

Abstract
High mobility group box chromosomal protein 1 (HMGB1) is a DNA-binding nuclear protein that can be released from dying cells and activated myeloid cells. Extracellularly, HMGB1 promotes inflammation. Experimental studies demonstrate HMGB1 to be a pathogenic factor in many inflammatory conditions including arthritis. HMGB1-blocking therapies in arthritis models alleviate disease and confer significant protection against cartilage and bone destruction. So far, the most successful HMGB1-targeted therapies have been demonstrated with HMGB1-specific polyclonal antibodies and with recombinant A box protein, a fragment of HMGB1. The present study is the first to evaluate the potential of a monoclonal anti-HMGB1 antibody (2G7, mouse IgG2b) to ameliorate arthritis. Effects of repeated injections of this antibody have now been studied in two conceptually different models of arthritis: collagen type II–induced arthritis (CIA) in DBA/1 mice and in a spontaneous arthritis disease in mice with combined deficiencies for genes encoding for the enzyme DNase type II and interferon type I receptors. These mice are unable to degrade phagocytozed DNA in macrophages and develop chronic, destructive polyarthritis. Therapeutic intervention in CIA and prophylactic administration of anti-HMGB1 monoclonal antibody (mAb) in the spontaneous arthritis model significantly ameliorated the clinical courses. Anti-HMGB1 mAb therapy also partially prevented joint destruction, as demonstrated by histological examination. The beneficial antiarthritic effects by the anti-HMGB1 mAb in two diverse models of arthritis represent additional proof-of-concept, indicating that HMGB1 may be a valid target molecule to consider for development of future clinical therapy.
INTRODUCTION
Chronic arthritides are inflammatory diseases associated with decreased quality of life and disability due to fatigue, pain and articular tissue destruction. Despite the clinical success of biologics in the treatment of chronic arthritis, there are still many patients with unmet medical needs. Thus, there are good reasons to search for novel therapeutic target molecules that can become the basis for improved treatment.
High mobility group box chromosomal protein 1 (HMGB1) is a nonhistone DNA-binding nuclear protein that displays both intracellular and extracellular activities. Inside the cell, HMGB1 exerts structural and transcriptional activities (1–3). Extracellular HMGB1 is a potent endogenous “danger signal” for the initiation of innate immunity. The translocation of HMGB1 from the inside to the outside of the cell is thus a critical event in host defense and inflammation and can occur via two separate mechanisms. HMGB1 may be actively released by inflammatory cells after stimulation with exogenous pathogen-derived molecules or endogenous inflammatory mediators as well as in response to ischemia (4–7). In addition, HMGB1 can be passively released during disintegration of primary necrotic cells or apoptotic bodies undergoing secondary necrosis (8,9). Although not fully resolved, it has been demonstrated that HMGB1-mediated functions are conveyed via several different receptors including toll-like receptor (TLR)-2, TLR4 and the receptor for advanced glycation end products (RAGE) (10–12), all implicated in the pathogenesis of arthritis. The following observations indicate a pathogenic role for HMGB1 in rheumatic diseases: HMGB1 is released at the site of joint inflammation (7,13,14); aberrant synovial HMGB1 expression is downregulated by intraarticular corticosteroid injections (15); intraarticular injection of rHMGB1 in mice induces destructive arthritis (16); therapeutic targeting of HMGB1 attenuates arthritis in animals and in particular ameliorates the structural damage (7,17–19); HMGB1 regulates the production and acts upstream of tumor necrosis factor (TNF), interleukin (IL)-1β, IL-6 and tissue-degrading proteases (20–22); and HMGB1 plays a pivotal role in the maturation of osteoclasts (23–25).
Experimental animal models of arthritis provide important tools for the development and evaluation of new therapeutic approaches, although all models differ from human disease. Collagen type II–induced arthritis (CIA) is the prototypical model for experimental arthritis that has been studied for many years. Recently, a novel spontaneous arthritis model was described with mice devoid of genes encoding for DNase II and interferon type I receptor (IFN-IR) (26). DNase II is an intracellular enzyme needed for macrophage degradation of DNA of engulfed apoptotic cells and expelled erythroid cell nuclei. Because of unexplained constitutive production of interferon β (IFN-β), DNase II−/− mice die as embryos but may be rescued by a deletion of the IFN-IR gene (27). DNase II−/− × IFN-IR−/− mice spontaneously develop destructive chronic polyarthritis at the age of 6–8 weeks with several features shared with rheumatoid arthritis (26). We previously reported beneficial results in these arthritis models by blocking HMGB1 with truncated HMGB1- derived A box protein, acting as a competitive HMGB1 antagonist, or by using polyclonal anti-HMGB1 antibodies (17,19). The HMGB1 A box protein is identical in all mammals, and therapy using this recombinant protein has generated beneficial results in a broad spectrum of preclinical disorders (reviewed in 28). However, the protein has a short survival time in circulation and may need to be modified to prolong its biological availability. HMGB1-specific polyclonal antibodies have also been efficacious in many experimental models, but cannot be applied in clinical studies. The present studies address the question of whether anti-HMGB1 monoclonal antibody (mAb) treatment may modulate arthritis when administered to mice with CIA or to DNase II−/− × IFN-IR−/− mice. The anti-HMGB1 mAb (2G7) used in our studies has previously demonstrated beneficial therapeutic effects in models for experimental sepsis and pancreatic islet graft transplantation (29,30). Furthermore, the 2G7 mAb performs excellently in vitro to detect natural HMGB1 in the HMGB1-specific Elispot assay developed by our group (31), in immunohistochemistry studies (32) and to neutralize rHMGB1-induced TNF production in cultured macrophages (11).
MATERIALS AND METHODS
Animals
All experiments were approved by the Stockholm North Ethical Committee, Sweden. Animals were housed in specific pathogen-free facilities at the Karolinska University Hospital (Stockholm, Sweden). The mice were housed with a maximum of six animals per cage and had free access to water and standard rodent chow. A 12-h light/dark circle was maintained at all times.
DNase II−/− × IFN-IR−/− mice were bred and generated as described previously (19,27). Because of the impaired fertility ability of DNase II−/− × IFN-IR−/− mice, heterozygous DNase+/− × IFN-IR−/− mice were kept for breeding. Determination of DNase II genotype was assessed using genomic DNA preparations from ear biopsies for detection of the DNase-null allele by polymerase chain reaction. Average weight at the start of experiments of female and male DNase II−/− × IFN-IR−/− mice was around 11 g. Female DBA/1 mice, with a weight of approximately 20 g, were supplied by Taconic (Ry, Denmark).
Induction of Collagen Type II Arthritis in DBA/1 Mice
Type II collagen (CII) obtained from bovine nasal cartilage and dissolved in 0.1 mol/L acetic acid at a concentration of 2 mg/mL was emulsified with an equal volume of Freund’s complete adjuvant. Then 100 μL of the emulsion containing 100 μg CII and 300 μg Mycobacterium tuberculosis was injected subcutaneously into the base of the tail of the mice. On day 28, the mice received a booster injection of 100 μg CII in 100 μL Freund’s incomplete adjuvant.
Evaluation of Arthritis
Mice were monitored daily for signs of erythema and swelling of the interphalangeal joints of the digits, the metacarpophalangeal joints and wrist in the forepaws and the metatarsophalangeal joints and ankle joints in the hindpaws. Severity of clinical arthritis in individual paws was scored on a scale of 0–3 as follows: 0 = no signs of arthritis, 1 = one type of joint affected, 2 = two types of joints affected, and 3 = the entire paw affected. Thus, the maximal score for each animal is 12, but mice reaching a total score of 9 were sacrificed because of ethical restrictions; therefore, 9 represents the highest possible score allowed in this study. The arthritis evaluation was performed by several observers blinded to the identity of the animals. The animals were sacrificed by CO2, 7 d (CIA study) or 5 wks (DNase−/− × IFN-IR−/− study) after initiation of treatment.
Administration of a Monoclonal Anti-HMGB1 Antibody in Two Arthritis Models
Prophylactic intervention with anti-HMGB mAb was initiated when the DNase II−/− × IFN-IR−/− mice were 5 wks old, which precedes expected onset of clinical disease. The animals were treated for 5 wks every second day with in-traperitoneal injections, 100 μg of a mono clonal anti-HMGB1 antibody (IgG2b 2G7, noncommercial antibody, developed at former Critical Therapeutics, Boston, MA, USA [now Cornerstone Therapeutics, Cary, NC, USA]) or buffer control. This antibody binds to an epitope within the amino acid region position 53–63 of the A box subunit.
Therapeutic intervention in CIA in DBA/1 mice with monoclonal anti-HMGB1 antibody 2G7 started when the animal presented with an arthritis score of ≥2. The animals were treated for 7 d with daily intraperitoneal injections of 70 μg 2G7 mAb or irrelevant antibody (MOPC-195, IgG2b from Sigma, St. Louis, MO, USA).
Microscopic Assessment of Joint Pathology
Paws were removed postmortem, dissected and fixed in 4% formaldehyde at 4°C overnight. The paws were then decalcified in EDTA buffer for 3–4 wks before dehydration and paraffin embedding. Serial sections were stained with hematoxylin and eosin to evaluate inflammatory cell infiltration and synovial hyperplasia, or stained with Safranin O (0.1%) to determine cartilage damage and bone erosions, assessing the most severely affected joint in each paw. Cell infiltration and synovitis were graded from 0 (no infiltration/normal synovium) to 3 (severely inflamed joint with maximal cellular influx/synovial hyperplasia). Cartilage destruction and matrix proteoglycan depletion were scored on a scale from 0 to 3, ranging from no abnormalities (score 0) to completely destroyed or proteoglycan-depleted cartilage (score 3). Bone erosions were graded on a scale from 0 (normal bone appearance) to 3 (fully eroded cortical bone) according to a previously published grading scheme (19). A mean score of the four paws were calculated; thus, the maximal score for each animal was 3. Joint pathology assessment was performed in a coded manner blinded to the identity of the tissue specimen and repeated twice. The agreement between the different evaluations was >95%, and the discrepancy was never more than one score step.
Statistical Analysis
Statistical analysis of clinical arthritis score was on the basis of calculated area under the curve values for individual animals in each group, and the differences between animals in the two studied groups were analyzed by two-tailed, non-parametric Mann-Whitney test. Differences in histopathology score between studied groups were analyzed by two-tailed, nonparametric Mann-Whitney test.
RESULTS
Anti-HMGB1 mAb Administration Ameliorated Arthritis in DNase II−/− × IFN-IR−/− Mice
Markedly increased systemic HMGB1 levels have previously been documented in sera of DNase II−/− × IFN-IR−/− mice, occurring even before onset of clinical disease and intraarticular HMGB1 release (19). Accordingly, treatment with anti-HMGB1 mAb was initiated 1–2 wks before expected clinical onset of arthritis in these mice. Prophylactic administration of the anti-HMGB1 mAb every second day for 5 wks to DNase II−/− × IFN-IR−/− animals significantly reduced the clinical severity, as revealed by lower clinical arthritis scores compared with controls (P = 0.02) (Figure 1). Control animals were treated with vehicle alone, since our previous study with A box injections in this model used this as a control (19) and we wanted to be able to compare results between the two studies. Joint histology demonstrated that intraarticular synovial inflammation and tissue destruction were also ameliorated in anti-HMGB1 mAb-treated animals versus controls. Previous studies have demonstrated that maximal synovial HMGB1 release in arthritis occurs in the invasive pannus tissue (17,19,32). It is thus of particular interest that administration of anti-HMGB1 mAb managed to counteract tissue destruction and cartilage proteoglycan depletion. The studied histological parameters included inflammatory cell influx, synovial hyperplasia, depletion of cartilage matrix and bone erosions, and all parameters were significantly reduced (P < 0.05) in anti-HMGB1 mAb-treated animals compared with controls (Figure 2). Paw specimens comprise several joints—including ankle/wrist, joints between tarsal/carpal and digital bones—with the histology scoring being based on studies of the most severely affected joint. Prophylactic intervention with the anti-HMGB1 mAb did not prevent the development of joint disease. However, the number of paw joints with nonproliferative synovial tissue containing only a few cell layers was greater in treated animals, and signs of cartilage and bone destruction were reduced compared with that of control mice. Synovial cell hyperplasia, inflammatory cell infiltration in synovitis and periarticular tissue were more evident in control mice.
Figure 1.
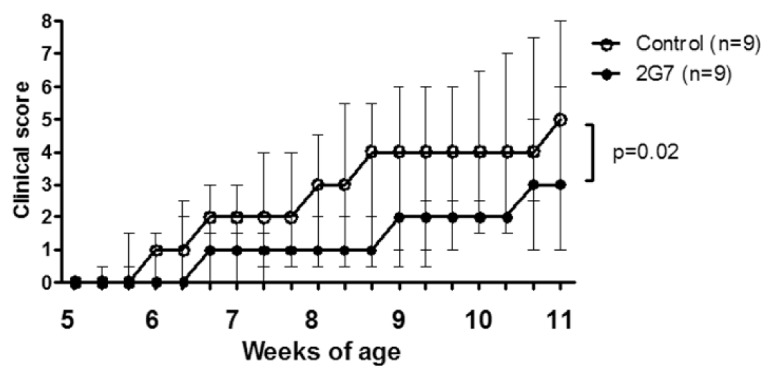
HMGB1 neutralization attenuates the course of spontaneous arthritis in DNase II−/− × IFN-IR−/− mice. Prophylactic therapy with anti-HMGB1 mAb 2G7 was initiated at the age of 5 wks in DNase II−/− × IFN-IR−/− mice with intraperitoneal injections every second day for 5 wks. Development of arthritis was significantly attenuated in 2G7-treated mice (n = 9) compared with mice treated with vehicle alone (n = 9). Values are median clinical score, and error bars represent interquar-tile range. Statistical analysis of clinical arthritis score is based on calculated area under the curve values for individual animals in each group. The differences between animals in the two studied groups were analyzed by a two-tailed, nonparametric Mann-Whitney test.
Figure 2.

HMGB1 blockade attenuates the histological severity of arthritis in DNase II−/− × IFN-IR−/− mice. Histopathology of joint specimens were evaluated, where signs of cell infiltration, cartilage destruction and bone erosions were determined and designated a score on a scale from 0 (normal appearance) to 3 (maximal pathology), as described in Materials and Methods. A significant lower score for all parameters was recorded in anti-HMGB1 mAb (2G7)–treated mice (n = 8) compared with control mice (n = 9). Representative micrographs illustrating articular joints stained with Safranin O after HMGB1 blocking therapy (B) and after control treatment with vehicle alone (C) are shown. Note destained cartilage layers (solid arrow) reflecting loss of matrix proteoglycans, massive cell infiltration and synovial hyperplasia invading into subchondral bone (dotted arrow) in control-treated articular tissue (C), compared with that of a 2G7-treated mouse, expressing more homogenous cartilage staining and preserved joint morphology (B). c, cartilage; js, joint space; s, synovial tissue. Original magnification 125×. **P < 0.01; ***P < 0.001. Differences in histopathology score between studied groups were analyzed by two-tailed, nonparametric Mann-Whitney test.
Successful Therapy with Anti-HMGB1 mAb in Collagen II–Induced Arthritis
Intraarticular inflammation in CIA has, similar to arthritis in DNase II−/− × IFN-IR−/− mice, previously been shown to be an HMGB1-dependent disease that can be successfully treated with polyclonal anti-HMGB1 antibodies (17). In this previous study, we used irrelevant antibody injections as a control and thus chose isotype antibody treatment as the control arm of this study to enable a comparison of monoclonal versus polyclonal anti-HMGB1 therapies in the same model. Although the clinical onset and disease progression in CIA in DBA/1 mice was more acute and dramatic in the joints, as compared with the spontaneous arthritis model, beneficial results were clearly demonstrated even when anti-HMGB1 mAb therapy started after onset of clinical disease in CIA (Figure 3). The mice received daily intraperitoneal injections with the anti-HMGB1 mAb or control antibodies for 7 d when they had reached an arthritis clinical score of ≥2. There was a significant reduction of clinical scores in the 2G7-treated animals (P < 0.05) compared with control-treated animals. In addition to the significant decrease of general disease severity in the 2G7-treated animals on the basis of area under the curve calculations, fewer animals in the 2G7 treatment group reached maximal disease severity (0 of 12 of the 2G7-treated animals compared with 3 of 7 of the control-treated animals).
Figure 3.
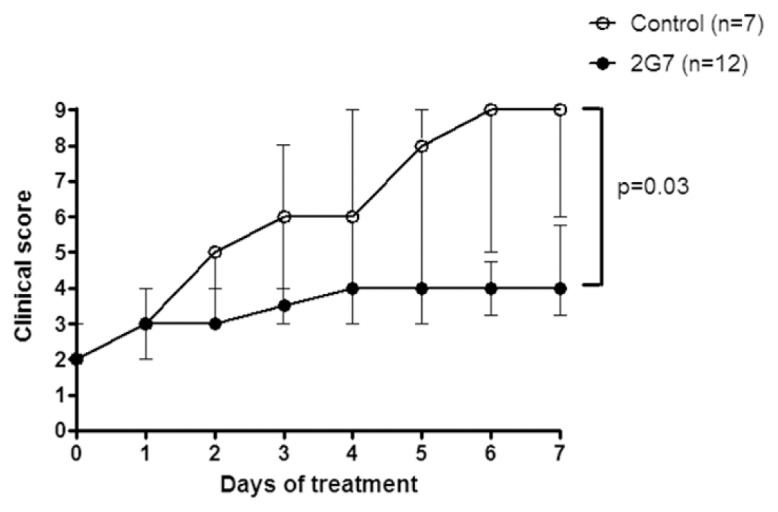
Therapeutic intervention with the monoclonal anti-HMGB1 antibody ameliorates clinical progression of collagen-induced arthritis. Mice were treated for 7 d with monoclonal anti-HMGB1 antibody 2G7, initiated when the animal reached a clinical arthritis score of ≥2. Disease development was significantly ameliorated in 2G7-treated mice (n = 12), compared with mice treated with irrelevant antibody (n = 7). Values are median clinical score, and error bars represents interquartile range. Statistical analysis of clinical arthritis score is based on calculated area under the curve values for individual animals in each group, and the differences between animals in the two studied groups were analyzed by two-tailed, nonparametric Mann-Whitney test.
DISCUSSION
This is the first study where a monoclonal antibody targeting HMGB1 has displayed significant effects in experimental arthritis. This result represents major progress, since anti-HMGB1 blocking monoclonal antibodies may have great clinical therapeutic potential. The generation of a good neutralizing anti-HMGB1 mAb has for many years proven difficult for several reasons. One obstacle is that stressed or dying cultured hybridoma cells producing the mAb will unfortunately release their nuclear HMGB1, which will then bind and block secreted anti-HMGB1 antibodies. Culture conditions are thus critical. Another explanation is that HMGB1 is a uniquely conserved molecule among species with 99% identity in mammals, rendering it less immunogenic. Therapeutic monoclonal anti-HMGB1 antibody intervention has only been successfully described in three previous publications. One mAb recognizing an epitope in the C-terminus of the HMGB1 molecule improved outcome in an experimental stroke model (33). The 2G7 mAb was previously demonstrated to improve survival in a gram-negative sepsis model (29) and to prevent TLR4-mediated early graft failure after pancreatic islet graft transplantation (30).
We selected the CIA and the DNase−/− × IFN-IR−/− mouse models to study the therapeutic performance of the 2G7 mAb, since we know from previous studies that they represent HMGB1- dependent diseases. CIA is the prototypical model for experimental arthritis used to study novel therapeutic approaches and it shares many clinical, histological and immunological features with human rheumatoid arthritis. These similarities include symmetric joint involvement, synovitis, cartilage and bone erosions and presence of rheumatoid factor (34). However, the clinical onset and disease progression in CIA is more acute and dramatic in the joints compared with those of rheumatoid arthritis patients. The clinical disease in DNase II−/− × IFN-IR−/− mice is milder than that of CIA and also has many similarities with human rheumatoid arthritis including symmetric, destructive polyarthritis with increased serum levels of anticyclic citrullinated peptide antibody, rheumatoid factor and matrix metalloproteinase-3 (26). DNase II−/− × IFN-IR−/− mice as well as CIA mice express high systemic levels of HMGB1 and in particular demonstrate extensive intraarticular HMGB1 release. Therapeutic HMGB1-specific intervention in the two models mediates beneficial results. Polyclonal anti-HMGB1 antibody and A box therapies have both been successfully applied in CIA (7,17). A box treatment has provided positive therapeutic results in DNase II−/− × IFN-IR−/− mice (19) with effects that are similar to those observed with the anti-HMGB1 mAb intervention in this study. Taken together, the results indicate that the 2G7 mAb represents a potent in vivo blocker of HMGB1. Both experimental models are also strongly TNF-dependent and have been demonstrated to respond favorably to TNF-blocking therapy. From a clinical point of view, we would have liked to include studies of the anti-HMGB1 mAb therapy in a model being HMGB1 but not TNF dependent, but we are not aware of any such model.
The anti-HMGB1 mAb used in the present study, in the published sepsis and islet graft transplantation studies (29,30) and the mAb studied in the stroke model (33) are rodent IgG antibodies that need to be humanized before evaluation in clinical trials. However, the beneficial therapeutic outcomes in these diverse conditions, including arthritis, sepsis, organ transplantation and stroke, indicate strong proof-of-concept in support of a further development of monoclonal anti-HMGB1 antibodies.
ACKNOWLEDGMENTS
This study was financially supported through the regional agreement on medical training and clinical research between the Stockholm County Council and Karolinska Institutet, the Swedish Medical Research Council, the Swedish Association against Rheumatism, Åke Wiberg’s Foundation, the Freemason Lodge Barnhuset in Stockholm, Kronprinsessan Lovisas Stiftelse and King Gustaf V’s Foundation.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Johns EW, Goodwin CHM, Walker JM, Sanders C. Chromosomal proteins related to histones. Ciba Found Symp. 1975;28:95–112. [Google Scholar]
- 2.Bustin M, Reeves R. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 3.Park JS, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284:C870–9. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 4.Gardella S, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;13:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonaldi T, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dumitriu IE, et al. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol. 2005;174:7506–15. doi: 10.4049/jimmunol.174.12.7506. [DOI] [PubMed] [Google Scholar]
- 7.Hamada T, et al. Extracellular high mobility group box chromosomal protein 1 is a coupling factor for hypoxia and inflammation in arthritis. Arthritis Rheum. 2008;58:2675–85. doi: 10.1002/art.23729. [DOI] [PubMed] [Google Scholar]
- 8.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 9.Bell CW, Jiang W, Reich CF, 3rd, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol. 2006;291:1318–25. doi: 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- 10.Yu M, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–9. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 11.Yang H, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–7. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hori O, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of RAGE and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 13.Kokkola R, et al. High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum. 2002;46:2598–603. doi: 10.1002/art.10540. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi N, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–81. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- 15.af Klint E, et al. Intraarticular glucocorticoid treatment reduces inflammation in synovial cell infiltrations more efficiently than in synovial blood vessels. Arthritis Rheum. 2005;52:3880–9. doi: 10.1002/art.21488. [DOI] [PubMed] [Google Scholar]
- 16.Pullerits R, et al. High mobility group chromosomal protein 1, a DNA-binding cytokine, induces arthritis. Arthritis Rheum. 2003;48:1693–700. doi: 10.1002/art.11028. [DOI] [PubMed] [Google Scholar]
- 17.Kokkola R, et al. Successful therapy in collagen-induced arthritis in mice and rats targeting extracellular HMGB1 activity. Arthritis Rheum. 2003;48:2052–8. doi: 10.1002/art.11161. [DOI] [PubMed] [Google Scholar]
- 18.Östberg T, et al. Oxaliplatin retains HMGB1 intranuclearly and ameliorates collagen type II-induced arthritis. Arthritis Res. Ther. 2008;10:R1. doi: 10.1186/ar2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Östberg T, et al. Protective targeting of high mobility group box chromosomal protein 1 in a spontaneous arthritis model. Arthritis Rheum. 2010;62:2963–72. doi: 10.1002/art.27590. [DOI] [PubMed] [Google Scholar]
- 20.Andersson U, et al. HMG-1 stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parkkinen J, et al. Amphoterin, the 30-kDa family of HMG1-type polypeptides: enhanced expression in transformed cells, leading edge localization, and interactions with plasminogen activation. J Biol Chem. 1993;268:19726–38. [PubMed] [Google Scholar]
- 22.Taguchi A, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–9. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Z, et al. HMGB1 regulates RANKL-induced osteoclastogenesis in a manner dependent on RAGE. J Bone Miner Res. 2008;23:1084–96. doi: 10.1359/JBMR.080234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamoah K, et al. High-mobility group box proteins modulate tumor necrosis factor-alpha expression in osteoclastogenesis via a novel deoxyribonucleic acid sequence. Mol Endocrinol. 2008;22:1141–53. doi: 10.1210/me.2007-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, et al. HMGB1 is a bone-active cytokine. J Cell Physiol. 2008;214:730–9. doi: 10.1002/jcp.21268. [DOI] [PubMed] [Google Scholar]
- 26.Kawane K, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006;443:998–1002. doi: 10.1038/nature05245. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- 28.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile and infectious inflammation. Annu Rev Immunol. 2011;29:139–62. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin S, et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med. 2006;203:1637–42. doi: 10.1084/jem.20052203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Q, et al. TLR4 mediates early graft failure after intraportal islet transplantation. Am J Transplant. 2010;10:1588–96. doi: 10.1111/j.1600-6143.2010.03151.x. [DOI] [PubMed] [Google Scholar]
- 31.Wähämaa H, et al. HMGB1-secreting capacity of multiple cell lineages revealed by a novel HMGB1 ELISPOT assay. J Leukoc Biol. 2007;81:129–36. doi: 10.1189/jlb.0506349. [DOI] [PubMed] [Google Scholar]
- 32.Palmblad K, et al. Morphological characterization of intraarticular HMGB1 expression during the course of collagen-induced arthritis. Arthritis Res. Ther. 2007;9:R35. doi: 10.1186/ar2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu K, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007;21:3904–16. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 34.Hegen M, Keith JC, Jr, Collins M, Nickerson-Nutter CL. Utility of animal models for identification of potential therapeutics for rheumatoid arthritis. Ann Rheum Dis. 2008;67:1505–15. doi: 10.1136/ard.2007.076430. [DOI] [PubMed] [Google Scholar]