

Abstract
Persons with neurodevelopmental disorders or autism spectrum disorder (ASD) often harbor chromosomal microdeletions, yet the individual genetic contributors within these regions have not been systematically evaluated. We established a consortium of clinical diagnostic and research laboratories to accumulate a large cohort with genetic alterations of chromosomal region 2q23.1 and acquired 65 subjects with microdeletion or translocation. We sequenced translocation breakpoints; aligned microdeletions to determine the critical region; assessed effects on mRNA expression; and examined medical records, photos, and clinical evaluations. We identified a single gene, methyl-CpG-binding domain 5 (MBD5), as the only locus that defined the critical region. Partial or complete deletion of MBD5 was associated with haploinsufficiency of mRNA expression, intellectual disability, epilepsy, and autistic features. Fourteen alterations, including partial deletions of noncoding regions not typically captured or considered pathogenic by current diagnostic screening, disrupted MBD5 alone. Expression profiles and clinical characteristics were largely indistinguishable between MBD5-specific alteration and deletion of the entire 2q23.1 interval. No copy-number alterations of MBD5 were observed in 7878 controls, suggesting MBD5 alterations are highly penetrant. We surveyed MBD5 coding variations among 747 ASD subjects compared to 2043 non-ASD subjects analyzed by whole-exome sequencing and detected an association with a highly conserved methyl-CpG-binding domain missense variant, p.79Gly>Glu (c.236G>A) (p = 0.012). These results suggest that genetic alterations of MBD5 cause features of 2q23.1 microdeletion syndrome and that this epigenetic regulator significantly contributes to ASD risk, warranting further consideration in research and clinical diagnostic screening and highlighting the importance of chromatin remodeling in the etiology of these complex disorders.
Introduction
Recent widespread implementation of chromosomal microarrays in research and clinical diagnostic testing of copy-number variations (CNVs) has catalyzed an explosion in the identification of genomic disorders, including microdeletion and microduplication syndromes. Among those recently discovered is the 2q23.1 region in which microdeletions result in a neurodevelopmental disorder (ND) previously described as pseudo-Angelman syndrome or autosomal-dominant mental retardation type 1 (MRD1 [MIM 156200]). The region was initially identified in 2003 from one of the first comparative genomic hybridization (CGH) surveys of developmental disorders,1 and 18 cases with the 2q23.1 microdeletion have since been described; the phenotypic features include intellectual disability, severe speech impairment, seizures, behavioral problems, microcephaly, mild craniofacial dysmorphism, small hands and feet, short stature, and broad-based ataxic gait.1–9
Advances in the resolution of genomic microarray technology and the power of next-generation sequencing now offer a route to more detailed definition of microdeletion syndrome regions. In some cases, such as deletion 2q33.1 (SATB2 [MIM 608148]), deletion 22q13.3 (Phelan-McDermid syndrome [MIM 606232]; SHANK3 [MIM 606230]), deletion 13q12.3 (Peters Plus syndrome [MIM 261540]; and B3GALTL [MIM 610308]), and others,10–13 individual genes have been implicated in generating the characteristic phenotype. Such findings might signify an important paradigm shift in the required resolution of diagnostic assessment, because small gene-specific microdeletions could result in a similar phenotype to that seen in the larger deletion syndrome, suggesting that diagnostic arrays should include not only coding regions but also noncoding exons in the assessment, particularly for dosage-sensitive genes. Consequently, we assembled a collaborative team of clinical diagnostic laboratories and research facilities in the United States, Canada, and Europe to characterize the genetic content and phenotypic outcomes associated with a spectrum of genetic alterations within the 2q23.1 deletion syndrome region. We demonstrate that MBD5 (MIM 611472), encoding the methyl-CpG-binding domain 5 protein, is a clear causal locus within the 2q23.1 deletion region and represents a previously unrecognized contributor to the genetic etiology of autism spectrum disorder (ASD).
Material and Methods
Subjects
We established an international collaboration to identify a cohort of individuals with deletion or translocation involving chromosome band 2q23.1 from chromosome analysis or microarray-based CGH at referring centers and collaborating institutions. When available, peripheral blood was collected, and DNA, RNA, and lymphoblastoid cell lines were prepared according to standard methods. Phenotypic information was obtained from diagnostic referral data, medical records, clinical reports, genetics clinic evaluations, neurodevelopmental and/or psychological evaluations, parent surveys, photos, previously published articles, and/or a clinical checklist sent to the referring physician. All samples and information were collected after informed consent was obtained and in accordance with local institutional review board approved protocols from Virginia Commonwealth University, Harvard Medical School, Children's Hospital of Boston, Case Western Reserve University, Partners HealthCare System, Istituto Di Ricovero e Cura a Carattere Scientifico (IRCCS) Associazione Oasi Maria Santissima, or Radboud University Medical Centre. In follow-up analyses, we sequenced 747 subjects who met diagnostic criteria for ASD from the Simon's Simplex Collection and the Autism Consortium of Boston. All subjects were assessed by structured clinical interview with the Autism Diagnostic Interview-Revised (ADI-R) or Autism Diagnostic Observation Schedule (ADOS).14–16 We surveyed CNV and exome data from large collaborative studies as comparison groups. The CNV data were obtained from multiple data sets of the International Schizophrenia Consortium (ISC). Rare CNVs were combined and cleaned to account for time and technology used according to the same procedure described previously.17–22 After filtering with a variant resolution of >100 kb, 7878 controls remained in the data set. Exome-sequencing data were obtained from the ongoing National Heart, Lung, and Blood Institute (NHLBI) Grand Opportunity (GO) Exome Sequencing Project (2043 individuals deeply phenotyped for disorders of the heart, lungs, and blood were sequenced for the MBD5 variant of interest). Data from this project were obtained from the Exome Variant Server in July 2011 (see Web Resources).
Phenotype Analysis
Data were collected for up to 70 characteristics related to subjects' medical and developmental history and neurological, behavioral, and physical characteristics (data not shown, Table 1). Short stature was defined by clinicians as height less than the 5th percentile or significantly shorter than siblings. Characteristics that were age dependent (behavior, sleep patterns, and craniofacial and skeletal abnormalities) were scored accordingly on the basis of development. Autistic-like symptoms were defined as either having reported autistic-like behaviors or were specifically evaluated with the ADOS, ADI-R, or given a diagnosis of Pervasive Developmental Disorder (PDD-NOS). Eye abnormalities and outer ear abnormalities varied among subjects and were grouped in the above broad categories. Data were analyzed from 65 patients (males and females). We compared 48 characteristics that were frequently reported to be associated with 2q23.1 deletion syndrome in 51 individuals with 2q23.1 deletions to 14 individuals with partial deletion and/or MBD5-specific alterations. All features were compared and scored as present, absent, or data not available. The frequency of each feature reflects the information reported by specialists in this study.
Table 1.
Phenotypic Comparison of MBD5 Alterations and 2q23.1 Deletions
| Common Features |
MBD5-Specific Disruption |
2q23.1 Deletion |
p Valuea | ||
|---|---|---|---|---|---|
| Frequency | Percentage | Frequency | Percentage | ||
| Neurological and/or behavioral | |||||
| Developmental delay | 14/14 | 100 | 51/51 | 100 | 1 |
| Motor delay | 6/6 | 100 | 25/25 | 100 | 1 |
| Language impairment | 5/7 | 71.4 | 24/24 | 100 | 0.427 |
| Ataxia | 0/3 | 0 | 15/21 | 71.4 | 0.042 |
| Infantile hypotonia | 3/4 | 75.0 | 14/15 | 93.3 | 0.386 |
| Infantile feeding difficulties | 2/3 | 66.6 | 14/14 | 100 | 1 |
| Seizures | 6/7 | 85.7 | 22/27 | 81.5 | 1 |
| Behavioral problems | 7/7 | 100.0 | 24/24 | 100 | 1 |
| Sleep disturbances | 3/6 | 50 | 11/15 | 73.3 | 0.354 |
| Short attention span | 4/4 | 100 | 11/11 | 100 | 1 |
| Self-injurious behavior | 1/3 | 50.0 | 14/23 | 60.8 | 1 |
| Stereotypic repetitive behavior | 3/5 | 60.0 | 15/17 | 88.2 | 0.210 |
| Aggression | 1/2 | 50 | 6/6 | 100 | 1 |
| Hyperphagia | 0/4 | 0 | 7/9 | 77.8 | 0.021 |
| Autistic-like symptoms | 14/14 | 100 | 51/51 | 100 | 1 |
| Growth abnormalities | |||||
| Postnatal growth retardation | 2/5 | 40.0 | 21/23 | 91.3 | 0.027 |
| Short stature | 2/5 | 40.0 | 19/22 | 86.4 | 0.056 |
| Craniofacial abnormalities | |||||
| Craniofacial manifestations | 4/6 | 66.6 | 26/26 | 100 | 0.030 |
| Microcephaly | 0/5 | 0 | 18/20 | 90.0 | 0.004 |
| Brachycephaly | 0/5 | 0 | 8/22 | 36.4 | 0.280 |
| Broad forehead | 2/5 | 40.0 | 14/16 | 87.5 | 0.063 |
| Synophrys | 1/5 | 20.0 | 8/17 | 47.1 | 0.360 |
| Thick/arched eyebrows | 1/1 | 100 | 15/20 | 75.0 | 1 |
| Eye abnormalities | 3/5 | 60.0 | 11/15 | 73.3 | 0.621 |
| Hypotelorism | 0/5 | 0 | 7/21 | 33.0 | 0.278 |
| Midface retrusion (hypoplasia) | 1/6 | 16.6 | 9/18 | 50.0 | 0.341 |
| Nasal abnormalities | 4/4 | 100 | 25/25 | 100 | 1 |
| Outer ear abnormalities | 2/5 | 20.0 | 14/16 | 87.5 | 0.025 |
| Wide mouth | 0/5 | 0 | 12/15 | 80.0 | 0.037 |
| Open mouth | 2/6 | 33.3 | 20/22 | 90.9 | 0.010 |
| Vermillion, thin upper lip | 2/2 | 100 | 15/20 | 75.0 | 1 |
| Vermillion, tented upper lip | 1/5 | 20.0 | 13/20 | 65.0 | 0.133 |
| Thick or everted lower lip | 1/3 | 33.3 | 12/18 | 66.6 | 0.530 |
| Downturned corners of the mouth | 0/3 | 0 | 13/18 | 72.2 | 0.042 |
| Dental abnormalities | 3/6 | 50.0 | 11/21 | 52.3 | 1 |
| Widely-spaced teeth | 1/2 | 50.0 | 8/17 | 47.1 | 1 |
| Large tongue, macroglossia | 0/5 | 0 | 4/21 | 19.0 | 0.552 |
| Short chin/micrognathia/retrognathia | 3/5 | 60.0 | 12/20 | 60.0 | 1 |
| Skeletal abnormalities | |||||
| Hand/foot anomalies | 4/6 | 66.6 | 23/24 | 95.8 | 0.094 |
| Small hands and feet | 1/5 | 20.0 | 18/24 | 75.0 | 0.036 |
| Clinodactyly, 5th finger | 1/5 | 20.0 | 15/22 | 68.2 | 1 |
| Brachydactyly | 0/4 | 0 | 9/22 | 40.9 | 0.263 |
| Short fifth digit of hands/feet | 0/4 | 0 | 7/17 | 41.2 | 0.090 |
| Sandal gap | 3/6 | 50 | 5/20 | 25.0 | 0.330 |
| Other Abnormalities | |||||
| Cardiovascular abnormalities | 0/5 | 0 | 2/18 | 11.1 | 1 |
| Urogenital abnormalities | 0/4 | 0 | 6/21 | 28.6 | 0.540 |
| Constipation | 5/5 | 100 | 9/10 | 90 | 1 |
| Localized hirsutism | 0/5 | 0 | 4/18 | 22.2 | 0.539 |
Uncorrected p value from Fisher's exact test. No results were significant following Bonferroni correction for multiple testing.
Genetic Studies
All subjects were analyzed for the purpose of clinical diagnostics with different oligonucleotide or bacterial artificial chromosome (BAC) array platforms available in academic or commercial diagnostic laboratories. All genomic positions are provided for human genome build NCBI36/hg18 because this was the common reference from the clinical diagnostic laboratories (UCSC Genome Browser, see Web Resources). Two translocation cases were analyzed by massively parallel paired-end sequencing on Illumina sequencing platforms (Illumina). DNA from DGAP142 was previously sequenced by a DNA capture method (CapBP) on the basis of cytogenetic analyses that had narrowed the breakpoint region to <1 Mb.23 DNA from SMS373 was sequenced with a customized whole-genome large insert jumping library. This method sequences the ends of fragments that were manipulated by molecular techniques to be separated by 3.5 kb inserts and yields very high physical coverage of the genome by the inserts between paired-end reads at minimal sequencing costs. In brief, 20 μg of DNA was sheared by a Covaris S2 and size selected around 3.5 kb. Cap adaptors with EcoP15I restriction sites were ligated to the ends, and fragments were circularized with an internal oligonucleotide adaptor containing a subject-specific barcode and a single biotinylated thymine. Restriction enzyme digestion was then performed, and biotinylated fragments containing the circularization junction were bound to streptavidin beads followed by standard library preparation with NEBnext reagents (New England Biolabs) and Illumina paired-end adaptors (see Talkowski et al.23 for a detailed description of all protocols and reagents).
To assess further the observed association between microdeletion of MBD5 (RefSeq NM_018328.4) and ASD, we performed a low-cost, high-throughput pooled PCR next-generation sequencing analysis of all MBD5 exons as previously described.24 In brief, we designed PCR amplicons spanning all coding and noncoding MBD5 exons, and we designed primers with a NotI restriction site (Table S1, available online). Samples were quantified by PicoGreen (Quant-iT) and normalized to equimolar concentration. PCR amplification was then performed for all MBD5 exons in all individuals and the products were again quantified by PicoGreen and normalized. Failed PCRs were repeated. As described by Calvo et al.,24 products were then concatemerized, size selected, randomly sheared by a Covaris S2, and size selected again, followed by Illumina paired-end library preparation of the sheared products (Illumina). Pooled libraries were quantified by qPCR with a serially diluted Illumina PhiX sample as a standard curve and sequencing was performed on two lanes of an Illumina HiSeq2000 with a targeted minimum read depth of 100× per exonic base (see Calvo et al.24 for complete details). The methodology was designed to be sensitive to detect variants with multiple occurrences in the case pools but was unlikely to have power to identify mutations in a single individual, though one such variant (p.1048Thr>Ile [c.3143C>T]) was discovered and validated. Following analysis (see below), confirmatory genotyping of all putative variants was performed with Sequenom iPLEX chemistry (Sequenom) on the 600 individuals in the pooled sequencing analysis as well as 147 additional samples available from the same resources (Simon's Foundation and Autism Consortium of Boston). To exclude any possible artifacts from pooled sequencing and genotypes, all mutations validated by Sequenom were again confirmed by capillary sequencing of both forward and reverse strands in the ASD case, both parents, and any available siblings.
In all sequencing experiments (breakpoint detection or mutation screening), reads were aligned with BWA25 or Novoalign (Novocraft). For translocations, BAM files were processed to identify rearrangement breakpoints by a freely available C++ program, BamStat (see Web Resources), developed to tabulate mapping statistics and output lists of anomalous read-pairs (defined as having ends that map to two different chromosomes, an abnormal insert size, or unexpected strand orientations).23 Mutation analysis was performed with the Genome Analysis Toolkit26,27 and Syzygy.28 Because the study design involved a case-only analysis, no association tests were performed in Syzygy, instead Syzygy was used exclusively to identify putative mutations for further analysis.
Expression Analyses
Samples available for expression analyses are highlighted in green in Figures 1 and 2. Lymphoblastoid cell lines (LCLs; human lymphocytes transformed with Epstein-Barr virus) from 2q23.1 deletion syndrome patients and controls were cultured according to standard methods. RNA was isolated from LCLs or lymphocytes collected from fresh blood and cDNA was prepared according to published methods.29 Quantitative RT-PCR was performed for mRNA expression of MBD5 and EPC2 as previously described.2 Briefly, predesigned assays on Demand Gene Expression Products (Fermentus, Glen Burnie, MD) Taqman minor groove binder (MGB) probes for MBD5 and EPC2 were used (Applied Biosystems). GAPDH (MIM 138400) was used as the endogenous control. All samples of cDNA were run in triplicate in 10 μl reaction volumes. Taqman Universal PCR Master Mix (Applied Biosystems), the probe, and deionized water were mixed together in a fixed ratio, and diluted cDNA (1:5) was then added to each well. PCR conditions were the Standard 7500 Run mode of the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems). Cycle threshold (CT) was determined during the geometric phase of the PCR amplification plots, as recommended by the manufacturer. Relative differences in transcript levels were quantified according to the ΔΔCt method and normalized to GAPDH. Acquired data were analyzed with 7500 Fast System SDS Software (Applied Biosystems). Standard error was generated for each sample as previously described, and significance (p < 0.05) was determined by a one-sample t test compared to theoretical mean of 1.0 (p < 0.001 for all deletion cases). Results are expressed as fold-change relative to 1.0 (normal control). EPC2 expression was evaluated to rule out positional effects because of singular deletion of MBD5 (Figure S1).
Figure 1.
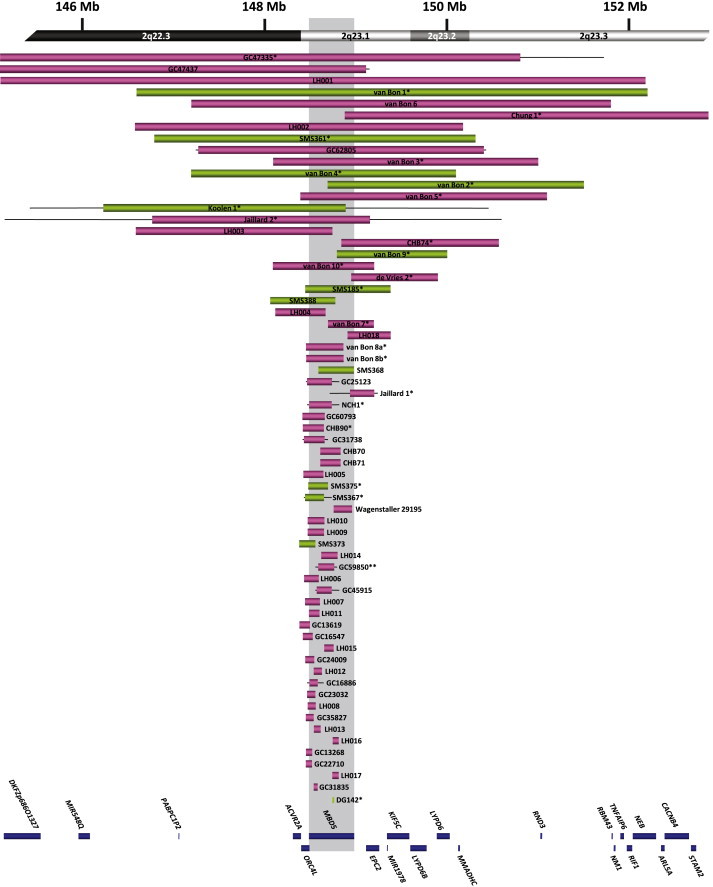
Delineation of the 2q23.1 Critical Region
Schematic representation of MBD5-containing deletions and translocation breakpoints in this report and those previously reported in the literature, arranged from largest to smallest. Boxes represent the minimum size of the deletions, and the horizontal lines extend through gaps in coverage to show the maximum deletion sizes. Green boxes represent individuals with MBD5 expression studies reported here. Single asterisks indicate cases known to be de novo; a double asterisk indicates a single inherited case in the cohort. Inheritance regarding all other cases is unknown. Genes within the region are represented by blue boxes, and the shaded region shows the location of MBD5.
Figure 2.
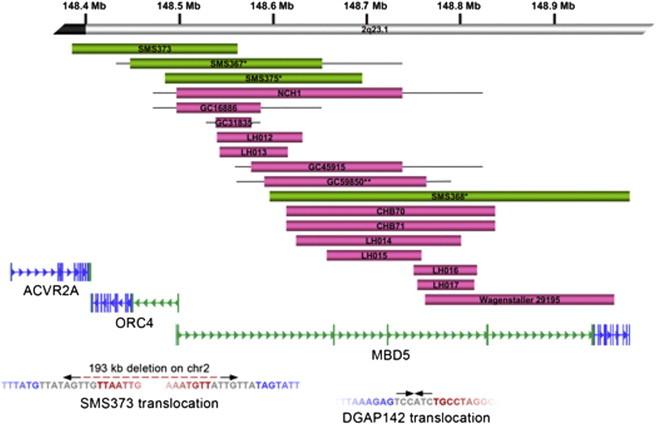
Deletions and Translocations Disrupting MBD5
(Top) Schematic representation of intragenic and partial deletions of MBD5. MBD5 genomic organization and mRNA (RefSeq Accession: NM_018328) are provided, including the large noncoding region (in green) between exon 1 and the coding sequence start site (in blue). Above the gene are the 18 microdeletions that partially and/or exclusively disrupt MBD5. These subjects had similar phenotypic features to individuals with deletions of the full 2q23.1 region (Table 1). Green bars represent those cases with expression data shown in Figure 4. Single asterisks indicate cases known to be de novo; the double asterisk indicates a single inherited case in the cohort. Inheritance regarding all other cases is unknown.
(Bottom) Translocation breakpoints in MBD5. Below the gene are paired-end sequencing data from two translocations found to disrupt MBD5. The arrows represent the orientation of sequence reads (inward facing for CapBP, outward facing for jumping libraries). The breakpoint on chromosome 2 in SMS373 was localized to the MBD5 noncoding region but determined to be an unbalanced rearrangement with derivative breakpoints separated by 193 kb on chromosome 2 and 106 kb on chromosome 10. DGAP142 yielded read pairs that localized the junction fragment to the same noncoding region23 with the loss of a single base at the chromosome 22 breakpoint in a region without annotated genes or functional sequences. Breakpoint sequences are given below the reads. The blue sequence represents the chromosome 2 breakpoint; the red sequence represents the chromosomal partner sequence, and the sequence at the breakpoint that does not map to either chromosome partner is inserted in gray. Breakpoints for both derivatives resulting in the 193 kb deletion of chromosome 2 are shown for SMS373. The second derivative of DGAP142 was balanced and identical to the reference.
Statistical Analyses
For the phenotype data, comparison of proportions in two-way contingency tables was performed with the Fisher's exact test as previously described.30,31 All reported p values are two-tailed. Given the known correlation between clinical variables, it is often difficult to assess the true number of independent tests performed. Though conservative, we performed Bonferroni correction for all pair-wise comparisons (Table 1). Statistical analysis for gene-expression data was performed with Prism 4 version 4.0b (GraphPad Software). A simple Fisher's exact test for allelic association was performed for the lone mutation tested.
Results
Analyses presented here sought to characterize genetic and phenotypic heterogeneity associated with the 2q23.1 microdeletion syndrome region and particularly to determine whether one gene or multiple genes in the region contribute to the abnormal phenotypes. Our analyses progressively focused on a single gene, MBD5, which we implicated as a single causal locus for the bulk of the characteristic phenotypic manifestations of the 2q23.1 microdeletion syndrome.
Refinement of the 2q23.1 Deletion Syndrome Critical Region
We identified 65 cases, including the 19 previously reported cases and 46 new cases, with microdeletions or translocation of 2q23.1 ranging from small deletions of 38 kb to >19 Mb, (Figure 1). Remarkably, after aligning the deletion regions, the smallest region of overlap (SRO) was defined to one gene, MBD5, that was within 2q23.1 and was common in all deletions and disrupted in both translocations (Figure 1). No other genes were included in the SRO for all deletions or were present in deletions that did not also involve MBD5. Fourteen (21.5%) of the 65 microdeletions and translocations were exclusively localized to the MBD5 locus, including several deletions that did not alter the MBD5 protein coding sequence because they were restricted to portions of the large noncoding region that contains multiple exons 5′ to the exon 6 translational start site (Figure 2, top).
The breakpoint capture sequencing previously performed for DGAP142, 46,XY,t(2;22)(q23.1;q13.31), identified a near perfectly balanced translocation between the long arms of chromosomes 2 and 22 that directly disrupted the 5′-noncoding region of MBD5 and did not impact any annotated genes, functional elements, or conserved sequences on 22q (Figure 2, bottom). Whole-genome sequencing of SMS373, 46,XY,t(2;10)(q23.1;q25.3), revealed an unbalanced translocation between the long arms of chromosomes 2 and 10 that also directly disrupted the 5′-noncoding region of MBD5 (Figure 2, bottom). Deletions were detected at the breakpoints of both chromosome partners; a 192,277 bp loss at the MBD5 breakpoint with insertion of 7 bp of foreign sequence that could not be uniquely aligned elsewhere in the genome (Figure 2, bottom) and a 106,412 bp deletion on chromosome 10 that included disruption of attractin-like 1 (ATRNL1 [MIM 612869]). Breakpoints in both cases were confirmed by PCR and Sanger sequencing.
Phenotype Analysis Indicates that Most Features of 2q23.1 Deletion Syndrome Are Associated with MBD5
Given the power of our sample and the common deletion of MBD5 in all cases (Figure 1), we were able to conduct a more comprehensive clinical characterization of 2q23.1 microdeletion syndrome, comparing 48 characteristic phenotypes between individuals with MBD5-specific deletions and those with broader 2q23.1 deletions to discriminate those phenotypes associated exclusively with MBD5 deletion (Table 1 and Figure 3). Results indicate that most (39/48 = 81.2%) phenotypic features of 2q23.1 deletion syndrome are consistent between both MBD5-specific alterations and larger 2q23.1 deletions. The frequently reported neurological, neurobehavioral, and craniofacial features associated with this syndrome were collectively observed in both cohorts. We report 13 out of 15 specific neurological and/or neurobehavioral features that were not significantly different between 2q23.1 deletion subjects and MBD5-specific disruption subjects, including developmental delay, motor delay, seizures, language impairment, and various behavioral problems (Table 1). In previous literature on 2q23.1, seizures and severe language impairment emerged as the two primary features associated with this disorder. In our assessment of severe language impairment in both cohorts, it is apparent that this feature is a common finding; 100% of 2q23.1 deletion cases and 71.4% of MBD5-specific disruption cases exhibit this phenotype. Seizures were also commonly reported in both groups at rates greater than >80%. We also set apart several craniofacial features, including thick or arched eyebrows, eye abnormalities, nasal abnormalities, thin upper lip, widely spaced teeth, and small chin, micrognathia, or retrognathia, present in statistically similar frequencies in both cohorts (Table 1). In addition, eye abnormalities that included esotropia, myopia, astigmatism, hypermetropia, and poor vision were observed in both cohorts. Individuals in both cohorts also exhibited either large prominent noses (older individuals) or small bulbous shaped noses (younger individuals) (see Figure 3). Note the thin upper lip observed in all 2q23.1 deletion subjects (Figure 3).
Figure 3.
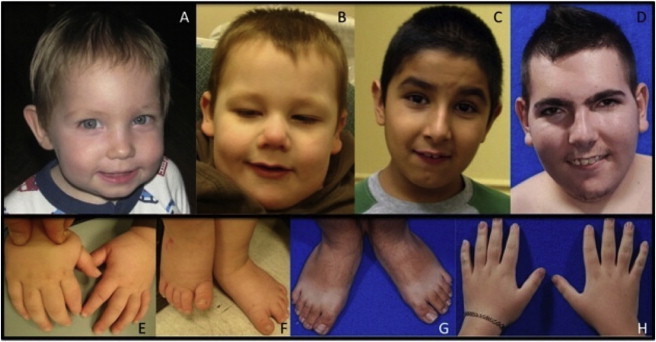
Clinical Features of Patients with Haploinsufficiency of MBD5
(A–D) The individuals presented have different 2q23.1 deletions (Figure 1). (A) SMS375, age 19 months. Note the microcephaly (45.8 cm, < 3rd percentile), broad forehead, bulbous nose, simple protruding ear lobes, and thin upper lip. (B) SMS373, age 2. Note the broad forehead, midface hypoplasia, high and broad nasal root, bulbous nose, fleshy ear lobes with bilateral Darwinian tubercle, and thin upper lip. (C) SMS367, age 7. Note low anterior hairline, midface retrusion (hypoplasia), large pronounced nose, and prominent columella. Note the space between the incisors. (D) SMS368, age 20. Note the bitemporal narrowing, synophrys, large pronounced nose, prominent ears, prominent columella, protruding upper teeth, and short neck.
(E) Hands of SMS373. Note the small plump hands, brachydactyly, and fifth finger clinodactyly.
(F) Feet of SMS373. Note the small feet and sandal gap between first and second toes.
(G) Feet of SMS368 illustrating small size and slight sandal gap between first and second toes.
(H) Hands of SMS368. Note the small hands with tapered fingers and the fifth finger clinodactyly.
Although it is apparent that a majority of the neurological and neurobehavioral features were consistent among 2q23.1 deletion and MBD5-specific disruption cases, key differences exist between the two cohorts; these include the presence in the 2q23.1 deletion cohort of ataxia, hyperphagia, postnatal growth retardation, a variety of craniofacial manifestations, and small hands and feet (Table 1). Hyperphagia is observed in 2q23.1 deletion cases (77.8%) and is not reported in any of the MBD5-specific disruptions (0%) (p = 0.021). Craniofacial manifestations overall also appear to be more frequent in 2q23.1 deletion patients (100%) than in MBD5-specific disruptions (71.4%) (p = 0.030). Specifically, five distinct craniofacial manifestations, microcephaly, external ear abnormalities, wide mouth, open mouth, and downturned corners of the mouth, appeared more commonly in 2q23.1 deletion subjects compared to MBD5-specific disruption subjects (Table 1), suggesting other genes could be involved in the etiology of these features. Further, external ear abnormalities such as prominent ears, small ears with large lobules, large simple cupped ears, low-set ears, or abnormal ears were observed at a higher frequency in patients with 2q23.1 deletions than in MBD5-specific disruptions (Table 1). The single feature that is significantly more prevalent in the 2q23.1 deletion cohort (75%) than in the MBD5-specific disruption cohort (20%) is small hands and feet (p = 0.036).
No patients with MBD5-specific disruptions were described as having brachycephaly, hypertelorism, macroglossia, short fifth digit of hand or feet, cardiovascular abnormalities, urogenital abnormalities, or localized hirsutism; however, the tests lack power because of the low number of cases observed. Some unique features that are reported in a few of the 2q23.1 deletion subjects are a high, narrow palate (5), genital abnormalities (5), a lower anterior hairline (4), pes planovalgus (3), long palpebral fissures (3) and mild metopic ridging (2).
Haploinsufficiency of MBD5
Prior to this study, we established haploinsufficiency of MBD5 in two larger 2q23.1 deletion cases (individuals SMS185 and SMS361);2 however, those cases had deletions involving other genes and also exhibited haploinsufficiency of EPC2 (Figure 1). Here, we assessed the impact of MBD5 promoter, 5′-noncoding exon, or coding exon deletions in 2q23.1 deletion subjects on MBD5 expression (Figure 4). We discovered that MBD5 mRNA expression levels were significantly reduced (22.5%–55.4%; p < 0.0001) across all 2q23.1 deletions studied when compared with the levels in normal controls (Figure 4). Because Williams et al.2 reported that EPC2 was haploinsufficient in the two large deletion cases, we also studied EPC2 mRNA levels in cell lines with MBD5-specific intragenic deletions and found normal levels of expression, indicating absence of positional effects from these events on EPC2 expression compared to controls (Figure S1). These data support haploinsufficiency of MBD5 as the only shared etiological finding among all cases evaluated, including those with alterations affecting the 5′-noncoding regions of the gene.
Figure 4.
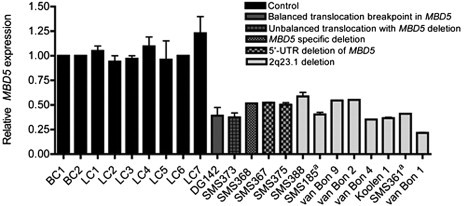
Reduced MBD5 Expression with MBD5 Deletions or Disruptions
Quantitative RT-PCR mRNA expression analysis of MBD5 is shown in lymphoblastoid cell lines or peripheral blood lymphocytes from individuals with MBD5 deletions (partial or complete) or disruptions of MBD5 and nine unaffected (no MBD5 deletion) subjects that were used as normal controls. Results were normalized to GAPDH expression. Relative expression values are based on the ΔΔCt value. Expression of all controls was normalized to one. Each bar represents mean (±standard error of the mean) of values from three to ten independent experiments. The error bars are present for each sample, but in some cases the error bars are too small to be seen. The data show a normal range of expression, 0.94- to 1.23-fold MBD5 expression, in lymphocytes (BC1-2) and lymphoblastoid cell lines (LCL1-7). Samples from cases described herein show 22%–55% expression of MBD5 (p < 0.0001 for all cases). aSMS185 and SMS361 were previously reported.2
Association of MBD5 with Autism
The genetic and phenotypic analyses suggested that the majority of 2q23.1 deletion cases exhibited autistic-like behaviors, some of which were referred with a clinical indication of ASD. As MBD5 has not been implicated previously in autism, we explored this hypothesis further. We estimated the frequency of the MBD5 microdeletion in ASD from a subset of the total clinical data for which we had clear evidence of the total number of subjects with ASD. From the Children's Hospital Boston data set, we identified three independent microdeletions in cases with ASD (a fourth case was a sibling who also carried the same microdeletion) from a total sample of 1786 ASD subjects (0.17%), and from a subset of the Signature Genomics data set with definitive phenotypic information and identical diagnostic array data, we observed a highly similar rate of four microdeletions of MBD5 from 2275 ASD cases (0.18%). A comparison to the 7878 controls screened for absence of psychiatric diagnosis found no deletions of the 2q23.1 segment, the full MBD5 locus, or any MBD5 coding exons, suggesting that haploinsufficiency of MBD5 is highly penetrant. A survey of the Database of Genomic Variants (DGV) found a very large duplication spanning many genes in the region but no deletions greater than 3 kb, all of which were confined to intronic sequences in the 5′-noncoding region.
Pooled sequencing of the MBD5 coding region and confirmatory individual sample genotyping in 747 cases diagnosed with ASD identified a previously described missense variant in the highly conserved and functionally critical methyl-binding domain (MBD), p.79Gly>Glu (rs34995577). Capillary sequencing confirmed an inherited mutation in six ASD cases (0.8% heterozygote frequency; 0.4% minor allele frequency; four of maternal inheritance, two of paternal inheritance). A second variant, p.1048Thr>Ile, was confirmed to be inherited in a single ASD case, but no other variants were observed more than once. The frequency of the p.79Gly>Glu variant in ASD subjects was significantly higher than the minor allele frequency in the ESP exome sequencing (3/2043 individuals sequenced, MAF = 0.07%), suggesting increased risk conferred to ASD cases by the mutant allele (p = 0.012, OR = 5.47, 95% confidence interval = 1.37–21.9). The G79 residue is conserved in human and mouse and highly conserved across MBD proteins, including MECP2 (MIM 300005), MBD1 (MIM 156535), MBD2 (MIM 603547), and MBD3 (MIM 603573).
Discussion
Our large-scale characterization of a microdeletion syndrome previously implicated in neurodevelopmental abnormalities revealed that a spectrum of genetic alterations results in highly similar phenotypic presentations, including small alterations localized to noncoding genomic regions. We identified 65 structural rearrangements spanning the 2q23.1 region, all of which disrupted a single gene in the critical region, MBD5, and included 14 independent structural variants (SVs) that solely interrupt MBD5. Extensive analysis of phenotypic features shows that partial or complete deletion of MBD5 leads to the core phenotype of intellectual disability, seizures, significant speech impairment, and behavioral problems observed in 2q23.1 deletion syndrome. Haploinsufficiency of MBD5 mRNA, including deletions of only the 5′-noncoding region, was confirmed in all cases evaluated and supports MBD5 as the major causative gene for 2q23.1 deletion syndrome and the core phenotype observed. Many of the 2q23.1 deletion cases exhibited ASD features, and we empirically estimated the frequency of MBD5 microdeletion and mutation of a highly conserved domain in MBD5 in ASD cases as well as comparison groups without ASD and found substantial risk conferred by MBD5 alteration. Our best estimates are that MBD5 deletion could impact one out of 500 to one out of 1000 ASD cases, and collectively, we find that up to 1% of ASD cases could have an alteration of MBD5 that confers genetic risk on the basis of the samples studied here.
MBD5 encodes a protein of the methyl-CpG-binding domain (MBD) family that also includes MECP2, a causative locus in Rett syndrome (RTT [MIM 312750])32 and is highly expressed in the brain, fetal testes, and fetal ovaries.32 As a member of the MBD protein family, the MBD5 protein has a 71 amino acid MBD from positions 11–81 of the protein. The p.79Gly>Glu variant, which is overrepresented in ASD in our study, changes amino acid 79 in the MBD from glycine to glutamate and is predicted by PolyPhen-2 (score = 0.995)33 to be damaging to the function of this highly conserved domain known to be involved in binding DNA at methylated CpGs. Specific cellular functions of other MBD family members have been well-described, but the function of MBD5 is currently not understood. A recent study suggested that MBD5 is unlikely to bind directly to methylated DNA but could contribute to the formation or function of heterochromatin.32 MBD5 could bind to DNA in a complex because it has been shown to interact directly with myocyte enhancer-binding factor 2C,34 encoded by MEF2C (MIM 600662), a gene known to play a crucial role in development and neurogenesis35 and to regulate expression of neuronal genes involved in formation of neuronal circuits and synaptic functions.10 Haploinsufficiency of MEF2C is associated with a microdeletion in 5q14.3 that results in an ASD phenotype, intellectual disability, seizures, and hypotonia,36 strikingly similar to the phenotype described here for haploinsufficiency of MBD5. Thus, MBD5 and MEF2C might function in a common pathway in neurogenesis, which, when disrupted, reveals characteristic neurodevelopmental phenotypes.
This analysis is the first comparative clinical study of this magnitude to define the role of a single gene in the clinical manifestations of 2q23.1 microdeletion syndrome and ASD and/or autism. Overall, ∼84% of phenotypic features evaluated were consistently observed in both MBD5-specific deletion and 2q23.1 deletion cases, suggesting that the overall syndrome phenotype is primarily due to MBD5 deletion. Developmental delay, motor delay, language impairment, seizures, behavioral problems, several craniofacial manifestations (see Table 1), and constipation were present in all or a majority of MBD5-specific disruptions and 2q23.1 deletions involving MBD5, supporting a significant role for MBD5 in these features. We also report that, unlike larger 2q23.1 deletion cases, MBD5-specific disruptions did not show microcephaly, certain neurological, growth, and craniofacial manifestations, or small hands and feet (Table 1). These findings suggest that altered gene dosage of other 2q23.1 genes probably modifies and contributes to the variable features in 2q23.1 deletion syndrome.
Our study specifically supports a role for MBD5 in ASD. Data from numerous autism CNV studies suggest that genetic etiology in at least 10% of autism patients can be attributed to genetic syndromes and known chromosomal anomalies, and estimates will increase as the resolution of screening tools improves.37 Further, both deletions and duplications, particularly involving chromosomal regions 16p13.11, 15q13.3, and 15q11.2, have been productive in identifying genomic architectural changes that are clearly associated with epilepsy and other developmental conditions; there are similar findings in both autism and schizophrenia cohorts, demonstrating enrichment of these and other copy-number regions compared to appropriate controls.38–41 Because ∼5%–15% of neurodevelopmental disorders, such as fragile X syndrome (MIM 300624; FMR1 [MIM 309550]), RTT (MECP2), Angelman syndrome (MIM 105830; UBE3A [MIM 601623]), Smith-Magenis syndrome (MIM 182290; RAI1 [MIM 607642]) and Potocki-Lupski syndrome (MIM 601883; RAI1), tuberous sclerosis complex (MIM 191100; 613254; TSC1 [MIM 605284], TSC2 [MIM 191092]), neurofibromatosis type 1 (MIM 162200; NF1 [MIM 613113]), and PTEN hamartoma syndrome (PTEN [MIM 601728]),42,43 are associated with a high prevalence of autism and are caused by single genes, they can offer tremendous insight into the pathogenesis of autism through the investigation of protein and gene pathway interactions. Adding MBD5 to the list of highly penetrant single genes that can contribute to ASD provides additional support for genes important in DNA methylation and chromatin remodeling in autism etiology. Other members of the MBD protein family have also been implicated in ASD and/or autism, most particularly MECP2, whose mutation or deletion can lead to RTT, seizures, intellectual disabilities, and/or autism.44,45 In support of these findings, existing mouse models for inactivation of many of the MBD family genes display autistic-like behavioral phenotypes that could be valuable for investigating the pathophysiology of autism-predisposing mutations and for identifying treatments.45,46 The data presented here further support a role for the MBD family in the etiology of ASD.
These analyses could represent a lower limit for the overall involvement of MBD5 in ASD because clinical data were limited for many individuals with detected microdeletions, the array resolution was too low on some platforms to adequately detect small alterations, and our sequencing screen for mutation was limited to alleles observed multiple times in the patient pools, meaning it was insensitive to any potential accumulation of single private mutations at a higher burden than in controls. The comparison of our data set to the available controls suggests large deletions of MBD5 are highly deleterious and penetrant, as no variations were observed from 7878 controls. Notably, the resolution of the CNV calls from the microarray studies was low, and only variants >100 kb could be confidently called. A higher resolution analysis could reveal smaller variants in controls, such as those seen in the 5′-noncoding region of the gene from DGV, which are intronic and less than 3 kb in size. Similarly, the p.79Gly>Glu variant is present in control populations at a significantly lower frequency than our cases. These data could suggest a mixed model of deleterious, fully penetrant deletions of MBD5 causing the syndromic disorder described here and reduced penetrance variants that significantly increase risk for ASD. A critical assessment of the allele frequency reported in additional ASD subjects and yet more controls will be required to determine the true effect size of the association reported here, which was limited to a relatively modest ASD cohort, but all variants were confirmed by multiple methods including capillary sequencing. It will also be of interest to evaluate ongoing autism exome sequencing to determine the total spectrum of MBD5 coding mutation burden, including those rare events that our methods were insensitive to detect, and to perform haplotype analyses of the region to determine if associated mutations are recurrent or identical by descent in the patient populations such as for the p.79Gly>Glu variant. Importantly, our clinical detection of ASD cases with these small alterations in the noncoding regions of MBD5 points to the need for greater sensitivity and analysis of current clinical diagnostic screening and proposes an alternative to the yet unexplained heritability in ASD from studies targeting large dosage changes or strictly de novo mutations identified from exome sequencing.
Our large-scale characterization of a microdeletion syndrome previously implicated in neurodevelopmental abnormalities reveals that a complex spectrum of genetic alterations, from point mutations and small deletions in noncoding regions through to larger chromosomal rearrangements, can result in highly similar phenotypic presentations, including ASD, because of effects on the same critical gene. These results suggest that the impact of both gross and quite subtle mutations at select loci could account for a significant portion of the overall disease risk in ASD and other neurodevelopmental disorders and that microdeletion syndrome regions provide a potential route to identifying these loci. Given similar findings supporting the importance of single genes in smaller studies of other recurrent microdeletion syndromes,10,12,47,48 small gene-specific SVs can result in a similar phenotype to that seen in the larger deletion syndrome. In addition to revealing the need for specific consideration of MBD5 in the molecular diagnosis of certain neurodevelopmental disorders, including ASD, our findings argue strongly for similar collaborative experimental approaches to screening of additional microdeletion syndromes for specific genetic factors contributing to neurodevelopmental abnormalities.
Acknowledgments
We are thankful to all of the subjects and families involved in this study. We are grateful for the assistance of Jennifer Defant, Santhosh Girirajan, Stephen Williams, Zalak Shah, and Tracey Oh in the collection of clinical data and in the preparation of this manuscript. We thank Pamela Sklar and the International Schizophrenia Consortium for contributing control data and Nancy J. Van Vranken for patient referral. We are also grateful to Manuel Rivas for assistance with the Syzygy analysis. We thank the Fondation Jérôme Lejeune (S.H.E.) for funding portions of this study, Developmental Genome Anatomy Project(GM061354), HD065286, National “973” project on Population and Health of China (2010CB529601), Science and Technology Council of Shanghai (09JC1402400 and 09ZR1404500), and the Simons Foundation Autism Research Initiative. M.E.T. was supported by a National Institutes of Health National Research Service Award (F32MH087123). The authors would also like to thank the NHLBI GO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the Women's Health Initiative Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). Fichera and Romano were supported by a grant from the “5 per mille” program.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BamStat and other sequence alignment tools: http://mappingtools.chgr.org/
Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA http://snp.gs.washington.edu/popgenSNP/
Genome Analysis Toolkit: http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Vissers L.E., de Vries B.B., Osoegawa K., Janssen I.M., Feuth T., Choy C.O., Straatman H., van der Vliet W., Huys E.H., van Rijk A. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams S.R., Mullegama S.V., Rosenfeld J.A., Dagli A.I., Hatchwell E., Allen W.P., Williams C.A., Elsea S.H. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur. J. Hum. Genet. 2010;18:436–441. doi: 10.1038/ejhg.2009.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagenstaller J., Spranger S., Lorenz-Depiereux B., Kazmierczak B., Nathrath M., Wahl D., Heye B., Glaser D., Liebscher V., Meitinger T., Strom T.M. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am. J. Hum. Genet. 2007;81:768–779. doi: 10.1086/521274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Bon B.W., Koolen D.A., Brueton L., McMullan D., Lichtenbelt K.D., Adès L.C., Peters G., Gibson K., Moloney S., Novara F. The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur. J. Hum. Genet. 2010;18:163–170. doi: 10.1038/ejhg.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koolen D.A., Vissers L.E., Nillesen W., Smeets D., van Ravenswaaij C.M., Sistermans E.A., Veltman J.A., de Vries B.B. A novel microdeletion, del(2)(q22.3q23.3) in a mentally retarded patient, detected by array-based comparative genomic hybridization. Clin. Genet. 2004;65:429–432. doi: 10.1111/j.0009-9163.2004.00245.x. [DOI] [PubMed] [Google Scholar]
- 6.Jaillard S., Dubourg C., Gérard-Blanluet M., Delahaye A., Pasquier L., Dupont C., Henry C., Tabet A.C., Lucas J., Aboura A. 2q23.1 microdeletion identified by array comparative genomic hybridisation: an emerging phenotype with Angelman-like features? J. Med. Genet. 2009;46:847–855. doi: 10.1136/jmg.2008.058156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Vries B.B., Pfundt R., Leisink M., Koolen D.A., Vissers L.E., Janssen I.M., Reijmersdal S., Nillesen W.M., Huys E.H., Leeuw N. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Gregori M., Ciccone R., Magini P., Pramparo T., Gimelli S., Messa J., Novara F., Vetro A., Rossi E., Maraschio P. Cryptic deletions are a common finding in “balanced” reciprocal and complex chromosome rearrangements: a study of 59 patients. J. Med. Genet. 2007;44:750–762. doi: 10.1136/jmg.2007.052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung B.H., Stavropoulos J., Marshall C.R., Weksberg R., Scherer S.W., Yoon G. 2q23 de novo microdeletion involving the MBD5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. Am. J. Med. Genet. A. 2011;155A:424–429. doi: 10.1002/ajmg.a.33821. [DOI] [PubMed] [Google Scholar]
- 10.Toro R., Konyukh M., Delorme R., Leblond C., Chaste P., Fauchereau F., Coleman M., Leboyer M., Gillberg C., Bourgeron T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld J.A., Ballif B.C., Lucas A., Spence E.J., Powell C., Aylsworth A.S., Torchia B.A., Shaffer L.G. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS ONE. 2009;4:e6568. doi: 10.1371/journal.pone.0006568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Durand C.M., Perroy J., Loll F., Perrais D., Fagni L., Bourgeron T., Montcouquiol M., Sans N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry. 2011 doi: 10.1038/mp.2011.57. Published online May 24, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aliferis K., Marsal C., Pelletier V., Doray B., Weiss M.M., Tops C.M., Speeg-Schatz C., Lesnik S.A., Dollfus H. A novel nonsense B3GALTL mutation confirms Peters plus syndrome in a patient with multiple malformations and Peters anomaly. Ophthalmic Genet. 2010;31:205–208. doi: 10.3109/13816810.2010.512355. [DOI] [PubMed] [Google Scholar]
- 14.Lord C., Rutter M., Goode S., Heemsbergen J., Jordan H., Mawhood L., Schopler E. Autism diagnostic observation schedule: a standardized observation of communicative and social behavior. J. Autism Dev. Disord. 1989;19:185–212. doi: 10.1007/BF02211841. [DOI] [PubMed] [Google Scholar]
- 15.Luyster R., Gotham K., Guthrie W., Coffing M., Petrak R., Pierce K., Bishop S., Esler A., Hus V., Oti R. The Autism Diagnostic Observation Schedule-toddler module: a new module of a standardized diagnostic measure for autism spectrum disorders. J. Autism Dev. Disord. 2009;39:1305–1320. doi: 10.1007/s10803-009-0746-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le Couteur A., Haden G., Hammal D., McConachie H. Diagnosing autism spectrum disorders in pre-school children using two standardised assessment instruments: the ADI-R and the ADOS. J. Autism Dev. Disord. 2008;38:362–372. doi: 10.1007/s10803-007-0403-3. [DOI] [PubMed] [Google Scholar]
- 17.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O'Donovan M.C., Craddock N., Norton N., Williams H., Peirce T., Moskvina V., Nikolov I., Hamshere M., Carroll L., Georgieva L., Molecular Genetics of Schizophrenia Collaboration Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 19.Sullivan P.F., Lin D., Tzeng J.Y., van den Oord E., Perkins D., Stroup T.S., Wagner M., Lee S., Wright F.A., Zou F. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol. Psychiatry. 2008;13:570–584. doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell S.M., Wray N.R., Stone J.L., Visscher P.M., O'Donovan M.C., Sullivan P.F., Sklar P., International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith E.N., Bloss C.S., Badner J.A., Barrett T., Belmonte P.L., Berrettini W., Byerley W., Coryell W., Craig D., Edenberg H.J. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol. Psychiatry. 2009;14:755–763. doi: 10.1038/mp.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferreira M.A., O'Donovan M.C., Meng Y.A., Jones I.R., Ruderfer D.M., Jones L., Fan J., Kirov G., Perlis R.H., Green E.K., Wellcome Trust Case Control Consortium Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Talkowski M.E., Ernst C., Heilbut A., Chiang C., Hanscom C., Lindgren A., Kirby A., Liu S., Muddukrishna B., Ohsumi T.K. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am. J. Hum. Genet. 2011;88:469–481. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calvo S.E., Tucker E.J., Compton A.G., Kirby D.M., Crawford G., Burtt N.P., Rivas M., Guiducci C., Bruno D.L., Goldberger O.A. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 2010;42:851–858. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rivas M. Deep Sequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011 doi: 10.1038/ng.952. Published online October 9, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Girirajan S., Truong H.T., Blanchard C.L., Elsea S.H. A functional network module for Smith-Magenis syndrome. Clin. Genet. 2009;75:364–374. doi: 10.1111/j.1399-0004.2008.01135.x. [DOI] [PubMed] [Google Scholar]
- 30.Girirajan S., Vlangos C.N., Szomju B.B., Edelman E., Trevors C.D., Dupuis L., Nezarati M., Bunyan D.J., Elsea S.H. Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet. Med. 2006;8:417–427. doi: 10.1097/01.gim.0000228215.32110.89. [DOI] [PubMed] [Google Scholar]
- 31.Edelman E.A., Girirajan S., Finucane B., Patel P.I., Lupski J.R., Smith A.C., Elsea S.H. Gender, genotype, and phenotype differences in Smith-Magenis syndrome: a meta-analysis of 105 cases. Clin. Genet. 2007;71:540–550. doi: 10.1111/j.1399-0004.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- 32.Laget S., Joulie M., Le Masson F., Sasai N., Christians E., Pradhan S., Roberts R.J., Defossez P.A. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE. 2010;5:e11982. doi: 10.1371/journal.pone.0011982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bandyopadhyay S., Chiang C.Y., Srivastava J., Gersten M., White S., Bell R., Kurschner C., Martin C.H., Smoot M., Sahasrabudhe S. A human MAP kinase interactome. Nat. Methods. 2010;7:801–805. doi: 10.1038/nmeth.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Radford J.C., Ragusa M.J., Shea K.L., McKercher S.R., Zaremba J.D., Soussou W., Nie Z., Kang Y.J., Nakanishi N. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:9397–9402. doi: 10.1073/pnas.0802876105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nowakowska B.A., Obersztyn E., Szymańska K., Bekiesińska-Figatowska M., Xia Z., Ricks C.B., Bocian E., Stockton D.W., Szczałuba K., Nawara M. Severe mental retardation, seizures, and hypotonia due to deletions of MEF2C. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2010;153B:1042–1051. doi: 10.1002/ajmg.b.31071. [DOI] [PubMed] [Google Scholar]
- 37.Buchanan J.A., Scherer S.W. Contemplating effects of genomic structural variation. Genet. Med. 2008;10:639–647. doi: 10.1097/gim.0b013e318183f848. [DOI] [PubMed] [Google Scholar]
- 38.Bremer A., Giacobini M., Eriksson M., Gustavsson P., Nordin V., Fernell E., Gillberg C., Nordgren A., Uppströmer A., Anderlid B.M. Copy number variation characteristics in subpopulations of patients with autism spectrum disorders. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2011;156:115–124. doi: 10.1002/ajmg.b.31142. [DOI] [PubMed] [Google Scholar]
- 39.Levinson D.F., Duan J., Oh S., Wang K., Sanders A.R., Shi J., Zhang N., Mowry B.J., Olincy A., Amin F. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry. 2011;168:302–316. doi: 10.1176/appi.ajp.2010.10060876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cichon S., Craddock N., Daly M., Faraone S.V., Gejman P.V., Kelsoe J., Lehner T., Levinson D.F., Moran A., Sklar P., Sullivan P.F., Psychiatric GWAS Consortium Coordinating Committee Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am. J. Psychiatry. 2009;166:540–556. doi: 10.1176/appi.ajp.2008.08091354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenfeld J.A., Ballif B.C., Torchia B.S., Sahoo T., Ravnan J.B., Schultz R., Lamb A., Bejjani B.A., Shaffer L.G. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet. Med. 2010;12:694–702. doi: 10.1097/GIM.0b013e3181f0c5f3. [DOI] [PubMed] [Google Scholar]
- 42.Kelleher R.J., 3rd, Bear M.F. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 43.Seno M.M., Hu P., Gwadry F.G., Pinto D., Marshall C.R., Casallo G., Scherer S.W. Gene and miRNA expression profiles in autism spectrum disorders. Brain Res. 2011;1380:85–97. doi: 10.1016/j.brainres.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 44.Nagarajan R.P., Hogart A.R., Gwye Y., Martin M.R., LaSalle J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1:e1–e11. doi: 10.4161/epi.1.4.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H., Yamagata T., Mori M., Yasuhara A., Momoi M.Y. Mutation analysis of methyl-CpG binding protein family genes in autistic patients. Brain Dev. 2005;27:321–325. doi: 10.1016/j.braindev.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Allan A.M., Liang X., Luo Y., Pak C., Li X., Szulwach K.E., Chen D., Jin P., Zhao X. The loss of methyl-CpG binding protein 1 leads to autism-like behavioral deficits. Hum. Mol. Genet. 2008;17:2047–2057. doi: 10.1093/hmg/ddn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauthier J., Siddiqui T.J., Huashan P., Yokomaku D., Hamdan F.F., Champagne N., Lapointe M., Spiegelman D., Noreau A., Lafrenière R.G. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 2011;4:563–573. doi: 10.1007/s00439-011-0975-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soysal Y., Vermeesch J., Davani N.A., Hekimler K., Imirzalioğlu N. A 10.46 Mb 12p11.1-12.1 interstitial deletion coincident with a 0.19 Mb NRXN1 deletion detected by array CGH in a girl with scoliosis and autism. Am. J. Med. Genet. A. 2011;155A:1745–1752. doi: 10.1002/ajmg.a.34101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.