

Abstract
Diabetes is associated with an increased risk of heart failure, in part explained by endoplasmic reticulum stress and apoptosis. Protein disulfide isomerase (PDI) prevents stressed cardiomyocytes apoptosis. We hypothesized that diabetes impairs PDI function by an alteration in its oxido-reductive state. Myocardial biopsies harvested from the anterolateral left ventricular wall from diabetic (n = 7) and nondiabetic (n = 8) patients were used to assess PDI expression and cardiomyocyte death. A mouse model of diabetes (streptozotocin injection, 130 mg/mL) was used to study PDI expression and its redox state after ischemia/reperfusion injury induced by 30-min occlusion of the left anterior coronary artery followed by reperfusion. Transthoracic echocardiography was performed to assess cardiac remodeling after 1 wk. Western blot analysis was used to analyze PDI expression, and methoxy-polyethyleneglycol-maleimide was used to assess its redox state. Dehydroascorbate (DHA) administration was used to restore the PDI redox state. Diabetic patients had a greater number of transferase-mediated dUTP nick-end labeling (TUNEL)-positive cells than nondiabetic patients despite a greater myocardial PDI expression suggesting altered PDI function. Diabetic mice had a worse postinfarction remodeling associated with an altered PDI redox state. DHA treatment restored functional PDI redox state and ameliorated post–myocardial infarction remodeling. An increase in PDI levels with a paradoxical decrease of its active form occurs in the diabetic heart after ischemia and may explain the lack of protective effects of PDI in diabetes. Restoration of PDI redox state prevents adverse remodeling. The potential significance of these findings deserves to be validated in a clinical setting.
INTRODUCTION
Ischemic heart disease is the most frequent cause of cardiovascular mortality in the United States and worldwide (1). Diabetes mellitus (DM) is recognized as a group of heterogeneous disorders with the common elements of hyperglycemia and glucose intolerance, due to insulin deficiency, impaired effectiveness of insulin action or both (2). Individuals with DM are at a significantly greater risk of developing coronary artery disease (3) and are at an increased risk of developing heart failure (4). These facts have led to the recognition of a distinct (and perhaps additive) disease process that directly affects the myocardium, a clinical entity termed “diabetic cardiomyopathy” (5). Perturbation of normal protein folding in DM leads to enhanced activation of the unfolded protein response in the diabetic heart, and failure of the unfolded protein response to reestablish protein folding leads to accumulation of dysfunctional proteins and to cell death (6,7). DM impairs the ability of the unfolded protein response to restore the physiological state by altering the function of its key components, one of which is the protein disulfide isomerase (PDI) (8). This enzyme is necessary for appropriate protein folding and prevention of protein misfolding during stress, as occurs during ischemic myocardial injury (9,10). In the liver of diabetic rodents, an altered redox state of PDI was reported that affects PDI’s function and leads to the accumulation of misfolded proteins in the endoplasmic reticulum (ER) lumen (9). We hypothesized that PDI protein expression may be altered in the diabetic heart because of the perturbation of protein folding and that diabetes can alter its redox state in the heart tissue, leading to increased susceptibility to damage. Here we analyzed (a) PDI expression and cell death in the heart of diabetic patients undergoing coronary artery bypass surgery and (b) PDI expression and redox state in a mouse model of diabetes and myocardial ischemia. Diabetic impairment of cardiac-protective mechanisms has been extensively reported. The aim of this study was to provide information regarding the lack of protective action of PDI during diabetes, starting from observational analysis in human samples of diabetic and nondiabetic patients and moving into a well-established mouse model of diabetes to detect the effects of hyperglycemia on PDI in the heart.
MATERIALS AND METHODS
Myocardial Sampling in Patients Undergoing Coronary Artery Bypass Surgery
The protocol for the harvesting and analysis of human samples was approved by an ethics committee at the Second University of Naples. Formal consent was obtained from all patients. Fifteen consecutive patients with angina pectoris or inducible myocardial ischemia and severe multivessel coronary artery disease, including disease of the left circumflex artery, requiring coronary artery bypass grafting as the mode of revascularization at a single center, were enrolled in the study: seven patients with DM (defined as any of the following: a chronic use of insulin or oral diabetic drugs, fasting glucose >200 mg/dL, or glycated hemoglobin >7.0%) and eight patients without DM. All procedures were performed by the same surgical team in a standardized fashion by a median sternotomy approach. A myocardial biopsy was harvested at the epicardial surface from the anterolateral left ventricular free wall, considered to be at ischemic risk, before institution of total cardiopulmonary bypass. Biopsies were collected in 10% paraformaldehyde phosphate-buffered solution.
Determination of PDI Expression and DNA Fragmentation in Cardiomyocytes
PDI expression was detected using a rabbit polyclonal antibody anti-PDI H-160 (Santa Cruz Technology, Santa Cruz, CA, USA) at 1:200 dilution. PDI expression was rated with a numeric score from +1 to +3 (+1 = mild; +2 = moderate; +3 = intense), proportional to the average staining intensity in the cardiomyocytes (10). Determination of apoptotic cardiomyo cytes by staining for transferase-mediated dUTP nick-end labeling (TUNEL) (for the detection of DNA fragmentation; Millipore, Billerica, MA, USA) was performed as previously described (10). The apoptotic rate was expressed as the number of apoptotic cardiomyocytes on all cardio myocytes per field. Immunohistochemistry and TUNEL scores were performed by two pathologists unaware of clinical data.
Experimental Model of DM in the Mouse
All animal experiments were conducted under the Guidelines on Humane Use and Care of Laboratory Animals for Biomedical Research, published by the National Institutes of Health (number 85-23, revised 1996). Adult male wild-type CD1 mice (Harlan, Indianapolis, IN, USA) were used for this experiment model. On their arrival, all animals were allowed to readjust to the housing environment for at least 3 d before any experiment, with freely accessible standard rodent food and water. Two groups of mice (10 mice/group) were treated with a single in-traperitoneal injection of 130 mg/kg streptozotocin (STZ) (Sigma-Aldrich, St. Louis, MO, USA), a dose known to induce diabetes due to cellular damage to the pancreatic β-cells. Blood samples were taken by puncturing the base of the ventral tail vein to measure nonfasting glucose level. Blood glucose levels were monitored at baseline and 3, 7, 21 and 28 d to follow the development of hyperglycemia and to monitor changes in glucose during all the experimental phases. Another two groups of mice (10 mice each) with matching age and weight were used as controls, being injected with a matching volume of vehicle solution (50 mmol/L sodium citrate, pH 4.0). After STZ treatment, nonfasting blood glucose levels were monitored at baseline and 3, 7, 21 and 28 d to follow the development and maintenance of hyperglycemia. The glucose plasma levels significantly increased by day 3, reaching a peak at day 7 and remaining increased until day 28.
Experimental Model of Myocardial Ischemia in the Mouse
Experimental acute myocardial infarction (AMI) was induced 21 d after DM induction in one STZ group and a matching control group. Ischemia/reperfusion (I/R) surgery was performed on day 21 as previously described (11). Briefly, the animals under anesthesia (70 mg/kg pentobarbital) underwent surgical opening of the chest and occlusion of the proximal left coronary artery for 30 min before reperfusion. Ten mice (five treated with STZ and five injected with vehicle) underwent a sham operation on day 21. On day 28, after echocardiographic analysis, the hearts were harvested and stored in liquid nitrogen for biochemical analysis. Before harvesting, the mice were injected with a high dose of pentobarbital (150 mg/kg) to induce overdose. The Institutional Animal Care and Use Committee of Virginia Commonwealth University approved the study.
Mouse Echocardiography
Doppler Echocardiography was performed using the Vevo770™ imaging system (VisualSonics, Toronto, Canada) and a 30-MHz probe in the mice under light anesthesia just before surgery (baseline echo) and 7 d after surgery, before sacrifice, as previously described (11). The M-mode cursor was positioned to measure anterior wall thickness (AWT) and posterior wall thickness (PWT) in diastole and systole and left ventricular end diastolic diameter (LVEDD) and left ventricular end systolic diameter (LVESD). Left ventricular fractional shortening (LVFS) was calculated as (LVEDD – LVESD)/LVEDD × 100.
Determination of PDI Expression and Red-Ox Status in the Mouse Heart
The samples were prepared for biochemical modifications to determine differences in the redox state of the PDI’s sulfidryl groups in the diabetic and non-diabetic samples, as previously described (12). All the chemicals used in the extraction protocol and in the sequence of reactions were purchased from Sigma-Aldrich. Methoxy-polyethyleneglycol-maleimide (mPEGmal) was used to covalently bind the reduced thiol groups in the sample that were initially engaged in disulfide bonds. The alkylation reaction with mPEGmal (pegylation) was performed using the method published by Appenzeller- Herzog and Ellgaard with some modifications (13). Briefly, the frozen tissues were powdered and incubated in ice-cold extraction buffer (80 mmol/L Tris-HCl, pH 7.3, 200 μmol/L phenylmethylsulfonyl fluoride) supplied with 20 mmol/L N-ethylmaleimide and 30% dimethyl sulfoxide (v/v). Samples were vortexed for 5 s, centrifuged for 1 min at 20,000g at 4°C, resuspended in the same buffer and incubated for 10 min on ice. After a second spinning, the tissue samples were placed in fresh extraction buffer without N-ethylmaleimide and homogenized passing the samples 10× through a 25-gauge needle syringe. After addition of sodium-dodecyl-sulfate (final concentration 0.1%), the samples were denatured for 10 min at 97°C and centrifuged 1 min at 20,000g at 25°C. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, final concentration 10 mmol/L) was added to the supernatants and incubated for 15 min to reduce the oxidized cysteines. mPEGmal (final concentration 15 mmol/L) was added and incubated for 1 h at room temperature. This sequence of reactions allowed the alkylation of the sulfhydryl groups gained in covalent bounds at the moment of harvest. Excess mPEGmal was removed by methanol/chloroform protein precipitation. The samples were resuspended in nonreducing Laemmly buffer 1× for Western blot analysis. Treatment of control tissue with 10 mmol/L dithiothreitol was performed for 5 min at 37°C in extraction buffer (80 mmol/L Tris-HCl, pH 7.3, 200 μmol/L phenylmethylsulfonyl fluoride) to assess the maximum reduction state of the proteins and to avoid a false- positive result. Protein analysis was performed using a rabbit anti–mouse polyclonal antibody raised against PDI (DL-11; Sigma-Aldrich) and monoclonal mouse anti–β-actin (clone C-2; Sigma-Aldrich). Densitometric analysis (Scion-Image, Scion Corporation, Frederick, MD, USA) was used to determine the protein expression of PDI and the differences in oxidized PDI, normalized for the β-actin quantity.
Induction of PDI by Ascorbic Acid and Tunicamycin in Cultured Cardiomyocytes
HL-1 cells were grown at 37°C (5% CO2 atmosphere) in Claycomb medium (Sigma-Aldrich), as previously described (10). In in vitro and in vivo models (14,15), ascorbic acid (AA) and tunicamicyn are known to modulate PDI activity and expression. We evaluated the ability of both the drugs to induce PDI expression in HL-1 cells and their effects on cardiomyocyte viability. In each experiment, the cells were incubated for 24 h with medium containing AA (0.2, 2 and 20 mmol/L) (Sigma-Aldrich) or tunicamycin (1, 2 and 3 μg/mL) (Sigma-Aldrich). PDI expression was determined by Western blot, as described above, using β-tubulin for the normalization. Trypan-blue dye (GIBCO, Carlsbad, CA, USA) was used to quantify the percentage of necrotic cells induced by the treatments in the cultured cardiomyocytes. Briefly, the medium was removed, the cells were washed and incubated with trypsin (0.05%, GIBCO) and the reaction was blocked with soybean trypsin inhibitor (GIBCO), as previously described (10). Harvested cells were pelleted at 500g for 5 min and resuspended in 0.005% trypan blue dye in PBS. Survival rate was expressed as percentage of try-pan blue–negative cells on the total amount of cells.
Treatment with DHA in Diabetic Mice
Given the ability of AA to enhance PDI expression and the known reductase activity of PDI (16) on the oxidized form of AA, dehydroascorbate (DHA), we explored the possibility that administration of DHA (for which cardiac intake is not significantly altered by diabetes [17]) would affect PDI expression and the redox state in diabetes, ultimately leading to reduced myocardial damage during I/R. One additional group of STZ-treated mice (n = 6) was treated daily by intraperitoneal injections with 200 mg/kg DHA (SigmaAldrich) starting 3 d before the surgical I/R procedure, continuing the treatment until day 7. Trans-thoracic echocardiography was performed before sacrifice, and the hearts were collected for PDI expression and redox state analysis.
Statistical Analysis
Statistical analysis was performed using the SPSS 11.0 package for Windows. Continuous variables are expressed as mean and standard error (SE). One-way analysis of variance was used to compare means between multiple (two or more) groups with the post hoc two-sided Dunnett test to specifically compare the between-subject effects versus controls in each group. The t test for unpaired data was used when comparing means between two groups only. Random-effects analysis of variance for repeated measures was used to compare pre- and postintervention echocardiographic parameters between the different groups with the post hoc two-sided Dunnett test to specifically compare the between-subject effects (streptozotocin and saline myocardial infarction groups). Correlations between two continuous variables were assessed using the Pearson test. Unadjusted two-tailed P values are reported throughout, with statistical significance set at the 0.05 level.
RESULTS
Demographic and Clinical Characteristics of the Patients
The clinical and pathological characteristics of the subjects are shown in Table 1. The median age of the patients was 60 years. All diabetic patients had diabetes for >1 year (mean 18 years). Diabetic patients had slightly greater BMI values and similar blood pressure values. Most of the patients had reduced left ventricular systolic function with a mean left ventricular ejection fraction of 38%. Tables 2 and 3 show biomarker values and the details of medical therapies of the diabetic versus nondiabetic patients. Diabetic patients had significantly higher values of blood glucose, insulin and glycated hemoglobin (Hb A1c) (all P values <0.001).
Table 1.
Clinical characteristics of the patients.
| Nondiabetic patients | Diabetic patients | |
|---|---|---|
| Number of patients | 8 | 7 |
| Age (years) | 60 ± 7 | 59 ± 8 |
| M/F | 4/4 | 4/3 |
| Diabetes duration (years) | — | 18 ± 7 |
| Body mass index (kg/m2) | 26 ± 0.6 | 28 ± 0.9 |
| Systolic blood pressure (mmHg) | 119 ± 6 | 121 ± 12 |
| Diastolic blood pressure (mmHg) | 80 ± 8 | 81 ± 9 |
| Heart rate (bpm) | 71 ± 3 | 75 ± 4 |
| Smokers [n (%)] | 6 (75) | 5 (71) |
| Angina pectoris [n (%)] | 4 (50) | 4 (57) |
| Hypertension [n (%)] | 3 (37) | 2 (28) |
| Left ventricular ejection fraction (%) | 38.6 ± 6.6 | 33.8 ± 7.6 |
| Left main coronary artery stenosis [n (%)] | 4 (50) | 4 (58) |
| Stenosis of left anterior descending artery and left circumflex artery [n (%)] | 4 (50) | 3 (42) |
Table 2.
Biomarkers.
| Nondiabetic patients | Diabetic patients | |
|---|---|---|
| Blood glucose (mg/dL) | 108 ± 15a | 232 ± 36 |
| Insulin (μU/mL) | 8.06 ± 3.3a | 10.6 ± 2.4 |
| Hb A1c (%) | 6.1 ± 0.7* | 8.3 ± 1.5 |
| Total cholesterol (mg/dL) | 264 ± 2 | 266 ± 2 |
| HDL cholesterol (mg/dL) | 49.5 ± 1.2 | 48.8 ± 1.6 |
| Triglycerides (mg/dL) | 198 ± 13 | 223 ± 14 |
| Serum creatinine (mg/dL) | 0.88 ± 0.03 | 0.89 ± 0.03 |
| Troponin I (ng/mL) | 1.0 ± 0.7 | 1.1 ± 0.9 |
Data are mean ± SE.
P < 0.001 versus diabetic patients.
Table 3.
Medical therapy.
| Nondiabetic patients | Diabetic patients | |
|---|---|---|
| β-Blocker | 3 (38) | 3 (42) |
| Diuretic | 2 (25) | 3 (42) |
| ACE inhibitors | 7 (87) | 6 (85) |
| Statin | 7 (87) | 6 (85) |
| Nitroglycerin | 7 (87) | 6 (85) |
| Sulfonylureas | — | 2 (28) |
| Metformin | — | 4 (57) |
| Insulin | — | 4 (57) |
Data are n (%).
PDI Expression and DNA Fragmentation in Human Cardiomyocytes
The immunohistochemical analysis of PDI in bioptic human heart samples collected from diabetic patients (Figures 1A–D) showed a marked difference in the staining of cardiomyocytes compared with nondiabetic controls. The expression score in the control hearts was heterogeneous, with samples showing different PDI staining (scores: +1 = 37.5%; +2 = 25%; +3 = 37.5%). In diabetic hearts, more samples showed an enhanced PDI, since none of the diabetic patients showed mild expression, with only two samples expressing moderate levels of PDI and six expressing high levels of PDI (scores: +1 = 0%; +2 = 28.6%; +3 = 71.4%), thus suggesting an upregulated PDI expression in the diabetic hearts. Cardiomyocyte death was assessed by TUNEL staining (Figures 1C–E) and showed a higher but not significantly different rate of TUNEL-positive cardiomyocytes in the diabetic patients’ hearts versus the control hearts (12 ± 2 versus 8 ± 2; P = 0.2). Moreover, in the control samples, the intensity of PDI staining was inversely proportional to the number of TUNEL-positive cells, thus supporting the concept that enhanced PDI expression may be protective in patients without diabetes (Figure 1E). In contrast, in the diabetic patients, there was no significant correlation between PDI expression and TUNEL-positive cell number. Comparing the two subgroups of patients with intense PDI expression, which would be expected to be protected by the enhanced PDI expression, the TUNEL-positive cardiomyocyte number was shown to be statistically significant lower in the nondiabetic versus diabetic patients (P < 0.05), suggesting perhaps a nonadequate protection of PDI in diabetic hearts. The degree of PDI expression in diabetic patients was directly correlated with plasma glucose and insulin levels (P < 0.05 for both correlations, data not shown), suggesting that the severity of diabetes/hyperglycemia may be related to PDI expression.
Figure 1.

PDI expression and cardiac cell death in nondiabetic and diabetic patients. (A–D) Bioptic myocardial tissue sections stained for PDI (A, B) and for TUNEL (C, D) to determine the ratio of PDI expression to cardiomyocyte cell death in nondiabetic (A, C) and diabetic (B, D) patients after myocardial ischemia. (E) Number of TUNEL-positive cells in samples with mild (+1), moderate (+2) and high expression of PDI. The values are expressed as percentage of TUNEL-positive cells per field on the total number of cardiomyocytes and are reported as interquartile ranges for both the groups of nondiabetic (n = 8) and diabetic (n = 7) patients.
STZ-Induced Diabetes Effects on PDI Myocardial Expression and Red-Ox Status
In this model of diabetes induced by STZ, the PDI protein expression in the mouse heart (Figures 2A, B) was not different comparing the diabetic, STZ-treated mice with the control mice. In contrast, the analysis of the PDI redox state showed a reduction of the most oxidized form, with a 70% reduction in the diabetic mouse hearts compared with the nondiabetic mice (P < 0.05) (Figures 2A, C).
Figure 2.
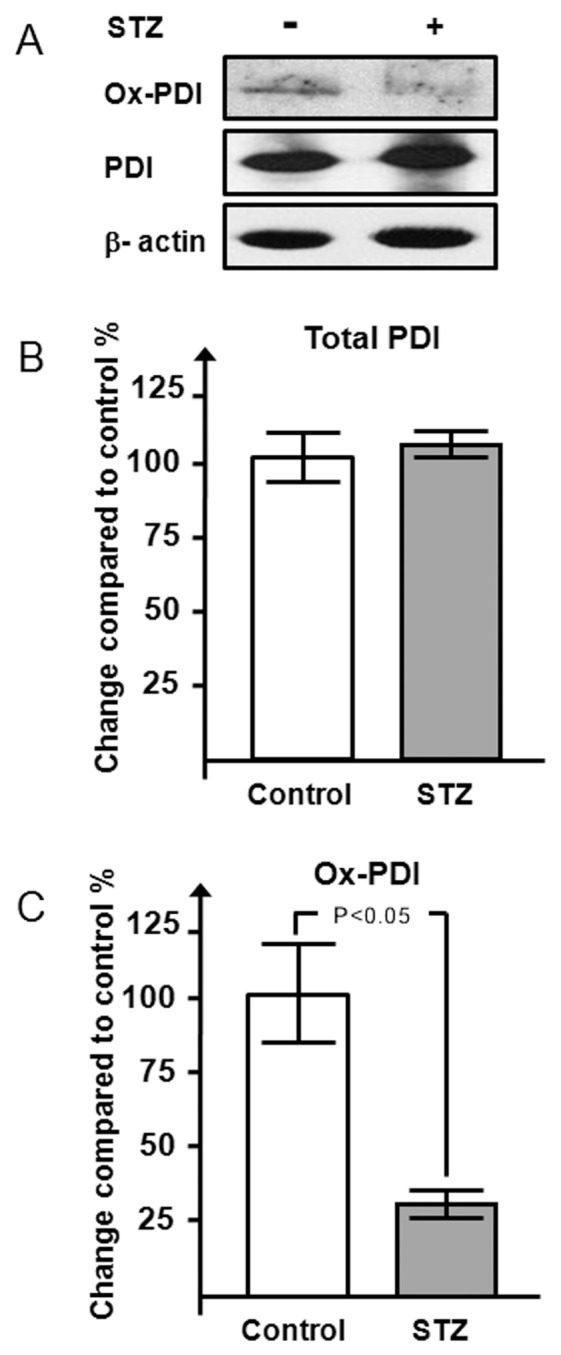
Effects of STZ-induced hyperglycemia on PDI redox state and expression in the mouse heart. Western blot (A) and densitometric analysis (B, C) of PDI and its oxidized form in STZ-treated mice (n = 6) versus vehicle-treated mice (n = 4) is shown. Results are expressed as mean ± SE percentage respect to control-vehicle–treated mice.
Ischemia-Induced Myocardial Damage and PDI Expression in Diabetic Mice
Seven days after surgery, cardiac function and remodeling were assessed by trans-thoracic echocardiography. The LVFS, at 7 d after AMI, was reduced by 16% in the STZ I/R mice (P = 0.05) (Figure 3A). The anterior wall diastolic thickness (AWDT) was significantly reduced by 40% in the STZ-treated group compared with the nondiabetic control group (0.40 ± 0.03 versus 0.70 ± 0.04 mm, P < 0.001), reflecting a greater degree of myocardial damage, with no changes between the sham and the STZ-sham operated group (P = NS). In the control group, the anterior wall systolic thickness (AWST) was markedly reduced compared with sham-operated mice (0.93 ± 0.09 versus 1.25 ± 0.04 mm; P < 0.05). Treatment with STZ further reduced the AWST (0.48 ± 0.06 versus 1.15 ± 0.04 mm in the STZ-sham group; P < 0.001), whereas the treatment with STZ alone did not produce any significant differences in AWT compared with the nondiabetic sham-operated mice (Figure 3B). On the other hand, the posterior wall diastolic thickness (PWDT) and posterior wall systolic thickness (PWST) were only slightly affected by I/R injury, as shown in Figure 3C.
Figure 3.

Echocardiographic assessment of left ventricular function and myocardial remodeling in diabetic mice 7 d after AMI. (A) LVFS of diabetic and nondiabetic mice after I/R injury or sham surgery (n = 4–7 per group). (B, C) Assessment on the anterior (B) and posterior (C) wall thicknesses (AWT and PWT) during diastole and systole (n = 4–7 per group). Values are expressed as mean ± SE.
Protein expression analysis (Figure 4A) showed that the I/R damage increased total PDI expression in both control- vehicle and STZ-treated mice (control sham 100 ± 13%; control STZ 112 ± 16%; control I/R 323 ± 29%; STZ I/R 301 ± 42%; P < 0.05 sham versus I/R for both control and STZ groups) (Figure 4B). The control vehicle-treated group following I/R had a normal quantity of oxidized PDI (93 ± 19% versus 100 ± 12% of control sham), whereas the diabetic I/R group experienced a reduction of PDI in the oxidized form (31 ± 9%; P < 0.05 versus control I/R), similar to that in the STZ diabetic noninfarct group (28 ± 5%).
Figure 4.
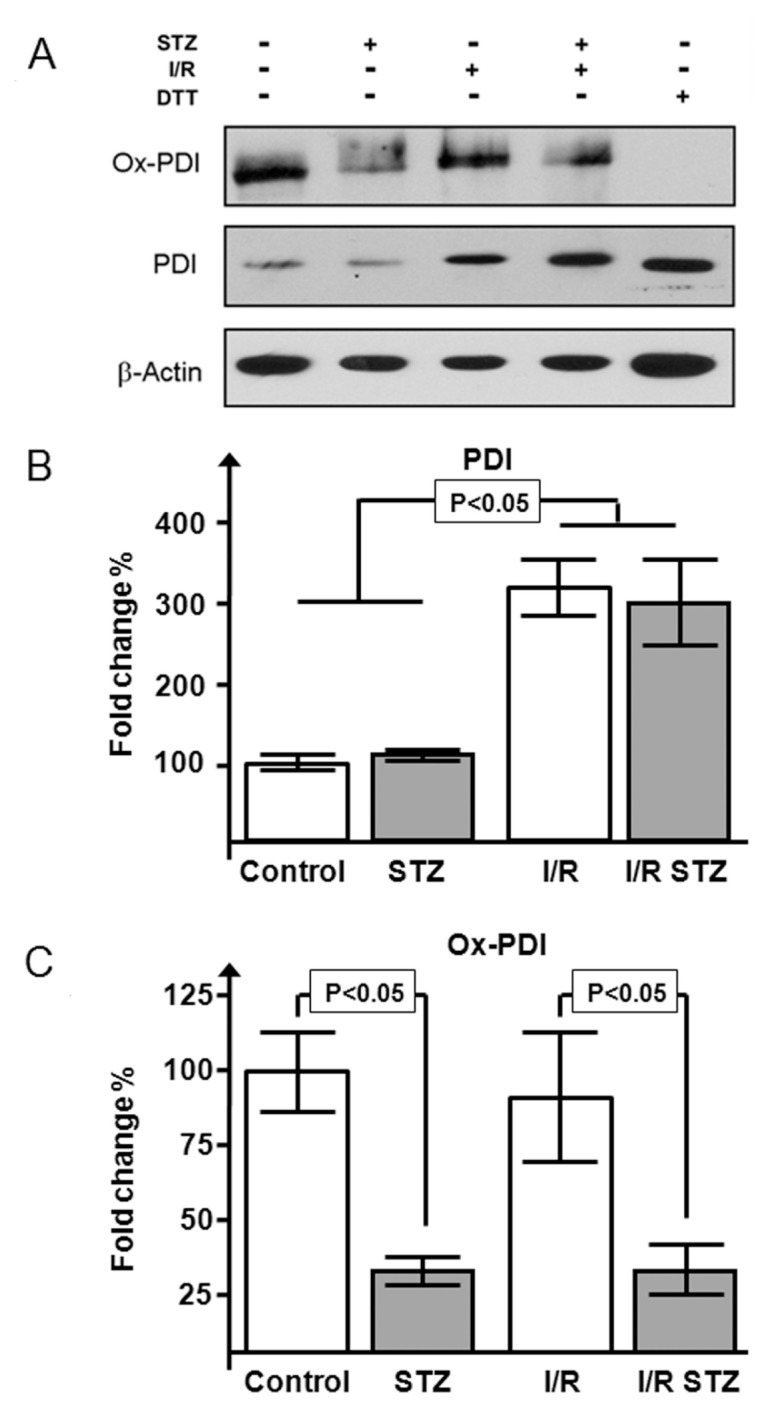
Effects of I/R injury on PDI expression and redox state in diabetic mice. Western blot (A) and densitometric analysis of PDI (B) and its oxidized form (C) in STZ-treated mice versus vehicle-treated mice after I/R (n = 4–6 per group) is shown. One dithiothreitol (DTT)-treated sample, being completely reduced, was used to exclude false-positive/nonspecific bands. Results are expressed as mean ± SE percentage with respect to control-vehicle–treated mice.
Pharmacological Induction of PDI in Cultured Cardiomyocytes
In an in vitro experiment, with the attempt to identify an agent able to increase the PDI expression maintaining a safe profile in cultured HL-1 cardiomyocytes, we tested the effects of increasing doses of AA (vitamin C) (0.2, 2 and 20 mmol/L) and tunicamicyn (1, 2 and 3 μg/mL) on PDI expression and HL-1 cells survival (Figures 5A, D). AA is essential for the hydroxylation of the collagen prolines, a reaction catalyzed by the PDI prolylhydroxylase complex. Tunicamicyn, on the other hand, has been shown to induce PDI expression via the enhancement of the unfolded protein response (15).
Figure 5.

PDI induction and cell viability after AA and tunicamicyn treatment in cultured cardiomyocytes. (A–C) Western blot (A) and densitometric analysis of PDI (B) in HL-1 cells after treatment with increasing doses (0.2, 2 and 20 mmol/L) of AA; cells were stained with trypan blue to exclude unhealthy cells (C). (D–F) Western blot (A) and densitometric analysis of PDI (B) were performed with increasing doses (1, 2 and 3 μg/mL) of tunicamicyn (Tun.) in HL-1 cells; trypan blue exclusion (C) was used to determine viable cells. Results are expressed as mean ± SE percentage with respect to control cells; n = 3 for each experimental point.
The lower dose of AA was inefficient to increase PDI protein levels (93 ± 8% versus 100 ± 7% of control), whereas the intermediate and the high dose enhanced PDI expression two-fold (P < 0.0002) (Figure 5B). Tunicamicyn significantly induced PDI expression at all the three doses tested (224 ± 14%, 231 ± 10% and 399 ± 12% for 1, 2 and 3 μg/mL, respectively; P < 0.0002 versus control) (Figure 5E). However, tunicamicyn caused a reduction of HL-1 cell viability (Figure 5F) at all the doses tested (85 ± 2% versus 96 ± 2% of control cells; P < 0.05), whereas all the concentrations of AA (Figure 5C) did not affect cardiomyocyte viability.
DHA Enhances Oxidized PDI Causing Reduction in Left Ventricular Wall Remodeling in the Heart of Diabetic Mice
Ascorbic acid (vitamin C) is an essential coenzyme for many reactions and can act as a free radical scavenger (18). DHA, the oxidized form of vitamin C, is transported via the facilitated-diffusion glucose transporters, GLUT 1, 3 and 4, being assimilated by cardiomyocytes (19,20). PDI was previously shown to possess a moderate DHA reductase activity (16,21), being able to convert DHA to AA, going from a reduced form to an oxidized form. Furthermore, in the ER microsomes of the liver of diabetic rats, an enhanced DHA reductase activity was observed (21). We explored the possibility that administration of DHA, for which myocardial uptake is apparently not altered by diabetes (17), could modify PDI expression and/or its redox state in our experimental model, reverting the detrimental effects of diabetes on post–myocardial infarction on the left ventricular wall remodeling. DHA treatment did not affect PDI protein expression during the AMI, yet it restored the oxidation of PDI (Figure 6A). Trans-thoracic echocardiography analysis (Figures 6B, C) showed that DHA administration preserved left ventricular function (P < 0.01 versus STZ I/R) and markedly preserved AWT in both systole and diastole (P < 0.01 versus STZ I/R mice).
Figure 6.

DHA ameliorates left ventricular function and remodeling reoxidizing PDI. (A) DHA effects on PDI oxidation after I/R injury. (B, C) Echocardiographic assessment of LVFS (B) and AWT in diastole and systole (C) in diabetic mice treated with DHA after I/R injury (n = 5–6 per group). Dashed boxes indicate the two groups analyzed by Western blot. Results are expressed as mean ± SE.
DISCUSSION
Most therapeutic approaches to limit tissue injury after myocardial ischemia are guided by the observation that the clinical outcome is improved with reduction of the initial ischemic injury (22,23). The fate of the cells in the region bordering the infarct is determined by the complex interactions between prosurvival and death factors (24–26). In diabetic patients, the risks of developing coronary artery disease and mortality after AMI are increased two- to four-fold compared with nondiabetic patients (3–5). Emerging data in the fields of both DM and AMI point to the role of ER stress (ERS) as one of the main events that can modulate cell tolerance to stress and survival after injury (6,8,10). Unsuccessful protein folding and ER dysfunction play an important role in the pathogenesis of several diseases, and the failure of ER chaperones appears to be important in the development of such pathologies (28). A large number of ERS-associated proteins have been shown to be involved in the development of several types of cardiomyopathies in both human samples and animal models (29). In a rat model of STZ-induced diabetes, ischemic damage induces the expression of glucose- responsive protein 78 (Grp 78, one of the major ERS inducers), which is associated with the expression and activation of caspase-12, an ER-activated caspase that induces apoptotic cell death when the ER is overloaded with misfolded proteins (30).
In a previous study, we found that PDI is upregulated in the viable peri-infarcted myocardium in the human heart and that adenoviral-mediated overexpression of PDI significantly reduced infarct size and cardiomyocyte apoptosis in the peri-infarct area and ameliorated the left ventricular remodeling compared with mice treated with an adenoviral control vector. These findings suggested that the unfolded protein response and ERS are activated after ischemic injury and that the increased expression of ER oxidoreductases and chaperones such as PDI plays a protective role against ischemic damage. In the current study, we analyzed the expression of PDI and the presence of TUNEL-positive cardiomyocytes in myocardial biopsies collected from diabetic and nondiabetic patients. Despite the low number of cases analyzed, we found an augmented cell death in the samples collected from diabetics compared with a general increase of PDI expression. This result is in contrast with the data derived from the analysis of the samples of the nondiabetic patients, in which PDI expression and the number of TUNEL-positive cells is inversely correlated. These data confirm our previous observation in which we found a greater expression of PDI associated with a lower apoptotic rate in autoptic samples (10). Among all the samples with the higher score of PDI staining (+3), the diabetics showed a significantly higher apoptotic rate. The enzyme PDI is a key moderator of the ERS, and it has been previously shown to be a potent antiapoptotic factor during acute ischemia (10). Mechanistically, this cardioprotection could be attributed to the ability of PDI to reduce ERS, dependent on the integrity of its active sites (10,12).
Altered PDI redox status, accompanied by ERS and accumulation of unfolded proteins with reduced cysteines in the ER lumen, has been previously reported (9). We used an experimental mouse model of diabetes to evaluate the effects of DM and severe hyperglycemia on cardiac remodeling and PDI expression and redox state after I/R injury to overcome the limitation linked to the availability of the human samples. In the normoglycemic mice, 7 d after surgery, we observed thinning of the anterior (infarct) wall. While STZ-induced diabetes per se did not affect cardiac dimensions in our mouse model, the myocardial damage due to I/R was significantly greater in diabetic versus normoglycemic mice. Mice with STZ-induced diabetes had an even more severe thinning of the anterior (infarcted) wall, reflecting a greater amount of damage in the infarct and peri-infarct areas. LVFS was markedly reduced by I/R and more so in the diabetic mice, despite a marked increase in PDI protein expression in both the groups.
We postulated that the function of PDI may be impaired in the diabetic hearts because of a change in the oxidation state of PDI cysteines. The two catalytic sites of PDI exist at equilibrium between both the oxidized and reduced form. We found that DM leads to an increase of the reduced form of PDI in the heart, which could lead to impaired folding of the substrate proteins and, hence, a lack of cellular protection. In the scenario of myocardial infarction, nutrient deprivation, increased oxidative stress, inflammation, protein synthesis and proper folding become crucial steps in the attempt to minimize cell structure damage, maintaining extracellular matrix integrity and cell function. Increased PDI protein expression and its reduced state could lead to an increased protein accumulation and ERS. This concept is supported by the antichaperone activity of PDI observed after pharmacological inhibition and during pathophysiological conditions such as amyotrophic lateral sclerosis (31).
Given the link between PDI and its DHA→AA reductase activity (16,20), we pretreated diabetic mice to attempt to restore PDI oxidation before I/R injury and continued the treatment after AMI. DHA treatment restored the oxidized state of PDI, which may be linked to a restored function of PDI.
There is evidence that early administration of antioxidant vitamins C and E in patients with AMI and concomitant DM reduces cardiac mortality (32) and that lower plasma concentrations of vitamin C seem to be associated with adverse changes in the microcirculation, peripheral arteries and ventricular repolarization (33).
We found that in the diabetic heart, PDI protein expression increase is not necessarily associated with cardiomyocyte survival, as shown by the human data; using a mouse model, we found that, in the diabetic heart, PDI is present in a mostly reduced form, which is disabled to perform disulfide bond formation in nascent proteins. We can speculate that, as is the case in the diabetic mouse, in the human samples from diabetic patients, PDI is in the reduced/nonactive form. Presently, it remains unresolved whether the discrepancy between increased PDI protein expression in the diabetic heart and decreased cardiomyocytes protection is a cause or a consequence of the more severe cardiac remodeling observed in diabetes. The demonstration of increased expression of a protective protein such as PDI does not imply a protective action; the activity of the protein (enzyme) must also be preserved. Whether a nonfunctional PDI may be even deleterious for the myocyte survival requires further investigation (34), even though the adenoviral overexpression of PDI point mutants unable to catalyze disulfide bond formation has been shown to be detrimental in HL-1 cells in normoxic and hypoxic conditions (10,12). DHA administration may be a viable strategy to restore PDI function. AA and DHA protect the heart of normoglycemic mice after I/R injury (19). We showed that, in diabetic mice, DHA ameliorates myocardial remodeling and maintains left ventricular function to a level comparable to the nondiabetics. This improvement is accompanied by the restoration of a normal level of oxidized PDI. In the microsomes collected from the liver of rats, other oxidoreductases have been found to be altered in their redox state by diabetes (9,35). We do not exclude that other ER-associated redox enzymes could be in an altered state in the diabetic heart and that DHA treatment could restore their function. Further studies are required to better analyze this issue.
In conclusion, PDI is an important component of the cellular response to is-chemic injury altered in diabetes by a dysregulation of its redox state. Restoration of the normal redox state of PDI by strategies such as DHA administration may result in a more favorable cardiac remodeling. Further studies are required to assess the therapeutic significance of targeting this pathway in a multidisciplinary approach to the therapy of the human diabetic heart.
ACKNOWLEDGMENTS
This work was supported by a Società Italiana di Cardiologia (SIC) training grant to S Toldo and by an American Heart Association Beginning Grant-in-Aid (Mid-Atlantic Affiliate) to A Abbate and by FUTURA-onlus to A Baldi.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.American Heart Association. Heart Disease and Stroke Statistics—2010 Update. Available at www.americanheart.org.
- 2.Harris M, Zimmet P. Classification of diabetes mellitus and other categories of glucose intolerance. In: Alberti K, Zimmet P, Defronzo R, editors. International Textbook of Diabetes Mellitus. 2nd ed. Chichester, U.K: Wiley; 1997. pp. 9–23. [Google Scholar]
- 3.Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 4.Rubler S, et al. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 5.Hayat SA, Patel B, Khattar RS, Malik RA. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci. 2004;107:539–57. doi: 10.1042/CS20040057. [DOI] [PubMed] [Google Scholar]
- 6.Xu J, et al. Diabetes- and angiotensin II- induced cardiac endoplasmic reticulum stress and cell death: metallothionein protection. J Cell Mol Med. 2009;13:1499–512. doi: 10.1111/j.1582-4934.2009.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakagawa T, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 8.Van der Kallen CJ, Van Greevenbroek MM, Stehouwer CD, Schalkwijk CG. Endoplasmic reticulum stress-induced apoptosis in the development of diabetes: is there a role for adipose tissue and liver. Apoptosis. 2009;14:1424–34. doi: 10.1007/s10495-009-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nardai G, et al. Diabetic changes in the redox status of the microsomal protein folding machinery. Biochem Biophys Res Commun. 2005;334:787–95. doi: 10.1016/j.bbrc.2005.06.172. [DOI] [PubMed] [Google Scholar]
- 10.Severino A, et al. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol. 2007;50:1029–37. doi: 10.1016/j.jacc.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Salloum FN, et al. Phosphodiesterase-5 inhibitor, tadalafil, protects against myocardial ischemia/reperfusion through protein-kinase g-dependent generation of hydrogen sulfide. Circulation. 2009;120:S31–6. doi: 10.1161/CIRCULATIONAHA.108.843979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toldo S, Severino A, Abbate A, Baldi A. The role of PDI as a survival factor in cardiomyocyte ischemia. Methods Enzymol. 2011;489:47–65. doi: 10.1016/B978-0-12-385116-1.00003-0. [DOI] [PubMed] [Google Scholar]
- 13.Appenzeller-Herzog C, Ellgaard L. In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid Redox Signal. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- 14.Ronchetti IP, Quaglino D, Jr, Bergamini G. Ascorbic acid and connective tissue. Subcell Biochem. 1996;25:249–64. doi: 10.1007/978-1-4613-0325-1_13. [DOI] [PubMed] [Google Scholar]
- 15.Bedard K, MacDonald N, Collins J, Cribb A. Cytoprotection following endoplasmic reticulum stress protein induction in continuous cell lines. Basic Clin Pharmacol Toxicol. 2004;94:124–31. doi: 10.1111/j.1742-7843.2004.pto940305.x. [DOI] [PubMed] [Google Scholar]
- 16.Wells WW, Xu DP, Yang YF, Rocque PA. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J Biol Chem. 1990;265:15361–4. [PubMed] [Google Scholar]
- 17.Minamizono A, Tomi M, Hosoya K. Inhibition of dehydroascorbic acid transport across the rat blood-retinal and -brain barriers in experimental diabetes. Biol Pharm Bull. 2006;29:2148–50. doi: 10.1248/bpb.29.2148. [DOI] [PubMed] [Google Scholar]
- 18.Barabás J, Nagy E, Degrell I. Ascorbic acid in cerebrospinal fluid: a possible protection against free radicals in the brain. Arch Gerontol Geriatr. 1995;21:43–8. doi: 10.1016/0167-4943(95)00654-4. [DOI] [PubMed] [Google Scholar]
- 19.Guaiquil VH, Golde DW, Beckles DL, Mascareno EJ, Siddiqui MA. Vitamin C inhibits hypoxia-induced damage and apoptotic signaling pathways in cardiomyocytes and ischemic hearts. Free Radic Biol Med. 2004;37:1419–29. doi: 10.1016/j.freeradbiomed.2004.06.041. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Schellhorn HE. New developments and novel therapeutic perspectives for vitamin C. J Nutr. 2007;137:2171–84. doi: 10.1093/jn/137.10.2171. [DOI] [PubMed] [Google Scholar]
- 21.Nardai G, et al. Protein-disulfide isomerase- and protein thiol-dependent dehydroascorbate reduction and ascorbate accumulation in the lumen of the endoplasmic reticulum. J Biol Chem. 2001;276:8825–8. doi: 10.1074/jbc.M010563200. [DOI] [PubMed] [Google Scholar]
- 22.Abbate A, Biondi-Zoccai GG, Van Tassell BW, Baldi A. Cellular preservation therapy in acute myocardial infarction. Am J Physiol Heart Circ Physiol. 2009;296:H563–5. doi: 10.1152/ajpheart.00066.2009. [DOI] [PubMed] [Google Scholar]
- 23.Baldi A, et al. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol. 2002;34:165–74. doi: 10.1006/jmcc.2001.1498. [DOI] [PubMed] [Google Scholar]
- 24.Abbate A, et al. Persistent infarct-related artery occlusion is associated with an increased myocardial apoptosis at post-mortem examination in humans late after an acute myocardial infarction. Circulation. 2002;106:1051–4. doi: 10.1161/01.cir.0000030936.97158.c4. [DOI] [PubMed] [Google Scholar]
- 25.Abbate A, Biondi-Zoccai GGL, Baldi A. Pathophysiologic role of myocardial apoptosis in post-infarction left ventricular remodeling. J Cell Physiol. 2002;193:145–53. doi: 10.1002/jcp.10174. [DOI] [PubMed] [Google Scholar]
- 26.Abbate A, et al. Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. J Am Coll Cardiol. 2003;41:753–60. doi: 10.1016/s0735-1097(02)02959-5. [DOI] [PubMed] [Google Scholar]
- 27.Abbate A, Bussani R, Amin MS, Vetrovec GW, Baldi A. Acute myocardial infarction and heart failure: role of apoptosis. Int J Biochem Cell Biol. 2006;38:1834–49. doi: 10.1016/j.biocel.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab. 2010;12:108–15. doi: 10.1111/j.1463-1326.2010.01282.x. [DOI] [PubMed] [Google Scholar]
- 29.Minamino T, Kitakaze M. ER stress in cardiovascular disease. J Mol Cell Cardiol. 2010;48:1105–10. doi: 10.1016/j.yjmcc.2009.10.026. [DOI] [PubMed] [Google Scholar]
- 30.Li Z, et al. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J Clin Biochem Nutr. 2007;41:58–67. doi: 10.3164/jcbn.2007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puig A, Gilbert HF. Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme. J Biol Chem. 1994;269:7764–71. [PubMed] [Google Scholar]
- 32.Jaxa-Chamiec T, Bednarz B, Herbaczynska-Cedro K, Maciejewski P, Ceremuzynski L MIVIT Trial Group. Effects of vitamins C and E on the outcome after acute myocardial infarction in diabetics: a retrospective, hypothesis-generating analysis from the MIVIT study. Cardiology. 2009;112:219–23. doi: 10.1159/000151239. [DOI] [PubMed] [Google Scholar]
- 33.Odermarsky M, Lykkesfeldt J, Liuba P. Poor vitamin C status is associated with increased carotid intima-media thickness, decreased microvascular function, and delayed myocardial repolarization in young patients with type 1 diabetes. Am J Clin Nutr. 2009;90:447–52. doi: 10.3945/ajcn.2009.27602. [DOI] [PubMed] [Google Scholar]
- 34.Atkin JD, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–65. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- 35.Nardai G, et al. Diabetic changes in the redox status of the microsomal protein folding machinery. Biochem Biophys Res Commun. 2005;334:787–95. doi: 10.1016/j.bbrc.2005.06.172. [DOI] [PubMed] [Google Scholar]