

Abstract
Deptor is an mTOR binding protein that affects cell metabolism. We hypothesized that knockdown (KD) of Deptor in C2C12 myocytes will increase protein synthesis via stimulating mTOR-S6K1 signaling. Deptor KD was achieved using lentiviral particles containing short hairpin (sh)RNA targeting the mouse Deptor mRNA sequence, and control cells were transfected with a scrambled control shRNA. KD reduced Deptor mRNA and protein content by 90%, which increased phosphorylation of mTOR kinase substrates, 4E-BP1 and S6K1, and concomitantly increased protein synthesis. Deptor KD myoblasts were both larger in diameter and exhibited an increased mean cell volume. Deptor KD increased the percentage of cells in the S phase, coincident with an increased phosphorylation (S807/S811) of retinoblastoma protein (pRb) that is critical for the G1 to S phase transition. Deptor KD did not appear to alter basal apoptosis or autophagy, as evidenced by the lack of change for cleaved caspase-3 and light chain (LC)3B, respectively. Deptor KD increased proliferation rate and enhanced myotube formation. Finally, in vivo Deptor KD (~50% reduction) by electroporation into gastrocnemius of C57/BL6 mice did not alter weight or protein synthesis in control muscle. However, Deptor KD prevented atrophy produced by 3 d of hindlimb immobilization, at least in part by increasing protein synthesis. Thus, our data support the hypothesis that Deptor is an important regulator of protein metabolism in myocytes and demonstrate that decreasing Deptor expression in vivo is sufficient to ameliorate muscle atrophy.
INTRODUCTION
Skeletal muscle serves as the largest protein reservoir in the body, and its mass represents a balance between rates of protein synthesis and degradation in the tissue. The process of protein synthesis is tightly regulated because of its high demand for cellular energy. Of the three regulatory steps involved in protein synthesis—translation initiation, elongation and termination—initiation plays the most significant role in regulating mRNA translation (1–3). At a molecular level, mTOR (mammalian target of rapamycin) kinase is a key regulator of translation initiation, being activated upon feeding and conversely inhibited in response to catabolic insults such as sepsis, excess glucocorticoids, alcohol or disuse atrophy (4–7). Exposure of muscle to growth factors and nutrients increases initiation via the mTOR pathway, thereby stimulating protein synthesis (3,8–10).
mTOR is sequestered within two distinct complexes: mTOR complex (mTORC)-1 and mTORC2. mTORC1 is composed of mTOR, raptor (regulatory-associated protein of TOR), LST8/G-protein β-subunit–like protein (GβL), proline-rich Akt substrate 40 kDa (PRAS40) and Deptor (DEP-domain containing partner of TOR) (11–14). In contrast, mTORC2 consists of mTOR, rictor (rapamycin-insensitive companion of mTOR), LST8/GβL, PRR5L (proline-rich protein 5–like), protor (protein observed with Rictor-1) and Deptor (5,15,16). As noted above, Deptor is a constituent of both mTOR complexes and is considered a negative regulator of mTOR function, since Deptor knockdown increases phosphorylation of signaling substrates downstream of both mTORC1 and mTORC2 (15). Conversely, overexpression of Deptor in cell culture models inhibits signaling pathways downstream of both mTOR-containing complexes. Additionally, in the absence of growth factors or in the presence of mTOR inhibitors, the mTOR-Deptor binding is strengthened, which thereby decreases mTOR activity and suppresses cap-dependent protein translation initiation (17).
Deptor is also a phospho-protein and as such can undergo posttranslational modification that affects its binding to mTOR. For example, in response to growth factor signaling, Deptor is phosphorylated and quickly degraded via the ubiquitin proteasome system pathway (15,16). Despite the few reports implicating Deptor as a regulator of translation initiation in cancer and transformed cells, there is a paucity of information related to its role in regulating other cellular functions, especially in skeletal muscle. Given the essential role mTOR plays in regulating protein translation initiation, cell cycle and proliferation, we posited that one or more of these mTOR functions are regulated by Deptor in myocytes. Therefore, the purpose of our current investigation was to examine changes in C2C12 myocyte protein synthesis, cell proliferation and cell cycle in response to Deptor knockdown (KD) using short hairpin (sh)-RNA–based in vitro experimental approaches. In addition, we previously reported that the inhibition of mTORC1 activity observed in response to sepsis or glucocorticoid excess was associated with an increase in Deptor protein level (4). Therefore, we also assessed whether in vivo Deptor KD by electroporation could ameliorate the decrease in muscle mass and protein synthesis seen in a catabolic condition associated with an elevation in Deptor.
MATERIALS AND METHODS
Cell Culture
C2C12 myoblasts (American Type Culture Collection, Manassas, VA, USA) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS), penicillin (100 IU/mL), streptomycin (100 μg/mL) (all from Mediatech, Herndon, VA, USA) under 5% CO2 at 37°C. To assess basal mTOR activity, experiments measuring protein synthesis and the phosphorylation of mTOR substrates were performed using 2% FBS without antibiotics- antimycotics for 8 h. 5-Aminoimidazole-4-carboxamide-1-β-d-ribonucleoside (AICAR; Toronto Research Chemicals, Toronto, Canada), when present, was added at a final concentration of 2 μmol/L. Insulin-like growth factor (IGF)-I (Genentech, San Francisco, CA, USA), when present, was used at a final concentration of 100 ng/mL for the last 1 h of the experiment. These doses maximally suppress and stimulate protein synthesis in C2C12 myocytes, respectively (18,19).
shRNA Interference
We used the lentiviral plasmid pLKO.1-mDeptor described by Peterson et al. (15), which targeted the mouse sequence 5′-CCG GCG CAA GGA AGA CAT TCA CGA TCT CGA GAT CGT GAA TGT CTT CCT TGC GTT TTT G-3′ (Addgene plasmid 21337; Addgene, Cambridge, MA, USA). The scramble shRNA used as a negative control was previously reported (20) with a hairpin sequence: CCT AAG GTT AAG TCG CCC TCG CTC TAG CGA GGG CGA CTT AAC CTT AGG (Addgene plasmid 1864). The plasmids were isolated using the Qiagen EndoFree Giga prep kit (Qiagen, Valencia, CA, USA). The actual DNA sequence was confirmed at the Pennsylvania State University College of Medicine DNA sequence core facility. Packaging plasmids psPAX2 and envelope protein plasmid pMD2.G were obtained from the Torono Lab (Addgene plasmid 12260 and 12259, respectively). HEK293FT cells (Invitrogen) were grown in DMEM; 80–85% confluent plates were rinsed once with Opti-MEM (Invitrogen) and then incubated with Opti-MEM for 4 h before transfection. psPAX2 and pMD2.G, along with either scramble or pLKO.1-mDeptor, were added after mixing with Lipofectamine 2000, as per the manufacturer’s instructions (Invitrogen). Opti-MEM was changed after overnight incubation with DMEM containing 10% FBS without antibiotics to allow cells to take up plasmids and recover. Culture media was collected at 36 and 72 h, and viral particles present in the supernatant were harvested after a 15-min spin at 1,500g to remove cellular debris. The supernatant was then further filtered using a 0.45-μm syringe filter. Supernatant-containing virus was either stored at −80°C for long-term storage or at 4°C for immediate use. Low-passage C2C12 cells at <60% confluence were infected twice overnight with 3 mL viral supernatant containing 8 μg/mL polybrene in serum-free–antibiotic-free DMEM. Fresh DMEM containing 10% FBS, antibiotics and 2 μg/mL puromycin (Sigma, St. Louis, MO, USA) was added the next day. Cells that survived under puromycin selection were either harvested (as stable cells) and stored or used immediately.
35S-Methionine Labeling
C2C12 myocytes were grown in six-well plates and treated as above. Protein synthesis was measured on 60% confluent myoblasts. For metabolic labeling, 10 μCi radiolabeled 35S-methionine (MP BioMedicals, Salon, OH, USA) was added to each well of a six-well plate for 1 h, and radiolabel incorporation into trichloroacetic acid (TCA) precipitable proteins was measured via liquid scintillation as previously described (5).
Immunoblot Analysis and Immunoprecipitation
After treatment, cells were rinsed two times with cold phosphate-buffered saline (PBS) and collected on ice in lysis buffer (20 mmol/L HEPES [pH 7.4], 2 mmol/L EGTA, 50 mmol/L, β-glycerophosphate, 0.3% 3[(3-cholamidopropyl) dimethylammonio]-propanesulfonic acid [CHAPS], 100 mmol/L KCl, 2 mmol/L EDTA, 50 mmol/L NaF, 0.5 mmol/L phenyl-methylsulfonyl fluoride [PMSF], 1 mmol/L benzamidine, 1 mmol/L sodium orthovanadate and 2 μg/mL leu-peptin). Lysates were sonicated for 10 min and then kept on a rocker for 30 min at 4°C before being clarified (14,000g for 10 min at 4°C). A portion of the resulting cell supernatant was used to determine protein concentration via a bicinchoninic acid assay (Pierce, Rockford, IL, USA). Sample buffer (5×) was added, and samples were loaded according to total protein content (20 μg) on polyacrylamide gels for separation by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to polyvinylidene fluoride (PVDF) membrane (Biotrace; PALL, Pensacola, FL), blocked in 5% nonfat dry milk and incubated overnight at 4°C with phospho-specific and total antibodies (all from Cell Signaling Technology, Boston, MA, USA, unless otherwise noted) for raptor (S792), mTOR (S2448), ribosomal protein S6 kinase-1 (S6K1; T389), PRAS40 (T246; Biosource, Camarillo, CA, USA), eukaryotic initiation factor (eIF) 4E-binding protein 1 (4E-BP1) (T37/46), Akt (S473), eIF4B (S422), 5′ AMP-activated protein kinase (AMPK; T172), cleaved caspase-3, poly (ADP- ribose) polymerase (PARP), microtubule-associated proteins light chain (LC)3B, retinoblastoma protein (pRb) (S807/S811), β-tubulin and p53. Antibodies for total pRb, p21, p27, cyclin- dependent kinase (cdk)-2, cdk-4, cdk-6 and β-actin (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA) and for Deptor (Millipore, Billerica, MA, USA) were also used. Excess primary antibody was removed by washing in 1 × [Tris-buffered saline (TBS)] + 0.1% Tween 20, and membranes were incubated with horseradish peroxidase–conjugated goat anti–rabbit or goat anti–mouse secondary antibody (Sigma). Blots were rinsed with 1 × TBS + 0.1% Tween 20 to remove excess secondary antibody and were treated with enhanced chemiluminescence (ECL plus) Western blotting reagents (Amersham, Piscataway, NJ, USA) and then developed using the Gnome (Syngene, Cambridge, UK). Uncompressed tiff images were analyzed using National Institutes of Health (NIH) ImageJ 1.6 software. After development, blots were stripped and incubated with antibodies for total proteins on blots that were probed earlier with phospho-specific antibodies. Antibodies against β-actin or β-tubulin served as an additional control for equal protein loading of samples.
RNA Extraction and Quantitation
Total RNA was isolated from C2C12 myocytes transfected with either scramble or Deptor shRNA using an RNeasy mini kit (Qiagen). For animal tissue, total RNA was isolated from 35 to 50 mg skeletal muscle using 1 mL Tri-reagent (Molecular Research Center, Cincinnati, OH, USA). The RNA pellet was reconstituted in 100 μL RNase-free water and subjected to RNA cleanup using an RNeasy mini kit. On the column, DNase I treatment was performed to remove any residual DNA contamination. RNA was eluted from the column with 30–50 μL RNase-free water, and the total RNA (1–2 μL) concentration was determined (NanoDrop 2000; Thermo Fisher Scientific, Waltham, MA, USA).
Reverse Transcription
Total RNA (1–5 μg) was primed with 200 ng oligo dT, 100 ng random primer and 0.5 mmol/L dNTP mix at 65°C for 5 min and then kept on ice for 1–2 min. cDNA was synthesized from the primed reaction using 200 units of superscript III reverse transcriptase, 40 units of RNase-Out, 1 × first-strand buffer and 5 mmol/L dithiothreitol in a 20-μL reaction (Invitrogen). The reaction was incubated at 25°C for 5 min and then at 50°C for 1 h, followed by 70°C for 15 min to inactivate the reverse transcriptase. To control for residual DNA contamination, each RNA sample was subjected to cDNA synthesis as described above without the superscript-III transcriptase. The cDNA was stored at −20°C.
Real-Time Quantitative Polymerase Chain Reaction
Real-time quantitative polymerase chain reaction (PCR) using primers shown in Table 1 was performed using 25–65 ng cDNA in a StepOnePlus system using TaqMan gene expression assays and the gene expression master mix (Applied Biosystems, Foster City, CA, USA). The cycling parameters were an initial 50°C for 2 min and 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Real-time PCR quantitation was on the basis of Ct values, where Ct is defined as the PCR cycle number that crosses an arbitrary signal threshold on the amplification plot. The comparative quantitation method 2−ΔΔCt was used in presenting gene expression of target genes in reference to an endogenous control, and 2−ΔCt was used in presenting expression of each housekeeping gene in validating the use of the endogenous control gene. ΔCt is expressed as the difference between the target and control samples (Ct target – Ct control).
Table 1.
Real-time PCR primers used in this study.
| Gene symbol | Gene name | Assay IDa | PubMed reference sequence |
|---|---|---|---|
| Actb | Actin, beta | Mm01205647_gl | NM_007393.3 |
| Gapdh | Glyceraldehydes-3-phosphate dehydrogenase | Mm999999915_gl | NM_008084.2 |
| Rpl32 | Ribosome protein L32 | Mm02528467_gl | NM_172086.2 |
| B2m | β-2 microglobulin | Mm00437762_ml | NM_009735.3 |
| Depdc6 (Deptor ) | DEP domain containing 6 | Mm01195339_ml | NM_145470.2 and NM_001037937.2 |
| Hprt | Hypoxanthine guanine phosphoribosyl transferase1 | Mm00446968_ml | NM_013556.2 |
Primers were purchased from Applied Biosystems. Assay ID represents specific details of primer sequences used.
Cell Cycle
Myoblasts were transfected with either a scramble (control) shRNA or an shRNA targeting Deptor. Cells were seeded in 10-cm dishes and used at 60% confluence (~24 h after seeding). Cells were trypsinized, washed with Dulbecco’s PBS (Invitrogen) and fixed in cold 70% ethanol overnight at −20°C. Cells were then stained with 100 μg/mL solution of propidium iodide buffer containing 0.1% Triton-X100 and 0.001% DNAse-free RNAse at 37°C for 30 min immediately before fluorescence-activated cell sorter (FACS) analysis. A total of 20,000 cells per sample were counted, and the cell cycle phase was measured by propidium iodide staining intensity using a Becton Dickinson FACS-Calibur flow cytometer (Becton Dickinson, Bedford, MA, USA) and ModFit software LT, version 3.2 (Verity Software, Topsham, ME, USA).
Cell Size and Proliferation
To determine cell size, stably transfected myoblasts were seeded in 10-cm dishes and used at 60% confluence. For cell number, cells were seeded at similar densities and counted at different time points. Myoblasts were trypsinized and suspended in DMEM with 10% FBS. Cells were then diluted in Isoton II solution (Beckman Coulter, Fullerton, CA, USA) and assayed using the Beckman Coulter Counter and particle size analyzer (Beckman Coulter). Cell size analysis was performed using the AccuComp® Z2 Coulter Counter software (Beckman Coulter).
MTT Assay
C2C12 myoblasts (~10,000/well) were grown in a 96-well plate for 24 h and then rinsed with PBS to remove the interfering phenol red from the DMEM. This step was followed by the addition of 50 μg methylthiazoletetrazolium (MTT; Sigma) per 100 μL PBS to cells in each well for 4 h at 37°C (21). MTT containing PBS was aspirated, and 100 μL DMSO was added to each well to dissolve the resulting formazan, and absorbance at 570 nm was quantified using a Spectra-max plate reader (Molecular Devices, Sunnyvale, CA, USA).
Cell Differentiation
Approximately 0.5 × 106 myoblasts transfected with scramble control or Deptor KD plasmids were seeded in 10-cm plates and photographed daily using a Nikon digital camera (Nikon, Tokyo, Japan) mounted on a binocular microscope using a 10× objective lens. Images were composed and edited in Photoshop 7.0 (Adobe Systems, San Jose, CA, USA). Background was reduced using contrast and brightness adjustments to enhance reprint, and all modifications were applied to the whole image. Similarly treated plates were collected at days 3, 5, 7 and 9 for Western blotting to measure myosin heavy chain (MHC) expression as a functional end point to measure differentiation biochemically, since myoblasts were allowed to fuse and form differentiated postmitotic myotubes by switching the media to 2% horse serum once the plates were confluent (days 3–4).
Cell Apoptosis and Autophagy Controls
Deptor KD and scramble control myoblasts of the same passage were treated with 2 μmol/L staurosporine in culture media for 4 h to induce apoptosis and were collected in the CHAPS media. To induce autophagy, C2C12 myoblasts were treated with Hanks’ balanced salt solution (HBSS) for 6 h, and cells were collected in CHAPS buffer.
Animals
Animal facilities and experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of The Pennsylvania State University College of Medicine and complied with the NIH Guide for Care and Use of Laboratory Animals. Adult male C57/BL6 mice (~23–28 g body weight; Charles River Breeding Laboratories, Cambridge, MA, USA) were maintained on a 12:12-h light–dark cycle, with water and food (Rodent Chow 8604; Harlan-Teklad, Madison, WI) provided ad libitum.
In Vivo Electroporation
Mice were briefly anesthetized with isoflurane (2–3% in O2) for plasmid injection, in vivo electroporation and subsequent hindlimb immobilization. Once mice were anesthetized, the lower hindlimbs were shaved to expose the skin, swabbed with 70% alcohol and air-dried to clean the injection site. Plasmid DNA (100 μg total in 100 μL 0.9% sterile saline) was slowly injected at multiple sites (approximately four) directly into the gastrocnemius through the skin via a 1-mL insulin syringe fitted with a 28-gauge needle (Becton Dickinson). A plasmid-targeting Deptor was injected into both the right and left gastrocnemius of one cohort of mice, whereas a second group of mice received bilateral injection of the scrambled control plasmid. Square-wave electric impulses generated by an electroporator (model ECM 830; BTX, San Diego, CA, USA) were delivered to muscle in both legs via caliper electrodes (model 384; BTX) coated with electrode conductive gel. The electrodes were applied to the lower hindlimb with only slight pressure to ensure proper contact. Pulse trains were delivered with a 200 V/cm field strength (eight pulses, 40 ms/pulse, 100-ms interval) on the basis of previously described parameters (22–24). Careful excision of the transfected muscle was performed 72 h after the electroporation procedure (see below). Parameters for in vivo electroporation were optimized in preliminary studies and are comparable to those reported by others (22–25).
Hindlimb Immobilization
Following in vivo electroporation, one hindlimb was wrapped with a single layer of surgical tape. Superglue was then applied to the tape and a 1.5-mL plastic microfuge tube without the lid was placed over the leg, maintaining the foot in a plantar-flexed position. The contralateral leg was not immobilized and functioned as the internal control. The contralateral leg does not undergo hypertrophy, and the validity of this unilateral hindlimb immobilization was previously reported (26,27). Food consumption of mice with the Deptor KD plasmid did not differ from mice that received control plasmid (control = 4.3 ± 0.3 g/d; Deptor KD = 4.2 ± 0.4 g/d). Thus, in vivo studies generated muscles in four experimental groups: (i) muscle injected with control plasmid that remained mobile, (ii) muscle injected with control plasmid but was then immobilized, (iii) muscle injected with Deptor KD plasmid that remained mobile and (iv) muscle injected with Deptor KD plasmid and was immobilized.
In Vivo Protein Synthesis
The in vivo rate of protein synthesis in the gastrocnemius (hereafter called muscle) was determined in control and Deptor KD–treated mice 3 d after plasmid electroporation and immobilization. Protein synthesis was determined using the flooding-dose technique, exactly as described (28,29). Mice were injected intraperitoneally with [3H]-l-phenylalanine (150 mmol/L, 30 μCi/mL, 1 mL/100 g body wt), and blood was collected 15 min later for determining the plasma phenylalanine concentration and radioactivity. Thereafter, muscles were rapidly excised, freeze-clamped and then stored at −70°C. Muscle was processed exactly as previously described (30,31). The rate of protein synthesis was calculated by dividing the amount of radioactivity incorporated into protein by the plasma phenylalanine-specific radioactivity, and the advantages and disadvantages of this method have been reviewed (32). The specific radioactivity of the plasma phenylalanine was measured by high-performance liquid chromatography analysis of supernatant from TCA extracts of plasma.
Statistics
Results for individual cell experiments (n ≥ 6) were replicated in at least three independent experiments and, when applicable, are presented as means ± SE calculated from the pooled data. Data were analyzed by an unpaired Student t test in two-group comparisons or with analysis of variance (ANOVA) and Tukeys posttest in multigroup comparisons to determine treatment effect when ANOVA indicated a difference among the means. GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA, USA) was used for analysis. Differences between groups were considered significant at P < 0.05.
All supplementary materials are available online at www.molmed.org.
RESULTS
Effect of Deptor Knockdown in C2C12 Myoblasts
shRNA directed toward Deptor in C2C12 myoblasts reduced Deptor protein levels by >90%, compared with scramble control values (Figures 1A, B). As expected, Deptor knockdown also reduced the Deptor mRNA content by ~95% in infected C2C12 cells (Figure 1C).
Figure 1.

Effect of Deptor KD in C2C12 myoblasts. (A) Representative Western blot of Deptor protein in control (scramble) and Deptor KD myoblasts. (B) Quantification of Western blot data. Values are means ± SE; n = 12 per group. P < 0.05 compared with time-matched scramble control values. (C) Real-time PCR showing decreased Deptor mRNA in myoblasts transfected with lentiviruses targeting Deptor. rpL32 served as the internal reference control gene, and its expression did not differ between control and Deptor KD cell (Scramble control = 100 ± 5; Deptor KD = 107 ± 7; P = NS). Values are means ± SE; n = 12 per group. P < 0.05 compared with time-matched scramble control values. (D) Effect of Deptor KD on myocyte protein synthesis. Values are means ± SE for n = 12 for each condition. For bar graphs, *P < 0.05, compared with control values. (E) Effect of Deptor KD on mTORC1 and mTORC2 signaling in C2C12 myoblasts. Representative Western blots for protein substrates involved in the mTORC1 and mTORC2 complex mediated signal transduction. Where absent, standard error bars are too small to be visualized.
The knockdown of Deptor increased global protein synthesis by ~50% under basal conditions (Figure 1D). To determine if the change in global protein synthesis involved mTOR-mediated signal transduction events, we performed Western blotting for mTOR substrates and binding partners. Deptor KD significantly (P < 0.05) increased phosphorylation of mTORC1 substrates S6K1 (T389) (control = 100 ± 5 arbitrary units (AU); Deptor KD = 151 ± 7 AU) and 4E-BP1 (T37/46) (control = 100 ± 10 AU; Deptor KD = 129 ± 4 AU). However, the increased phosphorylation of 4E-BP1 resulted from a concomitant and comparable increase in total 4E-BP1 protein expression in these myocytes. In addition, Deptor KD also increased (P < 0.05) the phosphorylation (T246) of PRAS40 (control = 100 ± 17 AU; Deptor KD = 193 ± 7 AU), an mTORC2 substrate (Figure 1E). To assess off-target effects of the shRNA-mediated Deptor KD, we performed Western blot analysis for proteins critical to the mTOR signaling pathway, including mTOR, raptor and S6K1 and found that there were no changes in the total protein content for these proteins (see Figure 1E and Figure 2B).
Figure 2.
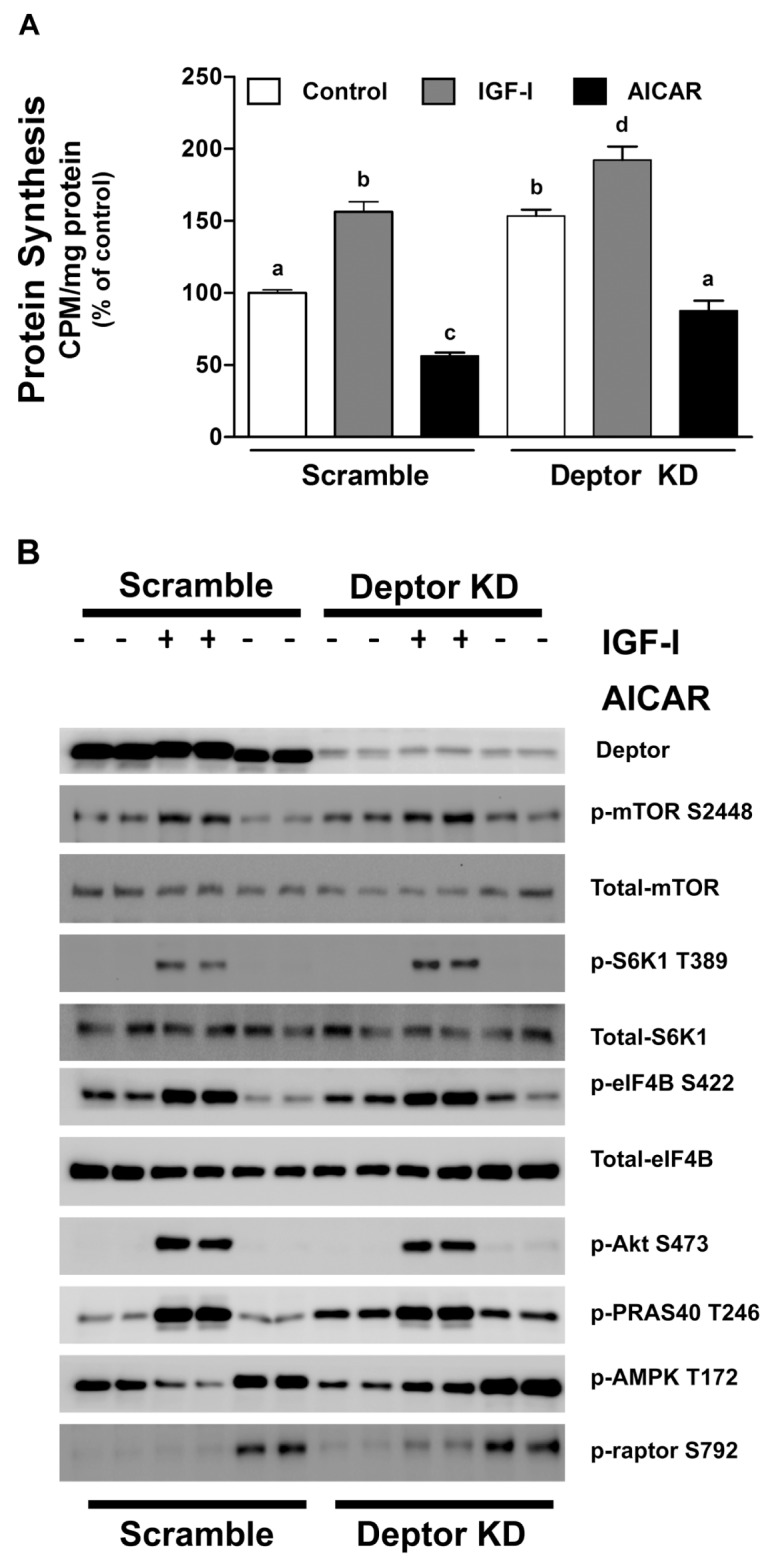
Effect of IGF-I and AICAR on scramble (control) and Deptor KD C2C12 myoblasts. Cells were transfected with scramble and Deptor KD–containing lentiviral particles and incubated with vehicle (control), IGF-I (100 ng/mL; 1 h) or AICAR (2 μmol/L; 8 h) and labeled with 35S-methionine. (A) Protein synthesis in myoblasts. Values are mean ± SE for n = 8–10 for each condition. Means not sharing the same superscript (a, b, c and d) are significantly different (P < 0.05). For quantification, data were normalized to scramble control values. (B) Representative Western blots for various total and phosphorylated proteins, where cells were treated as described above, except that the isotope was omitted. The blot is representative of at least three independent experiments with two to four replicates per experiment.
To determine whether Deptor KD altered myocyte responsiveness to external stimuli, cells were incubated with either an anabolic (IGF-I) or catabolic (AICAR) agent. As expected, incubation of control myocytes with IGF-I increased protein synthesis, whereas AICAR inhibited protein synthesis (Figure 2A). A comparable bidirectional response for protein synthesis toward IGF-I and AICAR was also seen in Deptor KD cells. To confirm the protein synthesis data, we performed Western blotting for mTOR and its substrates and binding partners. Cells with Deptor KD remained responsive to both types of stimuli, and their response was comparable to the scramble controls (Figure 2B). For example, IGF-I increased phosphorylation of mTOR (S2448), S6K1 (T389), eIF4B (S422), Akt (S473) and PRAS40 (T246), whereas AICAR increased phosphorylation of raptor (S792) and AMPK (T172) in both control and Deptor KD cells.
Deptor KD Increases Myoblast Size and Proliferation
On the basis of previous data, we hypothesized that Deptor KD would also increase myoblast size. Deptor KD increased the diameter (17.0 ± 0.1 μm) of low passage proliferating (~60% confluent) myoblasts, compared with scramble control cells (15.3 ± 0.1 μm), as measured using either the Coulter Counter particle size analyzer (Figure 3A) or FACS flow cytometry analysis (Supplementary Figure S1). Mean cell volume was also increased in myocytes with Deptor KD (Figure 3B). When both cell types were seeded at the same low initial density, the initial rate of proliferation between day 0 and day 4 did not differ between control and Deptor KD cells. However, at days 6 and 8, the cell number was greater in cells with Deptor KD, compared with time-matched control cells (Figure 4A). To exclude variations in the ability of the cell types to attach to the plates, cells were seeded and counted 4–8 h after seeding to allow for attachment. An equal number of cells were harvested after trypsinization in both the control and Deptor KD condition, suggesting no significant difference in the ability of these cells to attach to the culture plates (data not shown). To confirm that the proliferation rate of Deptor KD cells was faster, we used an independent colorimetric assay on the basis of the conversion of the MTT tetrazolium salt to its formazan product. Consistent with the above data, the MTT assay revealed that Deptor KD increased the rate of proliferation by ~20% (Figure 4B). Apoptosis poses another potential mechanism that may affect cell number and thus proliferation; however, Western analysis for the apoptotic markers cleaved caspase-3, and PARP did not differ between groups under our experimental conditions (Supplementary Figure S2).
Figure 3.
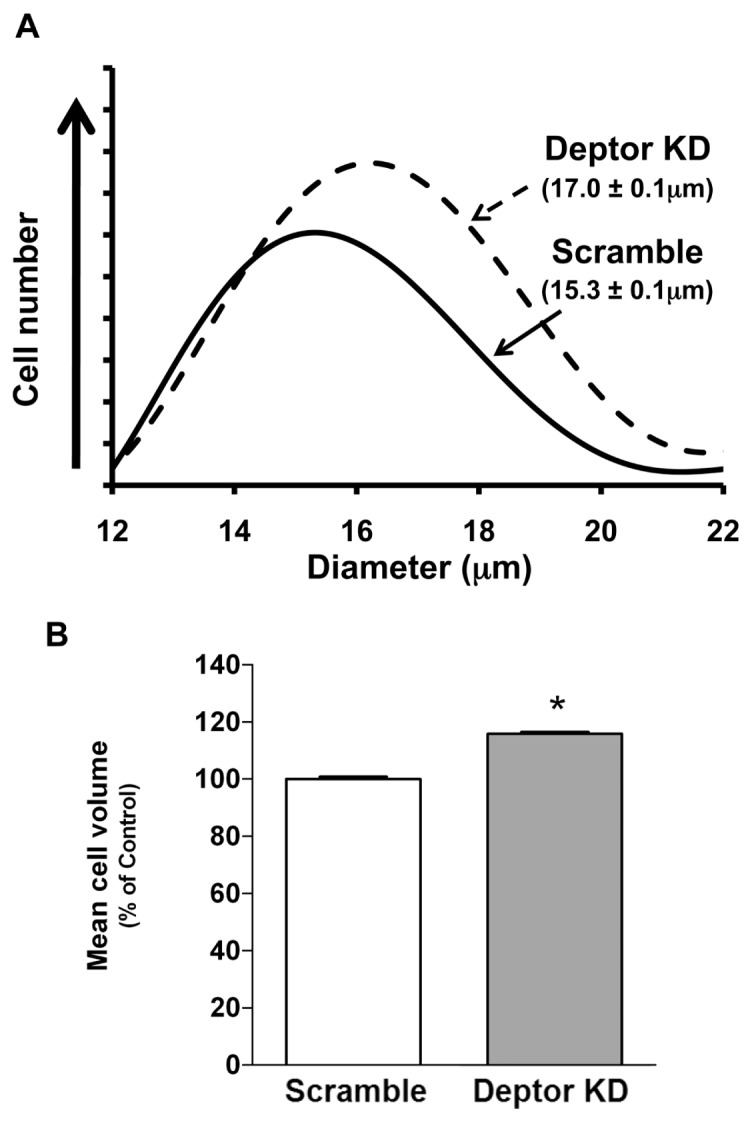
Effect of Deptor KD on cell size in C2C12 myoblasts. (A) Cell size, shown in parentheses, was measured using the Coulter Counter particle size analyzer; n = 8 for each condition. (B) Mean cell volume of myoblasts. Bar graph is mean ± SE; n = 7–9 for each condition, *P < 0.0001. Where absent, standard error bars are too small to be visualized.
Figure 4.
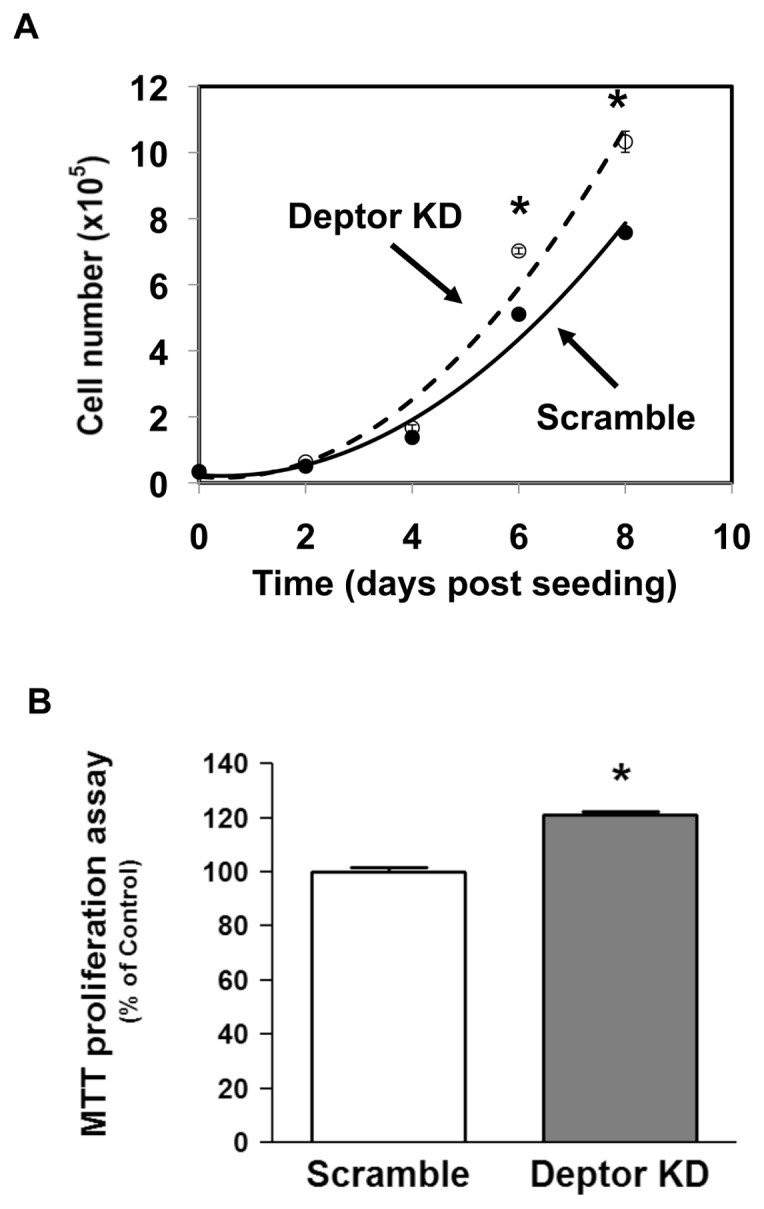
Effect of Deptor KD on C2C12 myoblast proliferation. Proliferation rate was determined in stably transfected myoblasts with scramble and Deptor KD. Myoblasts were seeded at the same density and counted using the Coulter Counter, as described in Materials and Methods. Time intervals are indicated; n = 6 for each treatment time point; experiments were repeated at least three times. *P < 0.05, compared with the time-matched control value. (B) Proliferation rate was measured using an independent MTT assay. Cells were treated with MTT for 4 h, and formazan produced was measured colorimetrically. Bar graph is mean ± SE; n = 16–18 for each condition, *P < 0.0001; experiments were repeated at least three times.
Deptor Knockdown Enhances Cell Cycle Progression
To address the underlying mechanism by which Deptor KD increased proliferation, we stained myoblasts with propidium iodide to study cell cycle. A smaller proportion of cells in G1/G0 of the cell cycle was detected in myocytes with Deptor KD, compared with the control values (Figures 5A, B; Table 2). We assessed whether proteins regulating the cell cycle, especially the G1 to S transition, were concomitantly altered. Figures 5C and D illustrate an increased S807/S811 phosphorylation of pRb consistent with increased progression from the G1 to S phase in myoblasts with Deptor KD. To further accentuate differences in the cell cycle, we also synchronized cells by serum starvation. After G1/G0 arrest, cells were released by the addition of serum and cells were analyzed once again at 16 h using flow cytometry. Figure 6A shows that Deptor KD dramatically increased the percent of cells in the S-phase of the cell cycle after serum stimulation, compared with time-matched scramble control myoblasts. This change in cell cycle after arrest and release was verified by Western analysis for pRb phosphorylation (Figure 6B). We also performed Western blot analysis for proteins crucial for cell cycle regulation (namely, p21, p27, p53, cdk-2, cdk-4, cdk-6 and cyclin D1) and found no change between control and Deptor KD myoblasts (Supplementary Figures S3A, B).
Figure 5.

Effect of Deptor KD on cell cycle in C2C12 myoblasts. Myoblasts were transfected with either control (scramble) shRNA or shRNA targeting Deptor. Myoblasts were grown in DMEM supplemented with 10% FBS for 18–24 h and stained with propidium iodide to assess cell cycle using FACS. Representative forward scatter histograms highlighting G1 and G2 phases of cell cycle for scramble control (A) and KD (B) are shown. The percentage of cells in each stage of the cell cycle for each treatment group is shown in the accompanying pie graphs. FL2-A, propidium iodide fluorescence. (C) Western blotting of samples (normalized to total protein content for loading) as described above. Blots were probed with total Deptor antibody to show the different groups (top band ) and with phospho-specific antibody to detect phosphorylation on S807/S811 of pRb protein (middle band ). The lower band shows β-tubulin to confirm equal loading. (D) Quantification of Western blot from C for phosphorylation on S807/S811 of the pRb protein.
Table 2.
Effect of Deptor KD on cell cycle in C2C12 myoblasts.
| Cell cycle | % G1 | % S | % G2 |
|---|---|---|---|
| Scramble | 65.1 ± 1.6 | 28.0 ± 0.8 | 6.9 ± 0.9 |
| Deptor KD | 56.7 ± 1.5a | 29.7 ± 0.7 | 13.6 ± 0.9a |
Myoblasts were transfected with either control (scramble) shRNA or shRNA targeting Deptor. Data are means ± SE for n = 12 for each condition.
P < 0.05 compared with time-matched control values.
Figure 6.
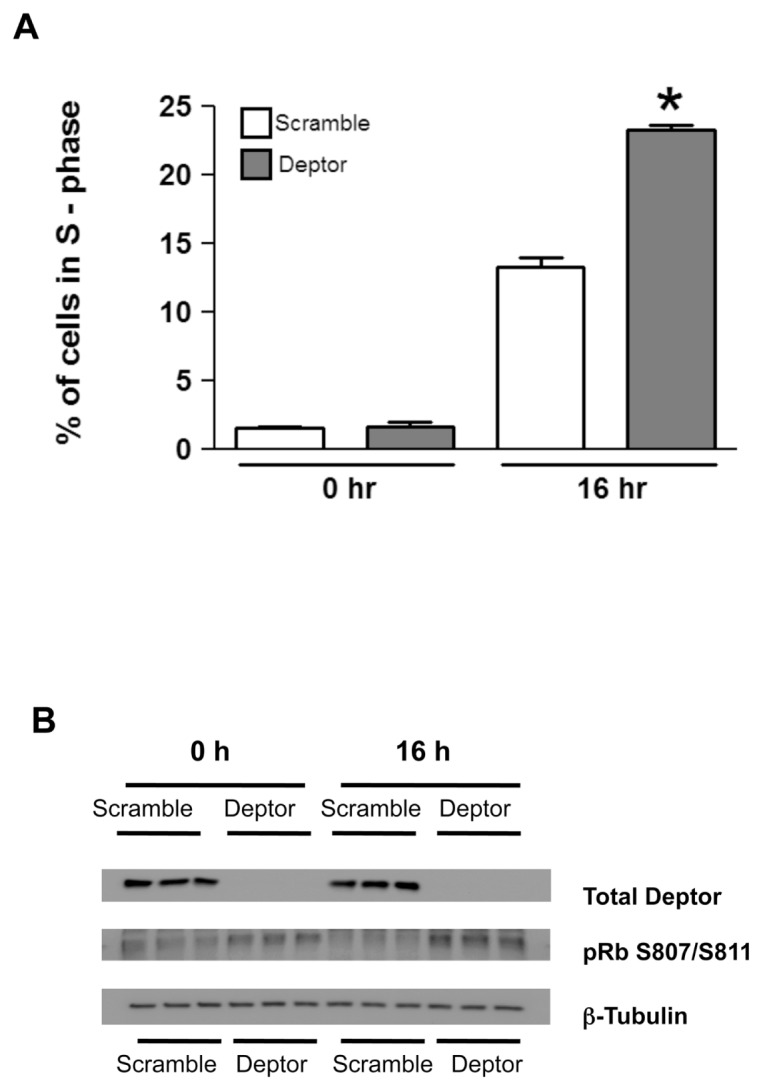
Effect of Deptor KD on cell cycle regulation. Control and Deptor KD myoblasts were grown in DMEM supplemented with 10% FBS overnight to 50–60% confluency. Myoblasts were then serum starved for 18–24 h in serum-free DMEM to arrest them in the G1 to G0 phase. Serum-starved myocytes were then released from the cell cycle arrest by the addition of fresh media containing 10% FBS. At 16 h, these proliferating cells were fixed. (A) Bar graph shows the percent of cells in the S-phase of the cell cycle in serum-starved cells (0 h) and after addition of serum (16 h); mean ± SE; n = 5–6 for each condition. *P < 0.05, compared with time-matched control values. (B) Western blot of samples (normalized to total protein content for loading) as described above. Blots were probed with total Deptor antibody (top band) and with phospho-specific antibody to detect phosphorylation on S807/S811 of pRb protein (middle band). The lower band was probed with β-tubulin to confirm equal loading.
Deptor KD Alters Myogenesis
Our data demonstrate the presence of a concomitant increase in proliferation and altered cell cycle in Deptor KD myoblasts. Because mTOR also regulates autophagy, which in turn plays an important role in cell differentiation (33,34), we determined the expression of proteins important in regulating autophagy and found that there was no difference in LC3B-II/LC3B-I ratio between control and Deptor KD myoblasts (control = 100 ± 8 AU; Deptor KD = 116 ± 10 AU) under normal physiological conditions (Supplementary Figure S4).
Next, we determined whether such changes in altered cell cycle and proliferation might be of physiological relevance to skeletal muscle development. In this regard, we seeded the same number of myoblasts and tracked their progression to form myotubes (Figures 7A, B). We observed that Deptor KD cells reached confluent status earlier than control cells. To quantitate these findings, cell lysates were collected at various stages of myocyte development to measure the expression of MHC, a protein expressed only in differentiated matured myotubes. In control cells, MHC expression was absent in myoblasts (day 3), and MHC expression in control cells was first detected on day 7 (Figure 7B). In contrast, in cells with Deptor KD, MHC could be detected on the blots by day 5. By day 9, both scramble control and Deptor knockdown cells exhibited MHC expression, with the content of MHC being increased in the Deptor KD cells (see Figures 7A, B). The protein content of the muscle transcription factor MyoD (used as an additional internal control) did not differ between scramble control and Deptor KD cells (Figure 7B).
Figure 7.
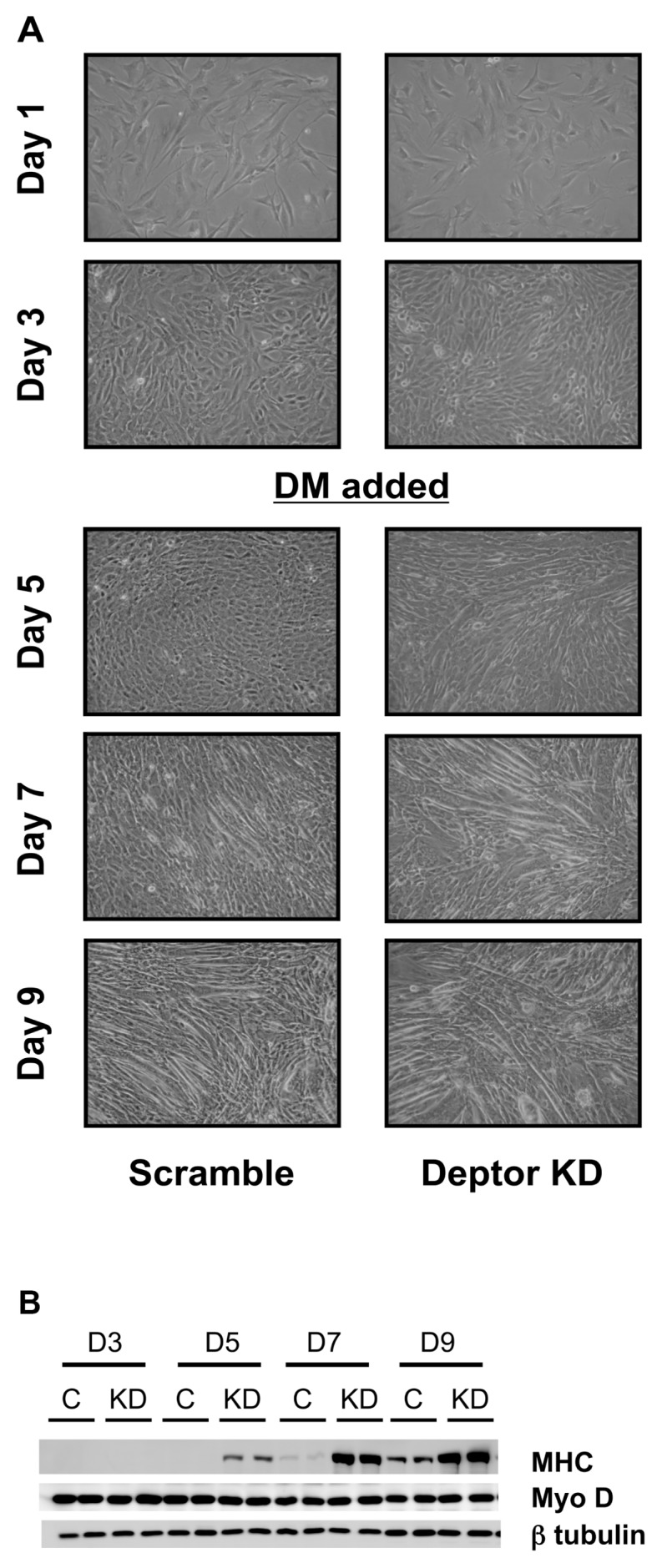
Effect of Deptor KD on C2C12 differentiation. (A) Myoblasts were transfected with either control (scramble) shRNA or shRNA targeting Deptor. Cells were plated at the same density and photographed daily (10× objective magnification) to visually record changes in cell proliferation (time to reach confluence) and formation of myotubes. On days 3–4, when the plates were confluent, the media were switched to 2% horse serum (DM = differentiation media) to induce myotube formation. Deptor KD in C2C12 myocytes enhances MHC protein expression. (B) Representative Western blots for Deptor protein, MHC and the muscle-specific transcription factor MyoD in samples treated as in A. β-Tubulin serves as a loading control.
Deptor KD In Vivo Prevents Muscle Loss Due to Immobilization
To determine the effect of Deptor KD on muscle growth in vivo, we electroporated the shRNA plasmid–targeting Deptor message into the gastrocnemius of C57/BL6 mice. Using this technique, Deptor mRNA content was decreased ~50% (Figure 8A). The hallmark of disuse atrophy is loss of muscle mass in the immobilized leg compared with the contralateral control leg. Figure 8B illustrates the decreased muscle mass after 3 days of hindlimb immobilization in the control animals and prevention of this atrophic response in the muscle where Deptor was decreased. In vivo–determined rates of protein synthesis were also quantitated in these same muscles (Figure 8C). In control muscle, there was no difference in protein synthesis in the gastrocnemius injected with scrambled control and Deptor KD plasmid. In contrast, in the immobilized leg, Deptor KD prevented the decreased rate of protein synthesis. The delta for decrement in protein synthesis between control versus immobilized muscle was significantly greater in control (1.27 ± 0.14 nmol Phe/h/mg protein) compared with Deptor KD (0.34 ± 0.16 nmol Phe/h/mg protein).
Figure 8.
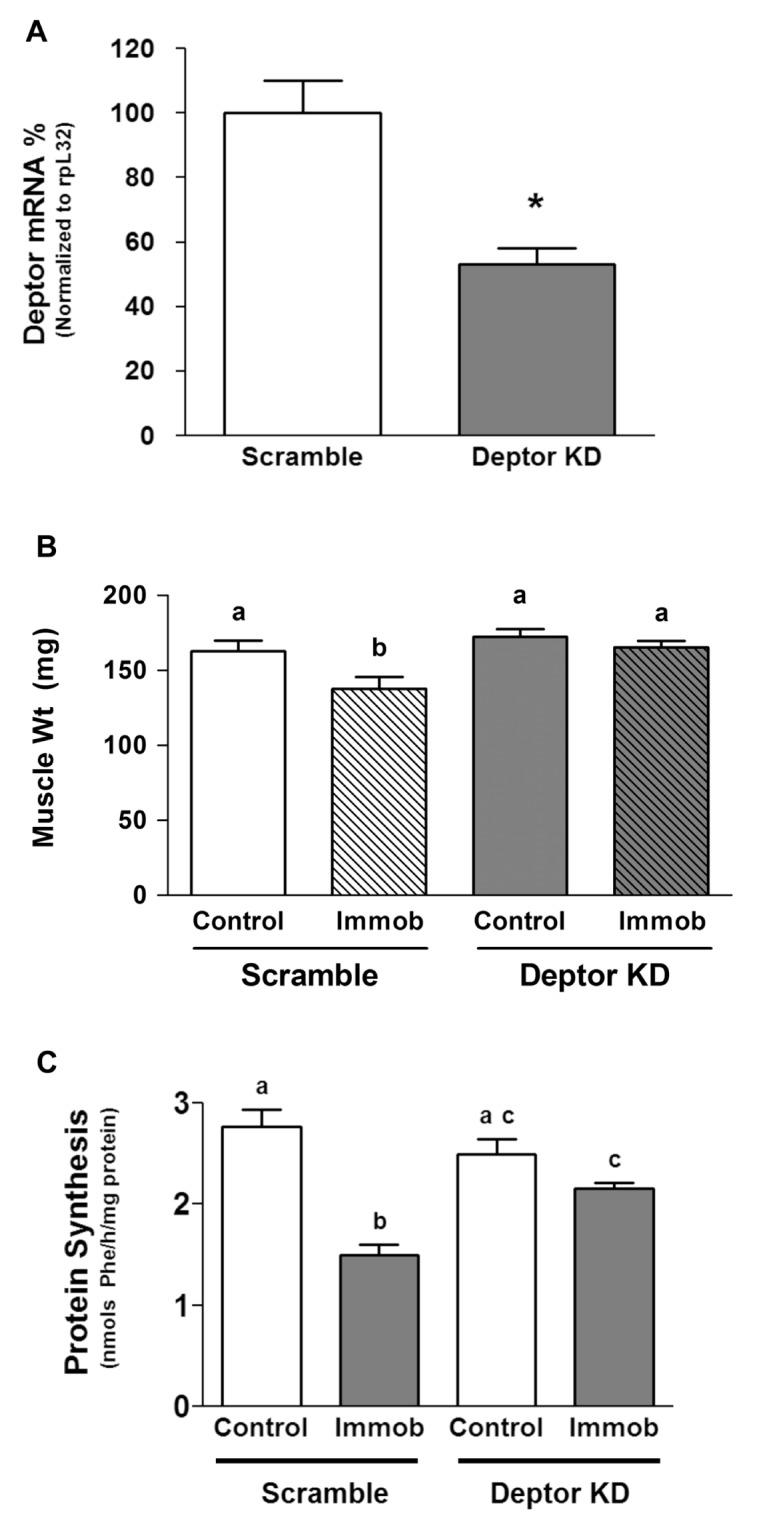
Effect of in vivo Deptor KD on skeletal muscle weight and protein synthesis. Gastrocnemius of C57/BL6 mice were electroporated with plasmids containing either scramble (control) shRNA or shRNA targeting Deptor mRNA. (A) Three days after electroporation, the animals were anesthetized and the muscle was excised and homogenized, and mRNA in the homogenate was quantified using real-time PCR. The bar graph shows Deptor mRNA content normalized to rpL32, which serves as an endogenous control. Data are mean ± SE; n = 5–6 for each condition. *P < 0.05, compared with control values. (B) Animals were treated as described above, and then immediately after electroporation, one hindlimb was immobilized to induce disuse atrophy, as described in Materials and Methods. At day 3, muscles were excised. B shows muscle weight and C shows the in vivo–determined rate of protein synthesis. For (B) and (C), values are means ± SE; n = 9–10 for each condition. Means with different superscripts (a, b and c) are significantly different (P < 0.05). Immob, immobilization.
DISCUSSION
Deptor was recently identified as an mTOR binding protein. Using RNA interference (RNAi), this mTOR-interacting protein was reported to negatively regulate both mTORC1 and mTORC2, as evidenced by increasing phosphorylation of known substrates. Whereas the role of Deptor has been studied in cancer and other transformed cell lines using short-term transient RNAi transfections (15,35), its role in regulating long-term mTOR-mediated events in skeletal muscle is not known. Moreover, as mTOR regulates multiple metabolic processes such as protein synthesis, cell size (growth), cell cycle, proliferation and development, we posited that decreasing Deptor would affect one or more of these mTOR-mediated events. Therefore, the purpose of our investigation was to ascertain the role of Deptor in mTOR-mediated events in skeletal muscle, and, to this end, we generated C2C12 myoblasts that had a stable KD of Deptor mRNA and protein expression. Deptor KD by ~90% in vitro increased protein synthesis by ~50% in myoblasts. Our data are consistent with the fact that Deptor is not the only regulator of protein synthesis. Protein translation initiation, being an energy-consuming process, has multiple regulatory mechanisms, and it is anticipated that KD of Deptor would activate such mechanism(s) that would restrain changes in protein synthesis. Such compensatory changes could in part explain the discordance between the percentage of Deptor KD and the percent increase in protein synthesis. Consistent with our data, rapamycin and Torin-1 (for example, both mTOR inhibitors) also suppress protein synthesis ~50% (36). Alternatively, whereas protein synthesis is regulated primarily at the translation initiation step, other steps such as elongation and termination also influence protein synthesis. Therefore, the observed difference in percent of Deptor KD and the percent increase in protein synthesis in vitro is within the expected range. Previous reports have used the phosphorylation of T389-S6K1 and T37/46-4E-BP1 (surrogate markers of mTORC1) and S473-Akt (mTORC2 substrate) to implicate Deptor as a putative negative regulator of mTOR, and such results are confirmed in the present study. This information appears to be the first report of Deptor KD on protein synthesis per se.
We postulated that Deptor KD would increase the anabolic and decrease the catabolic response of myocytes. Unfortunately, the data generated in this regard are equivocal and open to divergent interpretation. For example, whereas IGF-I increased protein synthesis in both control and Deptor KD cells, both the percent and absolute increase in protein synthesis appears reduced in KD cells. However, this conclusion has two caveats: (i) protein synthesis in the basal and IGF-I-stimulated condition was determined in different cells, and it is therefore not possible to calculate a standard error and hence perform a statistical analysis on the incremental change, and (ii) data interpretation may be further complicated by a “ceiling effect” present in the Deptor KD cells stimulated with IGF-I. Our supporting Western blot data, which show comparable phosphorylation of AKT, PRAS40 and S6K1 in control and Deptor KD cells, suggest that IGF-I responsiveness is largely unchanged between the two groups. In contradistinction, our data could also be interpreted to indicate that Deptor KD actually increases the maximal responsiveness of cells to IGF-I, since the absolute rate of protein synthesis is higher in Deptor KD cells than in control cells. Such an interpretation is possible because the IGF-I concentration used in the current study was purposefully selected to be maximally stimulating. Resolution of this issue will require complete dose- and time-response studies in both control and Deptor KD cells. Similar difficulties in data interpretation are encountered in evaluating the response of two groups to the catabolic agent AICAR.
Cell growth is a reliable indicator of increased mTOR activity in a variety of cell types (37,38); therefore, we studied the impact of decreasing the cellular content of Deptor. Typically under normal physiological conditions, cells must attain a genetically determined set size before they can replicate and divide, thus ensuring that the daughter cells are of an appropriate size after mitosis (39,40). This regulation of growth and cell division is lost under some disease states (for example, cardiac hypertrophy and cancer) or artificial (for example, use of certain pharmaceutical agents) conditions. mTOR plays an important role in regulating cell growth (40). In this context, knockdown of Deptor in MEF and HeLa cells increases cell size (15). Consistent with this report, Deptor KD in myoblasts also increased cell size and was associated with a coordinate increase in the rate of proliferation, compared with control cells. Thus, our data provide support to the previous report, suggesting that Deptor functions as a negative modulator of mTOR function in regulating cell size (15).
One potential explanation for the increased cell number in myoblasts with Deptor KD could be their resistance to undergo apoptosis, as previously demonstrated using a different cell line (15). In a subset of myeloma in which Deptor is overexpressed, this protein decreases apoptosis; this response is in contrast to its activity as a negative regulator of mTOR (15,35,41,42). Akt regulates and promotes cell survival via the serum/glucocorticoid regulated kinase 1 (SGK1) (mTORC2 substrate), and because Deptor KD activates Akt, we queried whether myocytes with Deptor KD were resistant to apoptosis. Activated mTOR phosphorylates and inhibits signals to the pro-apoptotic proteins, such as caspase-3, which play a critical role in induction of cellular apoptosis. Using the cleavage of caspase-3 and PARP, which are reliable markers of apoptosis, our data suggest there is no difference in the ability of control and Deptor KD myoblasts to undergo apoptosis under normal growth conditions. Therefore, our observed difference in proliferation rate could not be attributed to a change in apoptosis.
mTOR is also central in regulating cell cycle, as evidenced by the ability of the mTOR inhibitor rapamycin to arrest cells in the G1 phase (37,39,43–47). Furthermore, arresting cells in G1/G0 by serum starvation suggests that the mTOR signaling pathway regulates cell growth and cell cycle progression in response to nutrient availability (48–51). Propidium iodide staining of asynchronous myoblasts revealed that Deptor KD decreased the proportion of cells in the G1/G0 phase, compared with scramble control. Further, the proportion of Deptor KD myocytes in the active S phase was increased when determined under conditions in which the cell cycle was synchronized and then released from cell cycle arrest. Collectively, these data suggest Deptor is required for mTOR activity in regulating cell cycle and that Deptor KD enhances myoblast cycle progression, consistent with its role as a negative regulator of mTOR activity. However, we cannot exclude the possibility that the reduction of Deptor alters cell cycle kinetics by an undetermined mTOR-independent mechanism.
Because Deptor KD decreased the percentage of cells in the G1/G0 phase and concomitantly increased the number of cells in the active S phase, we focused on elucidating the potential mechanisms. mTOR can regulate cell cycle progression by a rapamycin-sensitive pathway by promoting RNA polymerase I and III activity via phosphorylation and inactivation of retinoblastoma protein (pRb) (52,53). In addition, rapamycin also prevents the mitogen-induced downregulation of the cyclin-dependent kinase inhibitor p27 (54). The pRb regulates G1 exit in the cell cycle. Upon mitogenic activation, pRb is phosphorylated, resulting in disruption of pRb binding to the E2F transcription factor, thus allowing transcription of proteins that are essential to cell cycle regulation and transition from G1 to S phase. We detected an increase in phosphorylation of pRb S807/S811 concurrent with increased progression from the G1 to S phase (55,56). Because Deptor KD increases Akt activity (15), we also studied this pathway, since it might affect the cell cycle by modulating glycogen synthase kinase (GSK) activity. Because GSK3 has been implicated in regulating cell cycle via cyclin D1, and cyclin D1 mediates pRb phosphorylation (57–60), we examined the effect of Deptor KD on these proteins. The role of cyclin D1, p53, p21, p27, cdk-2, cdk-4 and cdk-6 in mTOR-mediated cell cycle regulation is controversial. Whereas Muise-Helmericks et al. (57) implicated cyclin D1–mediated pRb phosphorylation, Faber and Chiles (61) reported changes in cell cycle regulation independent of these regulatory proteins. In contrast to previous observations by Muise-Helmericks et al., we did not detect a change in the total amount of p21, p27, p53, cdk-2, cdk-4, cdk-6 or cyclin D1 (Supplementary Figure S3), suggesting that under our specific experimental conditions and cell type, Deptor KD–mediated regulation of G1 to S phase does not involve a major role for these regulatory proteins. Our data support changes in cell cycle kinetics without changes in these regulatory proteins, as reported by Faber and Chiles (61). We cannot exclude the possibility that there may yet be other pathways involving pRb that are independent of cyclin D1. One such mechanism involves the stress-regulated mitogen-activated protein kinase p38, which phosphorylates pRb in a cell cycle–independent manner (62).
To determine the effect of Deptor KD on myotube formation and myogenesis and whether Deptor KD would enhance myoblast fusion, we monitored myotube formation. Time-lapse imaging and Western analysis for MHC (a marker for matured myotubes) indicated that knockdown of Deptor in C2C12 myoblasts enhanced myotube formation. Additionally, the lack of change in the MyoD content between the two groups suggests that differentiation stimulated by Deptor KD does not affect the expression of the MyoD transcription factor, thereby suggesting a different mechanism. Autophagy is another mTOR-regulated cellular event that plays an important role in differentiation of myoblasts to mature myocytes (63–66). Our results indicate that Deptor KD does not alter autophagy in C2C12 myoblasts under normal physiological conditions. Collectively, these changes suggest Deptor regulates muscle proliferation and differentiation via regulation of cell cycle regulatory proteins.
Finally, we also determined whether the reduction in Deptor in vivo was capable of altering muscle protein synthesis and mass. To this end, the gastrocnemius was electroporated with the same plasmid construct used in our cell culture model. Using this technique for gene transfer, Deptor mRNA was decreased ~50%. The exact mechanism for the smaller decrease in Deptor KD in vivo versus in vitro is not known but may include the following: (a) The in vitro KD of Deptor was determined in myocytes after stable integration using puromycin selection, whereas the in vivo KD was relatively transient (3 days). (b) Probably most important, not all fibers take up the shRNA targeting Deptor when transfected in vivo. (c) The in vitro response does not have the same hormonal, mechanical or neural influences, as would be present in vivo. Finally, the in vitro protein synthetic response was measured in myoblasts, whereas the in vivo response was determined primarily in myotubes, which are differentiated postmitotic cells. In contrast, reducing Deptor largely prevented the atrophic response produced by immobilization, and, in part, this response was mediated by an increased muscle protein synthesis. In summary, our data suggest that reducing Deptor is sufficient to prevent an atrophy-mediated decrease in muscle protein synthesis and muscle mass.
Understanding the role of Deptor in myocyte cell cycle and proliferation and the ability of this protein to regulate protein synthesis in vivo as described may prove important in designing new strategies to manage the muscle-wasting associated with catabolic insults such as sepsis, alcohol abuse and aging.
Supplemental Data
ACKNOWLEDGMENTS
We thank Margaret Shumate and Robert Frost for discussions and critical readings of the manuscript. We thank Danuta Huber, Anne Pruznak and Maithili Navaratnarajah for technical support. We also thank David Stanford of the Penn State Flow Cytometry Core Facility for help with cell cycle analysis imaging. This work was supported in part by grants from the National Institutes of Health (GM38032 and AA11290) (to CH Lang) and Pennsylvania Department of Health using Tobacco Settlement Funds (to AA Kazi). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Lin TA, et al. PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science. 1994;266:653–6. doi: 10.1126/science.7939721. [DOI] [PubMed] [Google Scholar]
- 2.Smith MR, et al. Modulation of the mitogenic activity of eukaryotic translation initiation factor-4E by protein kinase C. New Biol. 1991;3:601–7. [PubMed] [Google Scholar]
- 3.Kimball SR. Interaction between the AMP-activated protein kinase and mTOR signaling pathways. Med Sci Sports Exerc. 2006;38:1958–64. doi: 10.1249/01.mss.0000233796.16411.13. [DOI] [PubMed] [Google Scholar]
- 4.Kazi AA, Pruznak AM, Frost RA, Lang CH. Sepsis-induced alterations in protein-protein interactions within Mtor complex 1 and the modulating effect of leucine on muscle protein synthesis. Shock. 2011;35:117–25. doi: 10.1097/SHK.0b013e3181ecb57c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kazi AA, Lang CH. PRAS40 regulates protein synthesis and cell cycle in C2C12 myoblasts. Mol Med. 2010;16:359–71. doi: 10.2119/molmed.2009.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong-Brown LQ, et al. Alcohol and PRAS40 knockdown decrease mTOR activity and protein synthesis via AMPK signaling and changes in mTORC1 interaction. J Cell Biochem. 2010;109:1172–84. doi: 10.1002/jcb.22496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pruznak AM, Kazi AA, Frost RA, Vary TC, Lang CH. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribonucleoside prevents leucine-stimulated protein synthesis in rat skeletal muscle. J Nutr. 2008;138:1887–94. doi: 10.1093/jn/138.10.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anthony JC, et al. Contribution of insulin to the translational control of protein synthesis in skeletal muscle by leucine. Am J Physiol Endocrinol Metab. 2002;282:E1092–101. doi: 10.1152/ajpendo.00208.2001. [DOI] [PubMed] [Google Scholar]
- 9.Anthony JC, et al. Leucine stimulates translation initiation in skeletal muscle of postabsorptive rats via a rapamycin-sensitive pathway. J Nutr. 2000;130:2413–9. doi: 10.1093/jn/130.10.2413. [DOI] [PubMed] [Google Scholar]
- 10.Pham PT, et al. Assessment of cell- signaling pathways in the regulation of mammalian target of rapamycin (mTOR) by amino acids in rat adipocytes. J Cell Biochem. 2000;79:427–41. doi: 10.1002/1097-4644(20001201)79:3<427::aid-jcb80>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 11.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 12.Kim DH, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 13.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 14.Kim DH, Sabatini DM. Raptor and mTOR: subunits of a nutrient-sensitive complex. Curr Top Microbiol Immunol. 2004;279:259–70. doi: 10.1007/978-3-642-18930-2_15. [DOI] [PubMed] [Google Scholar]
- 15.Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu M, et al. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem. 2010;285:36387–94. doi: 10.1074/jbc.M110.169284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frost RA, Lang CH, Gelato MC. Transient exposure of human myoblasts to tumor necrosis factor-alpha inhibits serum and insulin-like growth factor-I stimulated protein synthesis. Endocrinology. 1997;138:4153–9. doi: 10.1210/endo.138.10.5450. [DOI] [PubMed] [Google Scholar]
- 19.Williamson DL, Bolster DR, Kimball SR, Jefferson LS. Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am J Physiol Endocrinol Metab. 2006;291:E80–9. doi: 10.1152/ajpendo.00566.2005. [DOI] [PubMed] [Google Scholar]
- 20.Sancak Y, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Das A, Desai D, Pittman B, Amin S, El-Bayoumy K. Comparison of the chemopreventive efficacies of 1,4-phenylenebis(methylene)seleno-cyanate and selenium-enriched yeast on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced lung tumorigenesis in A/J mouse. Nutr Cancer. 2003;46:179–85. doi: 10.1207/S15327914NC4602_11. [DOI] [PubMed] [Google Scholar]
- 22.Le Doux JM. Gene Therapy Protocols. Hu-mana Press; Totowa, N.J: 2008. [Google Scholar]
- 23.Corovic S, et al. The influence of skeletal muscle anisotropy on electroporation: in vivo study and numerical modeling. Med Biol Eng Comput. 2010;48:637–48. doi: 10.1007/s11517-010-0614-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gissel H. Effects of varying pulse parameters on ion homeostasis, cellular integrity, and force following electroporation of rat muscle in vivo. Am J Physiol Regul Integr Comp Physiol. 2010;298:R918–29. doi: 10.1152/ajpregu.00692.2009. [DOI] [PubMed] [Google Scholar]
- 25.Tuckow AP, Vary TC, Kimball SR, Jefferson LS. Ectopic expression of eIF2Bepsilon in rat skeletal muscle rescues the sepsis-induced reduction in guanine nucleotide exchange activity and protein synthesis. Am J Physiol Endocrinol Metab. 2010;299:E241–8. doi: 10.1152/ajpendo.00151.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krawiec BJ, Frost RA, Vary TC, Jefferson LS, Lang CH. Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am J Physiol Endocrinol Metab. 2005;289:E969–80. doi: 10.1152/ajpendo.00126.2005. [DOI] [PubMed] [Google Scholar]
- 27.Tucker KR, Seider MJ, Booth FW. Protein synthesis rates in atrophied gastrocnemius muscles after limb immobilization. J Appl Physiol. 1981;51:73–7. doi: 10.1152/jappl.1981.51.1.73. [DOI] [PubMed] [Google Scholar]
- 28.Vary TC, Lang CH. Assessing effects of alcohol consumption on protein synthesis in striated muscles. Methods Mol Biol. 2008;447:343–55. doi: 10.1007/978-1-59745-242-7_22. [DOI] [PubMed] [Google Scholar]
- 29.Lang CH, Lynch CJ, Vary TC. BCATm deficiency ameliorates endotoxin-induced decrease in muscle protein synthesis and improves survival in septic mice. Am J Physiol Regul Integr Comp Physiol. 2010;299:R935–44. doi: 10.1152/ajpregu.00297.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lang CH, Frost RA, Jefferson LS, Kimball SR, Vary TC. Endotoxin-induced decrease in muscle protein synthesis is associated with changes in eIF2B, eIF4E, and IGF-I. Am J Physiol Endocrinol Metab. 2000;278:E1133–43. doi: 10.1152/ajpendo.2000.278.6.E1133. [DOI] [PubMed] [Google Scholar]
- 31.Vary TC, Kimball SR. Sepsis-induced changes in protein synthesis: differential effects on fast- and slow-twitch muscles. Am J Physiol. 1992;262:C1513–9. doi: 10.1152/ajpcell.1992.262.6.C1513. [DOI] [PubMed] [Google Scholar]
- 32.Garlick PJ, McNurlan MA, Essen P, Wernerman J. Measurement of tissue protein synthesis rates in vivo: a critical analysis of contrasting methods. Am J Physiol. 1994;266:E287–97. doi: 10.1152/ajpendo.1994.266.3.E287. [DOI] [PubMed] [Google Scholar]
- 33.Srinivas V, Bohensky J, Shapiro IM. Au-tophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs. 2009;189:88–92. doi: 10.1159/000151428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeng M, Zhou JN. Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cell Signal. 2008;20:659–65. doi: 10.1016/j.cellsig.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 35.Boyd KD, et al. High expression levels of the mammalian target of rapamycin inhibitor DEPTOR are predictive of response to thalidomide in myeloma. Leuk Lymphoma. 2010;51:2126–9. doi: 10.3109/10428194.2010.509893. [DOI] [PubMed] [Google Scholar]
- 36.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 38.Thomas G, Hall MN. TOR signalling and control of cell growth. Curr Opin Cell Biol. 1997;9:782–7. doi: 10.1016/s0955-0674(97)80078-6. [DOI] [PubMed] [Google Scholar]
- 39.Fingar DC, et al. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–16. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–87. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de la Rubia J, Such E. DEPTOR expression and response to thalidomide: toward a new therapeutic target in multiple myeloma. Leuk Lymphoma. 2010;51:1960–1. doi: 10.3109/10428194.2010.527410. [DOI] [PubMed] [Google Scholar]
- 42.Proud CG. Dynamic balancing: DEPTOR tips the scales. J Mol Cell Biol. 2009;1:61–3. doi: 10.1093/jmcb/mjp012. [DOI] [PubMed] [Google Scholar]
- 43.Hentges KE, et al. FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proc Natl Acad Sci U S A. 2001;98:13796–801. doi: 10.1073/pnas.241184198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peponi E, et al. Activation of mammalian target of rapamycin signaling promotes cell cycle progression and protects cells from apoptosis in mantle cell lymphoma. Am J Pathol. 2006;169:2171–80. doi: 10.2353/ajpath.2006.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosner M, Fuchs C, Siegel N, Valli A, Hengstschlager M. Functional interaction of mammalian target of rapamycin complexes in regulating mammalian cell size and cell cycle. Hum Mol Genet. 2009;18:3298–310. doi: 10.1093/hmg/ddp271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Proud CG. Nutrient control of TORC1, a cell-cycle regulator. Trends Cell Biol. 2009;19:260–7. doi: 10.1016/j.tcb.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 47.Sehgal SN. Rapamune (RAPA, rapamycin, sirolimus): mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin Biochem. 1998;31:335–40. doi: 10.1016/s0009-9120(98)00045-9. [DOI] [PubMed] [Google Scholar]
- 48.Crespo JL, Hall MN. Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2002;66:579–91. doi: 10.1128/MMBR.66.4.579-591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asnaghi L, Bruno P, Priulla M, Nicolin A. mTOR: a protein kinase switching between life and death. Pharmacol Res. 2004;50:545–9. doi: 10.1016/j.phrs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 50.Oldham S, Hafen E. Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol. 2003;13:79–85. doi: 10.1016/s0962-8924(02)00042-9. [DOI] [PubMed] [Google Scholar]
- 51.Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117–26. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- 52.White RJ. Regulation of RNA polymerases I and III by the retinoblastoma protein: a mechanism for growth control. Trends Biochem Sci. 1997;22:77–80. doi: 10.1016/s0968-0004(96)10067-0. [DOI] [PubMed] [Google Scholar]
- 53.Hashemolhosseini S, et al. Rapamycin inhibition of the G1 to S transition is mediated by effects on cyclin D1 mRNA and protein stability. J Biol Chem. 1998;273:14424–9. doi: 10.1074/jbc.273.23.14424. [DOI] [PubMed] [Google Scholar]
- 54.Luo Y, et al. Rapamycin resistance tied to defective regulation of p27Kip1. Mol Cell Biol. 1996;16:6744–51. doi: 10.1128/mcb.16.12.6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ren S, Rollins BJ. Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell. 2004;117:239–51. doi: 10.1016/s0092-8674(04)00300-9. [DOI] [PubMed] [Google Scholar]
- 56.Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89:213–28. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 57.Muise-Helmericks RC, et al. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J Biol Chem. 1998;273:29864–72. doi: 10.1074/jbc.273.45.29864. [DOI] [PubMed] [Google Scholar]
- 58.Ong CS, Zhou J, Ong CN, Shen HM. Luteolin induces G1 arrest in human nasopharyngeal carcinoma cells via the Akt-GSK-3beta-Cyclin D1 pathway. Cancer Lett. 2010;298:167–75. doi: 10.1016/j.canlet.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 59.Yeste-Velasco M, et al. Glycogen synthase kinase-3 is involved in the regulation of the cell cycle in cerebellar granule cells. Neuropharmacology. 2007;53:295–307. doi: 10.1016/j.neuropharm.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 60.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faber AC, Chiles TC. Inhibition of cyclin-dependent kinase-2 induces apoptosis in human diffuse large B-cell lymphomas. Cell Cycle. 2007;6:2982–9. doi: 10.4161/cc.6.23.4994. [DOI] [PubMed] [Google Scholar]
- 62.Delston RB, Matatall KA, Sun Y, Onken MD, Harbour JW. p38 phosphorylates Rb on Ser567 by a novel, cell cycle-independent mechanism that triggers Rb-Hdm2 interaction and apoptosis. Oncogene. 2010;30:588–99. doi: 10.1038/onc.2010.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4:524–6. doi: 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- 64.Annovazzi L, et al. mTOR, S6 and AKT expression in relation to proliferation and apoptosis/autophagy in glioma. Anticancer Res. 2009;29:3087–94. [PubMed] [Google Scholar]
- 65.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–95. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ravikumar B, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.