scaffold to ISCU for the biogenesis of [Fe-S] centers in mitochondria, and the function of BOLA3 was previously unknown. Our results demonstrate that both play essential roles in the production of [Fe-S] centers for the normal maturation of lipoate-containing 2-oxoacid dehydrogenases, and for the assembly of the respiratory chain complexes.
Introduction
Pyruvate dehydrogenase complex (PDHc) deficiency most often occurs as an isolated enzyme defect caused by mutations in X-linked PDHA1 (MIM 300502) and produces a spectrum of clinical presentations ranging from fatal infantile lactic acidosis to mild psychomotor retardation.1,2 A smaller number of PDHc deficient individuals have mutations in PDHB (MIM 179060), PDHX (MIM 608769), dihydrolipoyl transacetylase (DLAT, MIM 608770), dihydrolipoyl dehydrogenase (DLD, MIM 238331), or PDP1 (MIM 605993), which controls the reactivation of phosphorylated PDHc.3–5 We have described two unrelated families with neonatal, severe deficiencies of PDHc associated with defects in oxoglutarate dehydrogenase complex (OGDHc) and with complexes I, II, and III of the respiratory chain, and we called this multiple mitochondrial dysfunctions syndrome (MIM 605711, Table 1). Using the technique of microcell-mediated chromosome transfer, we defined a candidate locus on chromosome 2p14 for both families.6 The locus covers 10 Mb of sequence in a region containing more than 50 genes, many of which have no defined function. We sequenced a number of genes as candidates for this syndrome and focused on genes with a high probability of targeting to the mitochondria (determined with Mitoprot7). Two mitochondrial genes involved in iron-sulfur ([Fe-S]) cluster assembly were likely candidates for containing mutations that could cause the multienzyme deficiency. This deduction was based on the presence of [Fe-S] clusters in protein components of all the affected respiratory chain enzyme complexes.
Table 1.
Relative Mitochondrial Enzyme Activities for NFU1, BOLA3, and ISCU Mutant Fibroblasts
| Enzyme | [Fe-S] Clusters | NFU1 Mutant Individuals6 | BOLA3 Mutant Individual6 | ISCU Mutant Individuals13–15 |
|---|---|---|---|---|
| Complex I (I+III) | [2Fe-2S] (×2), [4Fe-4S] (×6) | ∼30% | ∼30% | ∼70% |
| Complex II | [2Fe-2S], [3Fe-4S], [4Fe-4S] | 50% | 50% | ∼20% |
| Complex III | [2Fe-2S] | ∼60% | ∼60% | ∼40% |
| Complex IV | ∼50% in 2 sibs, normal in 1 sib | normal | normal | |
| Mitochondrial Aconitase | [4Fe-4S] | normal | normal | ∼30% |
| PDHc | requires covalently attached lipoate on E2 | ∼5−10% | ∼5−10% | ND |
| E2 | requires covalently attached lipoate on E2, but assay works with added lipoic acid | normal | ND | ND |
| E3 | normal | ND | ND | |
| OGDHc | lipoate requiring | ∼10% | ∼30% | ND |
ND is used as an abbreviation for not determined.
In this study, we show that two genes involved in the biogenesis of [Fe-S] clusters have profound effects on the respiratory chain and 2-oxoacid dehydrogenase complexes (Figure 1).
Figure 1.
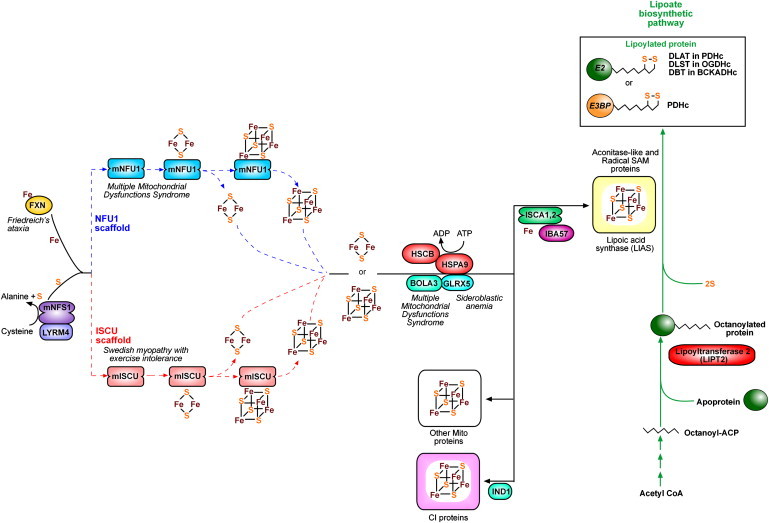
Proposed Scheme for the Interaction of Human [Fe-S] Genes within the Mitochondria
mNFS1 produces elemental sulfur from cysteine and makes it available to mISCU by interaction with this scaffold protein. LYRM4 is required for stabilization of mNFS1. Fe is provided by the chaperone frataxin (FXN) through an interaction with LYRM4 and mISCU, and [2Fe-2S] clusters are assembled upon each monomer of mISCU. These are then combined into [4Fe-4S] clusters. Ferredoxin provides the necessary reducing agents for these reactions from electrons initially provided by ferredoxin reductase (not shown). Cochaperones HSCB, HSPA9, and GLRX5 bind to ISCU and aid in the transfer of [2Fe-2S] clusters into recipient proteins. Only GLRX5 is required as a chaperone for [4Fe-4S] transfer. BOLA3 possibly functions by interacting with GLRX5. ISCA1,2 and IBA57 are required for the maturation of aconitase-like proteins and the catalytic activation of radical SAM enzymes (e.g., lipoic acid synthase). IND1 is a complex-I-specific assembly factor. mNFU1 is thought to act as an alternate scaffold, possibly for the assembly of a subset of [Fe-S] proteins.
The E3BP and E2 subunits have covalently bound lipoic acid, which requires a complex synthesis process involving transfer of an octanoyl-ACP (derived from fatty acid biosynthesis) onto an apoprotein by LIPT2. LIAS catalyzes the lipoylation of the octanoylated apo-protein by using SAM and sulfur (potentially produced by the action of mNFS1). Ferredoxin (FDX1) is thought to provide the reducing power. ISCA1,2 and IBA57 are required for catalytic activation of LIAS. Lipoylated E2 is present as dihydrolipoamide S-acetyltransferase (DLAT) in PDHc, dihydrolipoamide S-succinyltransferase (DLST) in OGDH and dihydrolipoamide branched chain transacetylase (DBT) in BCKADHc. Lipoylated E3-binding protein is present in PDHc. Diseases associated with defective mammalian proteins are noted in italics.
Materials and Methods
Families
Both families have been described in a previous publication.6 Briefly, the family with the NFU1 mutation comprises two affected males, one affected female, and one unaffected female (Figure 2). The parents are nonconsanguineous and of Mexican descent. The family with the BOLA3 mutation comprises one affected male and one unaffected male (Figure 3). The parents are first cousins and of East Indian descent. Enzymatic deficiencies of the respiratory chain and 2-oxoacid dehydrogenases are summarized in Table 1. All procedures were carried out with approval from The Hospital for Sick Children's research ethics board.
Figure 2.
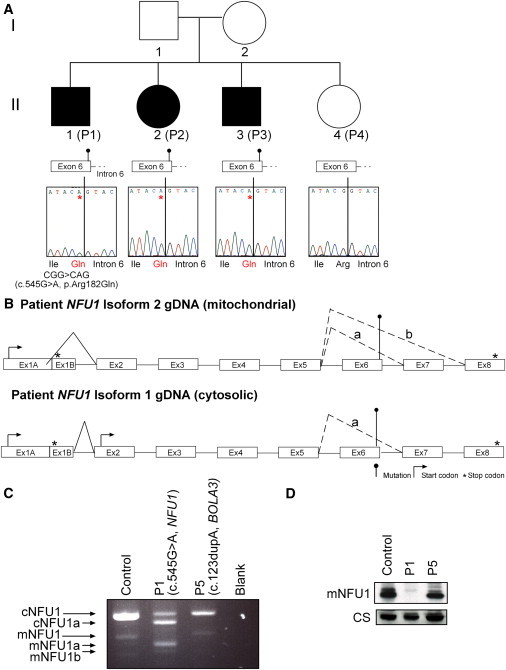
Analysis of NFU1 in Family 1
(A) Sequence chromatograms are shown for gDNA regions of NFU1. The c.545G>A (p.Arg182Gln) mutation can be seen in affected individuals P1–P3 (II-1, II-2 and II-3); there is no mutation in unaffected sibling P4 (II-4).
(B) The genomic structure of isoform 1 (cytosolic) and isoform 2 (mitochondrial) NFU1 is shown for individuals P1–P3 (II-1, II-2, and II-3). The c.545G>A mutation is identified. The normal alternate splicing that creates the two isoforms from the gDNA sequence is shown with solid lines, and the effect of the mutation on the splice site between exons 5 and 6 is demonstrated with dashed lines, yielding three possible transcripts.
(C) PCR amplification from individual P1 (II-1, NFU1 mutation), P5 (II-1 in Figure 3, BOLA3 mutation), and control cDNA. In the control and individual P5 cDNA, two transcripts representing the cytosolic and mitochondrial isoforms can be seen. In individual P1, there is almost no full-length mitochondrial NFU1, and two smaller transcripts are present (a and b). There is less cytosolic NFU1 in individual P1, with one transcript variant visible (a). The mutant transcripts a and b are all identified in (A) and were all verified by sequencing.
(D) Immunoblot of mitochondria showing the mitochondrial isoform of NFU1 with citrate synthase as a loading control.
Figure 3.
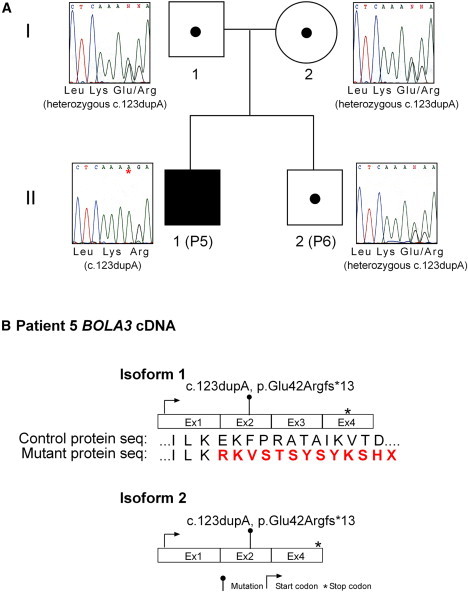
Analysis of BOLA3 in Family 2
(A) Sequence chromatograms are shown for gDNA regions of BOLA3. The single base pair duplication (c.123dupA) can be seen in individual P5 (II-1). The parents (I-1, I-2) and unaffected sibling P6 (II-2) are all heterozygous. There is no mutation in NFU1 mutant individual P1 (II-1 in Figure 2).
(B) The two isoforms of BOLA3 cDNA are shown with the mutation seen in P5 (II-1) highlighted.
Cell Culture
Cultured skin fibroblasts were grown from forearm skin biopsy (taken with informed consent) in α-MEM culture medium (11 mM glucose) and 20% fetal calf serum (Wisent, Saint-Jean-Baptiste de Rouville, Quebec, Canada). Lymphoblasts were grown in 10% fetal calf serum/RPMI. Primary human skin fibroblasts were immortalized with a retrovirus expressing the E7 gene of type-16 human papilloma virus and a retroviral vector expressing the protein component (htert) of human telomerase.8 These cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum.
Molecular Genetic Techniques
Genomic DNA was isolated from fibroblasts or blood with the Puregene genomic DNA isolation kit (Inter Medico). RNA was isolated with Trizol, and full-length NFU1 cDNA sequence was reverse transcribed with Superscript II reverse transcriptase and amplified with Platinum Hi-Fi Taq polymerase (all from Invitrogen). Oligonucleotide primers used are shown in Table S1, available online. All PCR products were sequenced directly by fluorescent sequencing methods (ACGT Corporation, Toronto, Canada).
Retroviral vectors containing the HA-tagged cDNA sequence of the cytosolic and mitochondrial versions of NFU1 and isoforms 1 and 2 of BOLA3 were created with the Gateway cloning system (Invitrogen) as previously described.9 We amplified cDNAs from these genes by using RNA isolated from fibroblasts with the RNeasy kit (QIAGEN), by OneStep RT-PCR (QIAGEN) with specific primers modified for cloning into Gateway vectors (flanked with attB1 and attB2-HA sequences). The PCR constructs were cloned into a Gateway-modified retroviral expression vector pLXSH. The fidelity of cDNA clones was confirmed by automated DNA sequencing. Retroviral constructs were used to transiently transfect a Phoenix packaging cell line with the HBS/Ca3(PO4)2 method (see Web Resources). Affected and control fibroblasts were infected 48 hr later by exposure to virus-containing medium in the presence of 4 μg/ml of polybrene.
Immunoblotting and Blue Native Gel Electrophoresis
For immunoblot analysis, fibroblasts were used to prepare mitochondria.10 twenty-five micrograms of mitochondria was resolved through a 12.5% stacking SDS/PAGE gel. The SDS/PAGE gel was electroblotted onto polyvinylidene fluoride membrane, blocked with 5% skim milk/Tris buffered saline with Tween (TBST), and probed with antibodies. Anti-human citrate synthase and anti-holoCOX were prepared by immunizing rabbits with purified citrate synthase or bovine heart cytochrome oxidase. The anti-NFU1 was a gift from Tracey Rouault, National Institutes of Health. Antibodies to complex I subunits 75 kDa (NDUFS1), 51 kDa (NDUFV1), and 49 kDa (NDUFS2) were prepared by immunizing rabbits with protein peptides, and complex I 39 kDa (NDUFA9) and ASHI (NDUFB8), complex II SDHA, complex III UQCRFS1, and UQCRC1 antibodies were purchased from Mitosciences (Eugene, OR, USA). CYTB antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Lipoate antibody was purchased from Cedarlane (Burlington, ON, Canada) and anti-GAPDH from Abcam (Cambridge, MA, USA). Immunoreactive proteins were visualized with Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life and Analytical Sciences). Blue native gel electrophoresis was carried out as previously described.11 We carried out densitometry measurements by using ImageJ software.12
Immunofluorescence
Cells were rinsed two times with PBS, fixed with PBS 4% PFA for 20 min, and rinsed three times with PBS. Permeabilization was performed with PBS 0.1% Triton for 15 min followed by three rinses with PBS. Nonspecific antigenic sites were blocked with PBS 4% BSA for 30 min, and primary antibody were applied in blocking solution for 2 hr at room temperature. After three rinses in PBS, secondary antibody in blocking solution was applied for 1 hr and followed by three rinses with PBS. Nuclei were counterstained with DAPI, and cover glasses were mounted in Fluoromount. The following antibodies were used: rabbit anti-NFU1 (1:1000), mouse anti-SDHA (1:1000), mouse anti-HA (1:5000), goat anti-mouse Alexa488 (1:2000), goat anti-rabbit Alexa488 (1:2000), goat anti-mouse Alexa594 (1:2000), and goat anti-mouse Alexa594 (1:2000).
Results
In the locus defined by chromosome transfer at 2p14, we searched for a gene that might have effects on both 2-oxoacid dehydrogenase complexes and respiratory chain complexes. Because the metabolic pathways involving [Fe-S] cluster assembly are a likely common cause for such defects, we sequenced NFU1 (MIM 608100), an essential protein in one of the two pathways for [Fe-S] assembly in both families (Figures 1 and 2A). [Fe-S] clusters function as the prosthetic groups of many proteins essential for intermediary metabolism and oxidative phosphorylation. They are constructed and assembled into proteins in a series of complex biochemical reactions that appear to run in two parallel pathways (Figure 1). Both ISCU (MIM 611911) and NFU1 can assemble [2Fe-2S] and [4Fe-4S] clusters, although only a human defect in ISCU has been noted before; this defect causes Swedish myopathy with exercise intolerance (MIM 255125).13–15 In family 1, a homozygous missense mutation, c.545G>A (p.Arg182Gln), was identified in exon 6 of NFU1 in the three affected siblings but not in their unaffected sibling, P4 (II-4). The mutation was not identified in seven different control samples or in the expressed sequence tag (EST) database (99 NFU1 ESTs).
NFU1 has two isoforms generated by alternative splicing, with specific subcellular localizations (Figure 2B).16 In isoform 2 (NM_001002755.1), exon 1B, which contains an in-frame stop codon, is spliced out, and the protein is predicted to be targeted to mitochondria by a leader sequence coded in exon 1A. Isoform 1 (NM_015700.2) contains exon 1B, and translation is predicted to start from a downstream AUG start codon in exon 2 and produce a protein that remains in the cytosol. The c.545G>A mutation thus affects both the mitochondrial and cytosolic isoforms of the protein, and is predicted to result in an p.Arg182Gln substitution, but only if exon 6 is spliced correctly to exon 7. It is, however, adjacent to the GU 5′ splice donor in intron 6, and RT-PCR analysis in Figure 2C shows that exon 6 is spliced out from the majority of transcripts. In fact the full-length mitochondrial transcript is not detected by this analysis. We have sequenced separate cloned RT-PCR transcripts of NFU1 from individuals P1–P3 (II-1, II-2, and II-3 in Figure 2) representing cNFU1a (cytosolic transcript missing exon 6), mNFU1 (mitochondrial transcript with c.545G>A mutation), mNFU1a (missing exon 6), and mNFU1b (missing exons 6 and 7). The only transcript that was not confirmed by sequencing was cNFU1 containing the c.545G>A mutation. Figure 2D shows the NFU1 protein product in mitochondria isolated from affected individuals P1 (II-1), P5 (II-1 in Figure 3), and control fibroblasts. There is no immunodetectable mitochondrial NFU1 protein in individual P1.
Mutations in the coding regions of NFU1, the promoter, or the intron/exon boundaries of the gene were not identified in the single affected individual from the second family (individual P5, II-1 in Figure 3), suggesting another gene defect in the same chromosomal region. Several candidate genes were tested and were chosen on the basis of chromosomal location, a high probability of mitochondrial import (determined with Mitoprot7), and tentative roles in metabolism or gene regulation (genes sequenced were AHSA2, BCS1L [MIM 603647], SMEK2 [MIM 610352], CCT4 [MIM 605142], SLC1A4 [MIM 600229], SPR [MIM 182125], SFXN5, FLJ30838, FAM161A [MIM 613596], LOC647074, MDH1 [MIM 154200], RAB1A [MIM 179508], and LOC388955). A homozygous single-base-pair duplication (c.123dupA; p.Glu42Argfs∗13) was identified in exon 2 of BOLA3 (MIM 613183, 4.7 Mb upstream of NFU1) in individual P5 (II-1 in Figure 3), resulting in a frameshift and predicting a premature stop codon (p.Glu42Argfs∗13). The parents and an unaffected sibling (individual P6, II-2 in Figure 3), were all heterozygous carriers for the mutation. The mutation was not identified in seven different control samples or in the EST database (68 BOLA3 ESTs).
The human protein BOLA3 is predicted to have a mitochondrial targeting sequence, and a 95% probability of import into the mitochondria.7 Human BOLA3 has two isoforms (NM_001035505.1 and NM_212552.2); isoform 2 lacks exon 3, which is present in isoform 1, resulting in different stop codons in the two transcripts (Figure 3B). Both isoforms are affected by the mutation in exon 2 in P5 (II-1 in Figure 3), and the amount of full-length transcript for either isoform does not appear to be reduced (data not shown). Respiratory chain complexes I, II, and III all have components that contain [Fe-S] clusters. For both families, several subunits of complex I (NDUFS1, which contains [Fe-S]; NDUFV1, which contains [Fe-S]; NDUFS2; NDUFA9; and NDUFB8) were analyzed and all showed a complete loss of protein except for NDUFS1, which had a 20% reduction in the NFU1 mutant compared to citrate synthase and 40% reduction in the BOLA3 mutant (Figure 4). Because NFU1 and ISCU have the same proposed role in [Fe-S] cluster synthesis, albeit in parallel pathways, we thought it prudent to compare the biochemical and clinical consequences of both defects. In contrast to the NFU1 and BOLA3 mutant fibroblasts, the ISCU mutant individual has normal NFU1; complex I NDUFS2, NDUFA9, and NDUFB8; and complex III UQCRC1 protein levels. However, complex III UQCRFS1, which contains an [Fe-S] cluster, was severely reduced in this individual (60% compared to citrate synthase). In the NFU1 and BOLA3 mutant individuals, complex II subunit SDHA was reduced by 69% and 73%, respectively, as well as UQCRFS1 (70% and 76% reduction) and UQCRC1 subunits of complex III (22% and 60% reduction), whereas cytochrome b expression was affected differently in the two individuals (13% increase and 30% reduction) (Figure 4).
Figure 4.
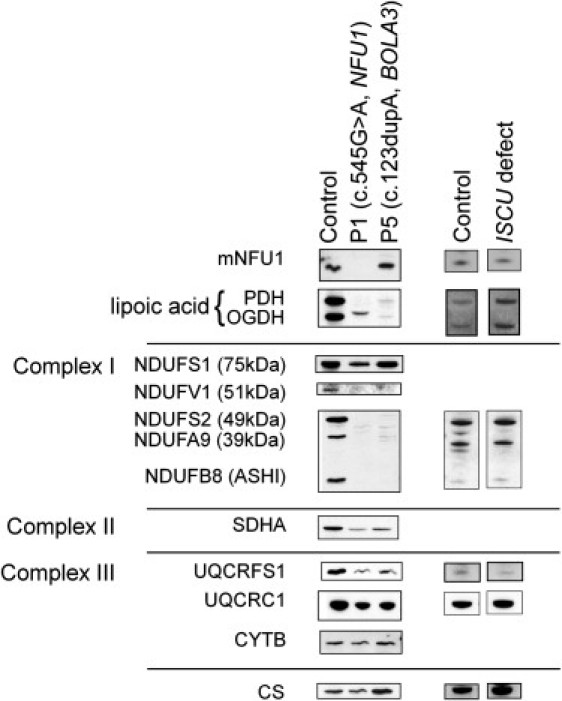
Immunoblot Analysis of the Oxoacid Dehydrogenases and Respiratory Chain Subunits in NFU1, BOLA3, and ISCU Mutant Fibroblasts
Immunoblots of 25 μg fibroblast mitochondria were immunoblotted with antibodies against NFU1, lipoate, complex I subunits (NDUFS1, NDUFV1, NDUFS2, NDUFA9, and NDUFA8), complex II subunit SDHA, complex III subunits (UQCRFS1, UQCRC1, and CYTB), and citrate synthase (CS).
In contrast PDHc and OGDHc constituent subunit proteins do not contain [Fe-S] clusters, but those complexes show profound enzyme deficiency.6 The common feature of these enzymes is the requirement for covalently attached lipoate on the E2 subunits of PDHc (dihydrolipoamide transacetylase [DLAT]) and OGDHc (dihydrolipoyl transsuccinylase [DLST] [MIM 126063]), and the E3BP subunit of PDHc.
We used an antibody to lipoate to identify the lipoate containing subunits of both PDHc and OGDHc complexes in fibroblast cell mitochondria. Anti-lipoate failed to detect the expected lipoylated E2 proteins of PDHc and OGDHc in both families with multiple mitochondrial dysfunctions syndrome (Figure 4). The individual with the NFU1 mutation has a single weak band at a different molecular weight, which could represent a modified form of lipoate. P5 (II-1 in Figure 3) showed no detectable lipoate labeling. We attempted to improve the defective enzyme activities in the affected fibroblasts by adding exogenous lipoate, but no effect was seen on PDHc activity after three days of culture in media supplemented with 30 μM lipoic acid.
To confirm the pathogenicity of the NFU1 and the BOLA3 mutations, we constructed retroviral expression vectors for both isoforms of each gene, transduced fibroblast cell lines, and tested for complementation of the biochemical defect by immunoblot, BN-PAGE electrophoresis (Figure 5), immunofluorescence (Figure 6), and PDHc enzyme activity measurements (Figure 7). Interestingly, the mitochondrial but not the cytosolic version of NFU1, completely rescued the respiratory chain and oxoacid dehydrogenase defects in immortalized fibroblasts from individual P1 (II-1 in Figure 2). Similarly, only the longer isoform of BOLA3 (BOLA3-1) restored all of the biochemical abnormalities in individual P5 (II-1 in Figure 3). For both defects, protein levels of complex I subunit NDUFA9, complex II subunit SDHA, and lipoate were all restored to normal levels (Figures 5A and 5C), as well as assembly of the full complexes I and II (Figures 5B and 5D). PDHc enzyme activity (both native and DCA-activated) was also restored to control levels (Figure 7). Immunofluorescence experiments in COS-7 cells and control fibroblasts (Figure 6A, a and b) confirmed that endogenous NFU1 mainly localizes into mitochondria. In accordance with immunoblot results, only the overexpression of the mitochondrial version of NFU1 is able to rescue the decreased level of SDHA observed in individual P1 (Figure 6A, compare d to c and e). Moreover, Figure 6B shows the long isoform of BOLA3 (BOLA3.1) localized into mitochondria and the short version (BOLA3.2) in the cytoplasm.
Figure 5.
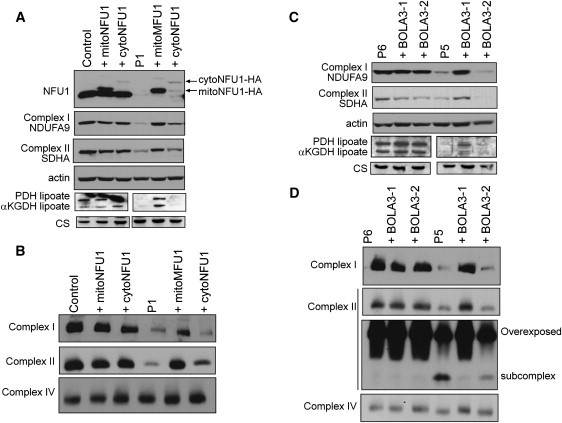
Rescue of the Biochemical Deficiencies in NFU1 and BOLA3 Mutant Fibroblasts by Retroviral Expression of the Wild-Type cDNAs
(A) Immunoblot of control and mutant NFU1 fibroblast mitochondria transduced with constructs expressing mitochondrial or cytosolic isoforms of NFU1were immunoblotted with antibodies against NFU1, lipoate, complex I subunit NDUFA9, complex II subunit SDHA, and actin.
(B) BN-PAGE analysis of the same samples as in (A).
(C) Immunoblot of control and mutant BOLA3 fibroblast mitochondria transduced with constructs expressing the BOLA3-1 and BOLA3-2 isoforms were immunoblotted with antibodies against NFU1, lipoate, complex I subunit NDUFA9, complex II subunit SDHA, and actin.
(D) BN-PAGE analysis of the same samples as in (C).
Figure 6.
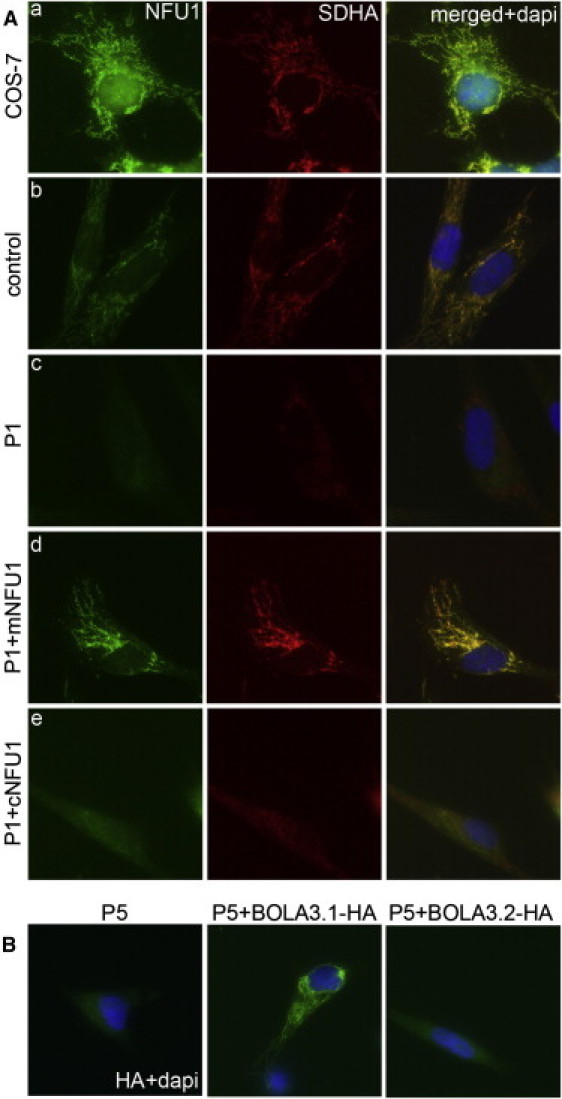
Immunofluorescence analysis of Fibroblast Cells Transduced with Retroviral Constructs Expressing the Cytosolic and Mitochondrial Versions of NFU1 and the Two Isoforms of BOLA3
(A) SDHA was used as a mitochondrial marker and as a marker of the respiratory chain defect. The predicted mitochondrial isoform of NFU1 localizes to mitochondria, and expression of the cDNA rescues the SDHA defect in individual P1 (II-1 in Figure 1) cells. The cytosolic isoform shows diffuse staining in mutant fibroblasts and does not rescue the complex II defect.
(B) BOLA3-1HA rescues the complex II defect of individual P5 and localizes to mitochondria, but BOLA3-2HA shows diffuse staining and does not rescue the defect.
Figure 7.
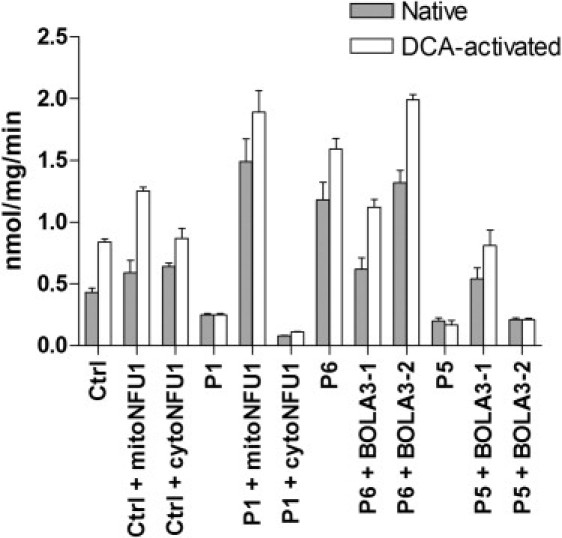
Pyruvate Dehydrogenase Enzyme Activities for Control and Mutant NFU1 and BOLA3 Fibroblasts
Native and DCA-activated PDHc enzyme activities were determined for control and mutant NFU1 fibroblast mitochondria transfected with mitochondrial and cytosolic transcripts of NFU1, and control and mutant BOLA3 fibroblast mitochondria transfected with BOLA3-1 and BOLA3-2 isoforms. Data are represented as mean ± SEM for four separate enzyme measurements.
Discussion
We have identified a family with a mutation in NFU1, which encodes an [Fe-S] cluster biogenesis protein. The c.545G>A mutation causes splice site instability leading to the production of several truncated transcripts and no mitochondrial NFU1 protein product. We have also identified a family with a mutation in BOLA3, a previously uncharacterized gene that the present results suggest is also involved in [Fe-S] cluster synthesis. How do these findings fit with our knowledge of [Fe-S] cluster biogenesis?
NFU1 as an Alternate Scaffold for [Fe-S] Cluster Biogenesis
[Fe-S] clusters function as the prosthetic groups of many proteins essential for intermediary metabolism and oxidative phosphorylation. They exist mainly as [2Fe-2S] and [4Fe-4S] clusters and participate in electron transfer reactions in which oxidation or reduction of oxygen or sulfur is desirable.17 [Fe-S] clusters are essential components of enzymes involved in the maturation of subunits of complexes I, II, and III as well in the synthesis of the enzyme-bound cofactor lipoate present in the 2-oxoacid dehydrogenases. [Fe-S] clusters are constructed and assembled into proteins in a series of complex biochemical reactions that appear to run in two parallel pathways.17 The process occurs in the mitochondrial matrix with the export of some clusters to form cytosolic and nuclear proteins (Figure 1).
The role of NFU1 in [Fe-S] cluster biogenesis has been deduced from its orthology to the bacterial protein NifU. NFU1 and ISCU in eukaryotes both evolved from different domains of NifU, a protein present in nitrogen-fixing bacteria. ISCU is thought to function as a scaffold for the assembly of [Fe-S] clusters, but the role of NFU1 is less clear. Several roles for NFU1 have been hypothesized, two of which are involved in [Fe-S] cluster biosynthesis. By demonstrating the ability of NFU1 to assemble [4Fe-4S] clusters and transfer them to apoproteins,16,18 it has been suggested that NFU1 could function as an alternate scaffold to ISCU for assembly of [Fe-S] proteins, thus providing parallel pathways (Figure 1).17,19,20
The c.545G>A mutation results in truncated transcripts for both mitochondrial and cytosolic NFU1 isoforms, and therefore could affect both protein products. Mammalian NFU1 is one of several [Fe-S] cluster genes (NFS1 [MIM 603485], ISCU, and NFU1) reported to have mitochondrial and cytosolic isoforms. In the individuals reported here, the c.545G>A mutation appears to be spliced out of virtually all of the transcribed forms of mitochondrial forms of NFU1, resulting in barely detectable levels of the protein (Figure 5A). Transfection of the cytosolic transcript does result in a partial restoration of complex I NDUFA9 and complex II SDHA subunit levels, but this appears to result from a low level of expression of the mitochondrial isoform from this construct, probably because of readthrough of the stop codon in exon 1B.
Inference of the Role of BOLA3 in [Fe-S] Cluster Synthesis
Mammalian BOLA3 belongs to a BolA-like family of proteins that is conserved from prokaryotes to eukaryotes. They were originally thought to be DNA-binding proteins (based on the presence of a helix-turn-helix motif), which create a round morphology in Escherichia coli cells when overexpressed.21 They have also been shown to be expressed under stress situations such as carbon starvation and oxidative stress.22
Little is known about the function of human BOLA3, but predictions can be made from the roles of ortholog proteins. BolA family members have been postulated to act as reductases, interacting with the mono-thiol glutaredoxin family.23 Genome-wide yeast two-hybrid studies have shown a physical interaction between cytosolic monothiol glutaredoxins (Grx) and BolA-like proteins in Saccharomyces cerevisiae and Drosophila melanogaster.24,25 In several genomes, genes encoding Grx and BolA-like proteins lie physically next to each other, and the two protein families show co-occurrence in organisms because only those that express BolA proteins possess monothiol Grx proteins.23 It is possible that BOLA3 is interacting with glutaredoxin 5, which along with several chaperones is involved in inserting [2Fe-2S] and [4Fe-4S] clusters into apoproteins (Figure 1).
Transfection of both isoforms of BOLA3 into deficient fibroblasts showed that only isoform 1 can restore protein steady-state levels, respiratory chain complex assembly, and enzyme activities (Figures 5B, 5D, and 7). The role of the shorter isoform is unknown. Immunofluorescence results show this isoform, but not BOLA3-2, localizes to mitochondria (Figure 6B).What is the nature of the 2-oxoacid dehydrogenase enzyme c defect in the mutant NFU1 and BOLA3 individuals?
Mutation of NFU1 would be expected to interfere with the pathway of [Fe-S] cluster synthesis resulting in a decrease in the assembly and activity of complexes I, II, and III. This does not however explain the profound deficit in PDHc activity (<10% control), in the face of apparently normal assembly of the protein components of PDH.6 The lack of immunoreactive lipoate in the PDH or 2OGDH complexes, however, is compelling and suggests that both NFU1 and BOLA3 are necessary for lipoate synthesis. That these are candidates for defects in lipoate synthesis is evident from work on yeast in which elimination of cysteine desulphurase (NFS1), iron sulfur assembly protein (ISA1), or IBA57 results in an inability to produce lipoate or lipoylated proteins.26,27 The E2 protein of PDHc, 2OGDH, and BCKADHc is initially octanoylated on lysine residues by lipoyl transferase 2 (LIPT2) in the biosynthetic pathway (Figure 1) or lipoyl transferase 1 (LIPT1) in the salvage pathway. This is subsequently transformed to lipoate by the addition of two sulfur atoms in a reaction catalyzed by lipoic acid synthase (LIAS), an enzyme possessing two [4Fe-4S] clusters with unusual cysteine-containing motifs CX3CX2C and CX4CX5C in the protein sequence.28–30 It is known that the E2 protein of the PDHc runs at a different mobility on SDS-PAGE gels when lipoylated (72 kDa) compared to its unlipoylated mobility (55 kDa). The mobility observed in individual P1 (II-1 in Figure 2) for the PDHc-E2 is less than 72 kDa, suggesting that the mobility has been altered by octanoylation. The intactness of the PDHc-E2 enzyme function is illustrated by the fact that fibroblasts from both affected individuals possess normal dihydrolipoyl transacetylase activity when measured by acetylation of reduced lipoic acid by acetyl CoA. However, because this assay is carried out with added reduced lipoic acid, the enzyme does not require enzyme-bound lipoate to function. Nevertheless, the activity of both PDHc and OGDHc in P5 the unsupplemented complex is greatly reduced, and there is virtually no such activity in individual P1. We hypothesize that the loss of mNFU1 and BOLA3-1 in these individuals might be specifically affecting the maturation of lipoic acid synthase, an [Fe-S] cluster protein, which in turn will impact the enzyme activities of PDHc and OGDHc (Figure 7).
Why Do NFU1 and BOLA3 Mutations Have a Different Phenotype from that Seen with ISCU Mutations?
The most profound differences between the three classes of defect is a loss of aconitase protein13 and enzyme activity in ISCU mutants and lipoate abnormalities in NFU1/BOLA3 mutants; there is an indication of less affected complex I in the ISCU mutant. Mitochondrial aconitase enzyme levels in the NFU1/BOLA3 mutant individuals were normal (Table 1) as were protein levels (data not shown). This suggests a divergence between the roles of NFU1/BOLA3 and ISCU, with NFU1 and BOLA3 specializing in proteins destined to become involved in the lipoate pathway. It had previously been suggested that aconitase-like proteins and radical S-adenosyl methionine (SAM) proteins (such as lipoate synthase) are assembled in the same pathway.26 Recent work shows that many more [Fe-S] cluster-containing proteins appear to have specific maturation or assembly factors, for example, human IND1 for complex I proteins,31,32 and E. coli ErpA, essential for an [Fe-S] protein involved in isoprenoid biosynthesis.33 It is therefore likely that the roles of NFU1 and BOLA3 in eukaryotes are not as simple as has so far been hypothesized with more specialization than previously thought. ISCU assembled [Fe-S] clusters appear to be preferentially channeled to aconitase and complex II, whereas NFU1- (and BOLA3-) dependent assembled clusters are more directed to complex I and lipoate synthesis.
We conclude that in humans there are parallel pathways for synthesis of [Fe-S] clusters, and there is a bias in each pathway for maturation of certain target proteins. How this is achieved will be the subject of further research.
Acknowledgments
We thank “MitoMarch for Kirkland” and the Canadian Institutes of Health Research (E.A.S.) for funding this project. E.A.S. is an International Scholar of the Howard Hughes Medical Institute. The authors declare no conflict of interests.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Phoenix Retroviral Producer Line Protocol, http://www.stanford.edu/group/nolan/protocols/pro_helper_dep.html
References
- 1.Cameron J.M., Levandovskiy V., Mackay N., Tein I., Robinson B.H. Deficiency of pyruvate dehydrogenase caused by novel and known mutations in the E1alpha subunit. Am. J. Med. Genet. A. 2004;131:59–66. doi: 10.1002/ajmg.a.30287. [DOI] [PubMed] [Google Scholar]
- 2.Lissens W., De Meirleir L., Seneca S., Liebaers I., Brown G.K., Brown R.M., Ito M., Naito E., Kuroda Y., Kerr D.S. Mutations in the X-linked pyruvate dehydrogenase (E1) alpha subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum. Mutat. 2000;15:209–219. doi: 10.1002/(SICI)1098-1004(200003)15:3<209::AID-HUMU1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 3.Kerr D.S., Wexler I.D., Tripatara A., Patel M.S. Human defects of the pyruvate dehydrogenase complex. In: Patel M.S., Harris R.A., editors. Alpha-keto acid dehydrogenase complexes, R.T. Birkhäuser; Basel: 1996. pp. 249–269. [Google Scholar]
- 4.Maj M.C., MacKay N., Levandovskiy V., Addis J., Baumgartner E.R., Baumgartner M.R., Robinson B.H., Cameron J.M. Pyruvate dehydrogenase phosphatase deficiency: Identification of the first mutation in two brothers and restoration of activity by protein complementation. J. Clin. Endocrinol. Metab. 2005;90:4101–4107. doi: 10.1210/jc.2005-0123. [DOI] [PubMed] [Google Scholar]
- 5.Robinson B.H. Lactic acidemia (Disorders of pyruvate carboxylase, pyruvate dehydrogenase) In: Scriver C.R., Sly W.S., Valle D., editors. The metabolic and molecular bases of inherited disease, B.A. McGraw-Hill; New York: 2001. pp. 2275–2295. [Google Scholar]
- 6.Seyda A., Newbold R.F., Hudson T.J., Verner A., MacKay N., Winter S., Feigenbaum A., Malaney S., Gonzalez-Halphen D., Cuthbert A.P., Robinson B.H. A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am. J. Hum. Genet. 2001;68:386–396. doi: 10.1086/318196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claros M.G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 8.Lochmüller H., Johns T., Shoubridge E.A. Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res. 1999;248:186–193. doi: 10.1006/excr.1999.4407. [DOI] [PubMed] [Google Scholar]
- 9.Antonicka H., Sasarman F., Kennaway N.G., Shoubridge E.A. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;15:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 10.Pitkänen S., Raha S., Robinson B.H. Diagnosis of complex I deficiency in patients with lactic acidemia using skin fibroblast cultures. Biochem. Mol. Med. 1996;59:134–137. doi: 10.1006/bmme.1996.0078. [DOI] [PubMed] [Google Scholar]
- 11.Antonicka H., Ostergaard E., Sasarman F., Weraarpachai W., Wibrand F., Pedersen A.M., Rodenburg R.J., van der Knaap M.S., Smeitink J.A., Chrzanowska-Lightowlers Z.M., Shoubridge E.A. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am. J. Hum. Genet. 2010;87:115–122. doi: 10.1016/j.ajhg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abramoff M.D., Magelhaes P.J., Ram S.J. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 13.Hall R.E., Henriksson K.G., Lewis S.F., Haller R.G., Kennaway N.G. Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J. Clin. Invest. 1993;92:2660–2666. doi: 10.1172/JCI116882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haller R.G., Henriksson K.G., Jorfeldt L., Hultman E., Wibom R., Sahlin K., Areskog N.H., Gunder M., Ayyad K., Blomqvist C.G. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Invest. 1991;88:1197–1206. doi: 10.1172/JCI115422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mochel F., Knight M.A., Tong W.H., Hernandez D., Ayyad K., Taivassalo T., Andersen P.M., Singleton A., Rouault T.A., Fischbeck K.H., Haller R.G. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 2008;82:652–660. doi: 10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong W.H., Jameson G.N., Huynh B.H., Rouault T.A. Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proc. Natl. Acad. Sci. USA. 2003;100:9762–9767. doi: 10.1073/pnas.1732541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rouault T.A., Tong W.H. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishio K., Nakai M. Transfer of iron-sulfur cluster from NifU to apoferredoxin. J. Biol. Chem. 2000;275:22615–22618. doi: 10.1074/jbc.C000279200. [DOI] [PubMed] [Google Scholar]
- 19.Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–838. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 20.Lill R., Dutkiewicz R., Elsässer H.P., Hausmann A., Netz D.J., Pierik A.J., Stehling O., Urzica E., Mühlenhoff U. Mechanisms of iron-sulfur protein maturation in mitochondria, cytosol and nucleus of eukaryotes. Biochim. Biophys. Acta. 2006;1763:652–667. doi: 10.1016/j.bbamcr.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Aldea M., Hernández-Chico C., de la Campa A.G., Kushner S.R., Vicente M. Identification, cloning, and expression of bolA, an ftsZ-dependent morphogene of Escherichia coli. J. Bacteriol. 1988;170:5169–5176. doi: 10.1128/jb.170.11.5169-5176.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos J.M., Freire P., Vicente M., Arraiano C.M. The stationary-phase morphogene bolA from Escherichia coli is induced by stress during early stages of growth. Mol. Microbiol. 1999;32:789–798. doi: 10.1046/j.1365-2958.1999.01397.x. [DOI] [PubMed] [Google Scholar]
- 23.Huynen M.A., Spronk C.A., Gabaldón T., Snel B. Combining data from genomes, Y2H and 3D structure indicates that BolA is a reductase interacting with a glutaredoxin. FEBS Lett. 2005;579:591–596. doi: 10.1016/j.febslet.2004.11.111. [DOI] [PubMed] [Google Scholar]
- 24.Giot L., Bader J.S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y.L., Ooi C.E., Godwin B., Vitols E. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 25.Ito T., Tashiro K., Muta S., Ozawa R., Chiba T., Nishizawa M., Yamamoto K., Kuhara S., Sakaki Y. Toward a protein-protein interaction map of the budding yeast: A comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gelling C., Dawes I.W., Richhardt N., Lill R., Mühlenhoff U. Mitochondrial Iba57p is required for Fe/S cluster formation on aconitase and activation of radical SAM enzymes. Mol. Cell. Biol. 2008;28:1851–1861. doi: 10.1128/MCB.01963-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Onder O., Yoon H., Naumann B., Hippler M., Dancis A., Daldal F. Modifications of the lipoamide-containing mitochondrial subproteome in a yeast mutant defective in cysteine desulfurase. Mol. Cell. Proteomics. 2006;5:1426–1436. doi: 10.1074/mcp.M600099-MCP200. [DOI] [PubMed] [Google Scholar]
- 28.Cicchillo R.M., Booker S.J. Mechanistic investigations of lipoic acid biosynthesis in Escherichia coli: Both sulfur atoms in lipoic acid are contributed by the same lipoyl synthase polypeptide. J. Am. Chem. Soc. 2005;127:2860–2861. doi: 10.1021/ja042428u. [DOI] [PubMed] [Google Scholar]
- 29.Cicchillo R.M., Iwig D.F., Jones A.D., Nesbitt N.M., Baleanu-Gogonea C., Souder M.G., Tu L., Booker S.J. Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43:6378–6386. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- 30.Miller J.R., Busby R.W., Jordan S.W., Cheek J., Henshaw T.F., Ashley G.W., Broderick J.B., Cronan J.E., Jr., Marletta M.A. Escherichia coli LipA is a lipoyl synthase: In vitro biosynthesis of lipoylated pyruvate dehydrogenase complex from octanoyl-acyl carrier protein. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 31.Bych K., Kerscher S., Netz D.J., Pierik A.J., Zwicker K., Huynen M.A., Lill R., Brandt U., Balk J. The iron-sulphur protein Ind1 is required for effective complex I assembly. EMBO J. 2008;27:1736–1746. doi: 10.1038/emboj.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheftel A.D., Stehling O., Pierik A.J., Netz D.J., Kerscher S., Elsässer H.P., Wittig I., Balk J., Brandt U., Lill R. Human ind1, an iron-sulfur cluster assembly factor for respiratory complex I. Mol. Cell. Biol. 2009;29:6059–6073. doi: 10.1128/MCB.00817-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loiseau L., Gerez C., Bekker M., Ollagnier-de Choudens S., Py B., Sanakis Y., Teixeira de Mattos J., Fontecave M., Barras F. ErpA, an iron sulfur (Fe S) protein of the A-type essential for respiratory metabolism in Escherichia coli. Proc. Natl. Acad. Sci. USA. 2007;104:13626–13631. doi: 10.1073/pnas.0705829104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.