

Abstract
Early reperfusion after myocardial ischemia that is essential for tissue salvage also causes myocardial and vascular injury. Cardioprotection during reperfusion therapy is an essential aspect of treating myocardial infarction. Angiopoietin-1 is an endothelial-specific angiogenic factor. The potential effects of angiopoietin-1 on cardiomyocytes and vascular cells undergoing reperfusion have not been investigated. We propose a protective mechanism whereby angiopoietin-1 increases the integrity of the endothelial lining and exerts a direct survival effect on cardiomyocytes under myocardial ischemia followed by reperfusion. First, we found that angiopoietin-1 prevents vascular leakage through regulating vascular endothelial (VE)-cadherin phosphorylation. The membrane expression of VE-cadherin was markedly decreased on hypoxia/reoxygenation but was restored by angiopoietin-1 treatment. Interestingly, these effects were mediated by the facilitated binding between SH2 domain–containing tyrosine phosphatase (SHP2) or receptor protein tyrosine phosphatase μ (PTPμ) and VE-cadherin, leading to dephosphorylation of VE-cadherin. siRNA against SHP2 or PTPμ abolished the effect of angiopoietin-1 on VE-cadherin dephosphorylation and thereby decreased levels of membrane-localized VE-cadherin. Second, we found that angiopoietin-1 prevented cardiomyocyte death, although cardiomyocytes lack the angiopoietin-1 receptor Tie2. Angiopoietin-1 increased cardiomyocyte survival through integrin-β1–mediated extracellular signal–regulated kinase (ERK) phosphorylation, which inhibited caspase-9 through phosphorylation at Thr125 and subsequently reduced active caspase-3. Neutralizing antibody against integrin-β1 blocked these protective effects. In a mouse myocardial ischemia/reperfusion model, angiopoietin-1 enhanced cardiac function and reduction in left ventricular–end systolic dimension (LV-ESD) and left ventricular–end diastolic dimension (LV-EDD) with an increase in ejection fraction (EF) and fractional shortening (FS). Our findings suggest the novel cardioprotective mechanisms of angiopoietin-1 that are achieved by reducing both vascular leakage and cardiomyocyte death after ischemia/reperfusion injury.
INTRODUCTION
Prolonged periods of myocardial ischemia cause irreversible loss of cardiomyocytes and vasculature, which are vital in maintaining cardiac integrity and function (1–3). Thus, early reperfusion is the paramount strategy for myocardial salvage (4). However, reperfusion itself also causes myocardial damage, resulting in ventricular remodeling and heart failure (4,5). In addition to “cardiomyocyte death,” reperfusion leads to interstitial edema, which is primarily due to increased “vascular permeability,” subsequently compounding myocardium loss (2,3). Protection of cardiovascular cells from ischemia/reperfusion (IR) injury is the prime focus for optimizing therapeutic strategies.
Angiopoietin-1 (Ang-1) is a strong antipermeability factor that binds to the endothelial-specific Tie2 receptor and induces Tie2 phosphorylation (6,7). Ang-1/Tie2 signaling regulates vessel formation and maintenance of endothelial integrity (8,9). The Ang-1/Tie2 system is also implicated in hypoxic myocardium pathology, where Ang-1 expression is reduced (10). Conversely, overexpression of Ang-1 in acute- infarcted myocardium resulted in reduction in edema and infarct sizes and preservation of ejection fractions (11). These improvements were attributed to the ability of Ang-1 to prevent vessel permeability expressing the Tie2 receptor (6,7). Recently, fibroblasts, skeletal myocytes and cardiomyocytes lacking Tie2 receptor expression were shown to bind Ang-1, potentially via integrins (12,13). However, the detailed mechanism of how Ang-1 affects cardiomyocytes through integrins and maintains vascular integrity in the infarcted myocardium has not been clarified.
Herein, we present novel mechanisms and the signaling pathway by which Ang-1 maintains vascular integrity and protects cardiomyocytes from IR injury. Specifically, we showed that Ang-1 promotes endothelial integrity through vascular endothelial (VE)-cadherin dephosphorylation and cardiomyocyte survival through the integrin-β1/phospho ERK (pERK)/caspase-9(Thr125)/caspase-3 axis. Therefore, we propose that Ang-1 is an important therapeutic candidate to preserve myocardial function after reperfusion injury.
MATERIALS AND METHODS
In Vivo Mouse Myocardial IR Model and Evaluation of Vascular Leakage
All animal experiments were performed under approval from the Institutional Animal Care and Use Committee of Seoul National University Hospital. To make the mouse IR model, male C57BL/6 mice (8–10 wks) were anesthetized with an intraperitoneal injection of tiletamine/zolazepam (10 mg/kg, Zoletil®; Virbac Laboratories, Carros, France) and Xylazine (5 mg/kg; Rompun; Bayer, Leverkusen, Germany). Under mechanical ventilation (Harvard Apparatus), the heart was exposed through the left-sided thoracotomy. The left anterior descending artery was ligated using 8-0 polypropylene sutures with a small piece of polyethylene tube, which was inserted in a knot. After 45 min, the polyethylene tube was removed for reperfusion injury. Subsequently, the chest wall was closed after draining out all bloody fluid. For controls, a sham surgery was performed the same way, with the exception of coronary artery ligation. Adenovirus-expressing cartilage oligomeric matrix protein–angiopoietin-1 (COMP-Ang-1) or β-galactosidase was injected intravenously through the tail vein 10 d before surgery (1 × 109 plaque-forming units) (14). We measured the circulating COMP-Ang-1 at 24 h after IR, and the concentration of circulating COMP-Ang-1 was approximately 3.2 μg (data not shown).
Evaluation of vessel leakage
At 24 h after IR, cardiac vessel leakage was quantified by intravenous injection of 1% Evans blue (Sigma-Aldrich, St. Louis, MO, USA) 15 min before sacrifice (15,16). The excised hearts were retrogradely perfused through aorta with phosphate-buffered saline (PBS) to wash residual nonleaked Evans blue in coronary vasculature. To quantify the Evans blue leakage, heart sections were placed in formamide and incubated at 55°C for 2 h, and then the optical density of the supernatant at 610 nm was measured.
Cell Culture and Hypoxia/Reoxygenation Injury Condition
Cardiomyocyte preparation
Neonatal rat cardiomyocytes were isolated from hearts of 2-day-old Fischer-344 rats. Cardiomyocytes were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 containing 15 mmol/L HEPES; 5% horse serum; insulin, transferrin, and selenium (ITS; Gibco; Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Gibco Invitrogen). Cardiomyocyte purity was confirmed by staining with anti–troponin T (Santa Cruz) and actinin (Sigma), whereas fibroblast was confirmed with an anti–fibroblast-specific marker (AF5110-1; Acris) and vimentin (Sigma).
Endothelial cells and hypoxia/reoxygenation injury condition
Human umbilical vein endothelial cells (HUVECs) (Lonza, passages 4–6) were grown in endothelial growth medium (EGM bullet kit; Clonetics) containing 10% fetal bovine serum at 37°C. Hypoxic conditions were achieved using a BBL GasPac System (Becton Dickinson, Lexington, KY, USA), which can catalytically reduce oxygen levels to <1% oxygen within 15 min, as previously described (17,18). Cells were then placed under normoxic conditions, supplied with fresh medium and continuously incubated for reoxygenation treatment. As a recombinant Ang-1 protein, a potent Ang-1 variant (COMP-Ang-1) was used (14). In blocking experiments, Tie-2Fc (10 μg/mL; R&D Systems, Minneapolis, MN, USA), blocking antibodies against integrin-β1 (50 μg/mL; Becton Dickinson) or integrin-β3 (50 μg/mL; Becton Dickinson) were added to the cells, 30 min before COMP-Ang-1 (cAng-1) treatment in reoxygenation conditions.
FITC-Dextran Permeability Assay
Permeability across the HUVECs cultured on 0.1% gelatin-coated transwells (0.4-μm pore size; Corning Costar; Corning, NY, USA) was measured by adding 0.1 mg FITC-conjugated dextran (molecular weight, ~70,000; Sigma) to the upper chamber. After 10 min, the fluorescence of the lower chamber was determined by a fluorescence spectrofluorometer (Tecan Spectra Fluor) at an excitation wavelength of 490 nm and an emission wavelength of 515 nm.
Western and Immunoprecipitation Analysis
Cells were lysed for 20 min in lysis buffer containing protease inhibitors (Roche, Indianapolis, IN, USA). Mouse hearts were grinded in liquid nitrogen and then lysed using protein lysis buffer. Total protein (10~30 μg) was immunoblotted with specific primary antibodies overnight at 4°C. α-Tubulin or β-actin was used as an internal control.
Immunoprecipitation analysis
Total protein (500 μg) incubated with primary antibodies overnight at 4°C (Protein G-Agarose; Roche) was incubated at 4°C for 90 min with gentle mixing. The beads were then washed twice with lysis buffer.
Membrane fraction for VE-cadherin
Cells were solubilized with cytoskeleton-stabilizing buffer containing protease inhibitor (Roche) and then the soluble fraction was collected (19). Insoluble pellets were further incubated at 95°C with cytoskeleton-stabilizing buffer containing 1% Triton X-100/1% sodium dodecyl sulfate. We used antibodies as follows: VE-cadherin, SHP2, Tie2, phospho-tyrosine, PTPβ/VE-PTP, PTPμ, B-cell lymphoma–extra large (Bcl-XL), total glycogen synthesis kinase-3β (GSK-3β) (Santa Cruz), phospho-AKT, pERK, total extracellular signal–related kinase (ERK), pGSK-3β(Ser9), caspase-9, cleaved caspase-9, cleaved caspase-3 (Cell Signaling, Danvers, MA, USA), caspase-9(Thr125) (Novus), total AKT (Becton Dickinson), phospho-VE-Cadherin (Y658, Biosource), integrin-linked kinase (ILK) (Chemicon), α-tubulin and β-actin (Calbiochem).
Immunofluorescence Staining
Cells were grown on the 0.1% gelatin-coating cover glasses and fixed with 100% methanol for 20 min at −20°C. After blocking with 1% bovine serum albumin/PBS for 1 h, cells were labeled with anti–VE-cadherin followed by Cy3- conjugated secondary antibody (Jackson). Cardiomyocytes were fixed with 4% paraformaldehyde (PFA) and labeled with anti–caspase-3 followed by Alexa-555 secondary antibody. The nuclei were stained with di aminido phenyl indol (1 μg/mL in PBS), mounted using a fluorescent mounting medium (DAKO) and examined using a fluorescence microscope (Olympus IX71) and confocal microscopy (Carl Zeiss).
RNA Interference and Reverse Transcriptase–Polymerase Chain Reaction
Cells were grown up to 80% confluence, and small interfering RNAs (siRNAs) (50 μmol/L) against SHP2, PTPβ, PTPμ or control siRNA (Dharmacon) were transfected using Metafectene-Pro (Biontex). Total RNA was isolated using a TriZol reagent (Invitrogen), and reverse transcriptase–polymerase chain reaction was performed using avian myeloblastosis virus reverse transcriptase (Promega) with the following primers: human SHP2, 5′-GCCTGTTTCA GATTACATCAATGC-3′ (forward); 5′-TTTGGGAACGTCAATATCGC-3′ (reverse); human PTPβ, 5′-ACATCCCTGGCAACAACTTC-3′ (forward); 5′-TGGCCACACCGTATAGTGAA-3′ (reverse); human PTPμ, 5′-CCGGTGCATGGACATCCTGCC-3′ (forward); 5′-CTTGTACTGATCCAGGAGGTC-3′ (reverse); GAPDH, 5′-CCTCTGGAAAGCTGTGGCGT-3′ (forward); 5′-TTGGAGGCCATGTAGGCCAT-3′ (reverse).
Assays for Cell Death
Lactate dehydrogenase assay
The absorbance of lactate dehydrogenase in serum-free media was measured at 340 nm, following the manufacturer’s instructions (Roche).
Propidium iodide (PI) staining
Cells were harvested, washed with PBS (plus 5 mmol/L ethylenediaminetetraacetic acid [EDTA]) and fixed with 70% ethanol for 24 h at −20°C. Cells were collected and resuspended in PBS containing 5 mmol/L EDTA. After 50 μg/mL RNase A digestion (Roche), cells were stained with PI (50 μg/mL) and analyzed with an FACSCalibur (Becton Dickinson).
TUNEL assay
Cells were fixed with 4% PFA and treated with proteinase K (20 μg/mL) for 10 min, and then terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) analysis (Roche) was performed. DNA fragmentation was visualized by staining with 0.25% diaminobenzidine/0.075% H2O2. TUNEL+ cells were counted in 10 different microscopic fields from at least three different culture plates.
Ultrastructural Analysis by Electron Microscopy and Immunohistochemical Staining
Electron microscopy analysis
Mouse hearts were fixed in 0.1 mol/L sodium cacodylate buffer (pH 7.3) containing 4% PFA and 1.5% glutaraldehyde for 2 h, transferred to 5% glutaraldehyde overnight and then transferred to 1% osmium tetroxide for 1 h. Blocks were washed, dehydrated in a graded ethanol series and embedded in Epon/Araldite resin. Ultrathin sections were stained with uranyl acetate/lead citrate and were viewed using a transmission electron microscope (TEM, JEM-100CX, JEOL).
Immunohistochemical staining
Tissues were fixed in 4% PFA, dehydrated in a graded sucrose series, embedded and frozen in optimal cutting temperature (OCT) compound and serially sectioned to 5–10 μm at −20°C. We performed immunohistochemistry using a Vector Labs immunohistochemistry kit, Caspase-9(Thr125) (Novus, Littleton, CO, USA), pERK and cleaved caspase-3 (Cell Signaling). At least three animals were used for each group.
Physiological Study of Left Ventricular Function and Masson’s Trichrome Staining
Four weeks after IR surgery, cardiac function was evaluated by M-mode echocardiography using Aplio XG (15 MHz; Toshiba, Japan). We measured left ventricular–end systolic dimension (LV-ESD) and left ventricular– end diastolic dimension (LV-EDD) at the mid-ventricular papillary muscle level with parasternal long-axis and short-axis two-dimensional echocardiographic guidance, and then ejection fraction (EF) and fractional shortening (FS) were calculated. After functional analysis, mouse hearts were excised, fixed, embedded and stained by Masson’s trichrome staining to determine infarct size and cardiac remodeling. The infarct area was measured by an image analysis system (Scion Image; Scion Corporation) in five different sections from each animal.
Statistical Analysis
Quantification of band intensity was analyzed using TINA 2.0 (RayTest) and normalized to the intensity of α-tubulin. The results are expressed as means ± standard deviation. The differences between the groups were compared by the unpaired t test or one-way analysis of variance, followed by post hoc analysis with the Bonferroni test. P values ≤0.05 were considered statistically significant. All statistical analyses were performed using SPSS 17.0 (SPSS, Chicago, IL, USA).
All supplementary materials are available online at www.molmed.org.
RESULTS
Treatment of cAng-1 Reduced Vascular Leakage and Facilitated Membrane Localization of VE-Cadherin in Endothelial Cells
We first examined whether Ang-1 could reduce vascular leakage in an in vivo myocardial IR model (Supplementary Figure S1). We used the COMP-Ang-1 (cAng-1) protein, which is stable and potent compared with the native Ang-1 (14,20). Vascular leakage, measured by intravenous injection of Evans blue, was evident in IR/Ad-β-gal hearts compared with the sham animals (P < 0.01), but it was significantly reduced by cAng-1 treatment in IR/Ad-cAng-1 hearts (P < 0.01) (Supplementary Figure S1). We next investigated the role of cAng-1 in HUVECs under hypoxia/reoxygenation (H/Reoxy), an in vitro condition mimicking in vivo IR (Figure 1 and Supplementary Figure S2). Using the passage of FITC-dextran through endothelial monolayers as a measure of permeability, we found that cAng-1 significantly reduced endothelial leakage by H/Reoxy in a dose-dependent manner (P < 0.05) (Supplementary Figure S2a), and this effect was blocked by Tie-2Fc, a soluble Tie2 receptor (Figure 1A). In this experiment, we found 200 ng/mL cAng-1 was the most effective concentration in terms of permeability reduction (Supplementary Figure S2), and we used this concentration in the next step.
Figure 1.
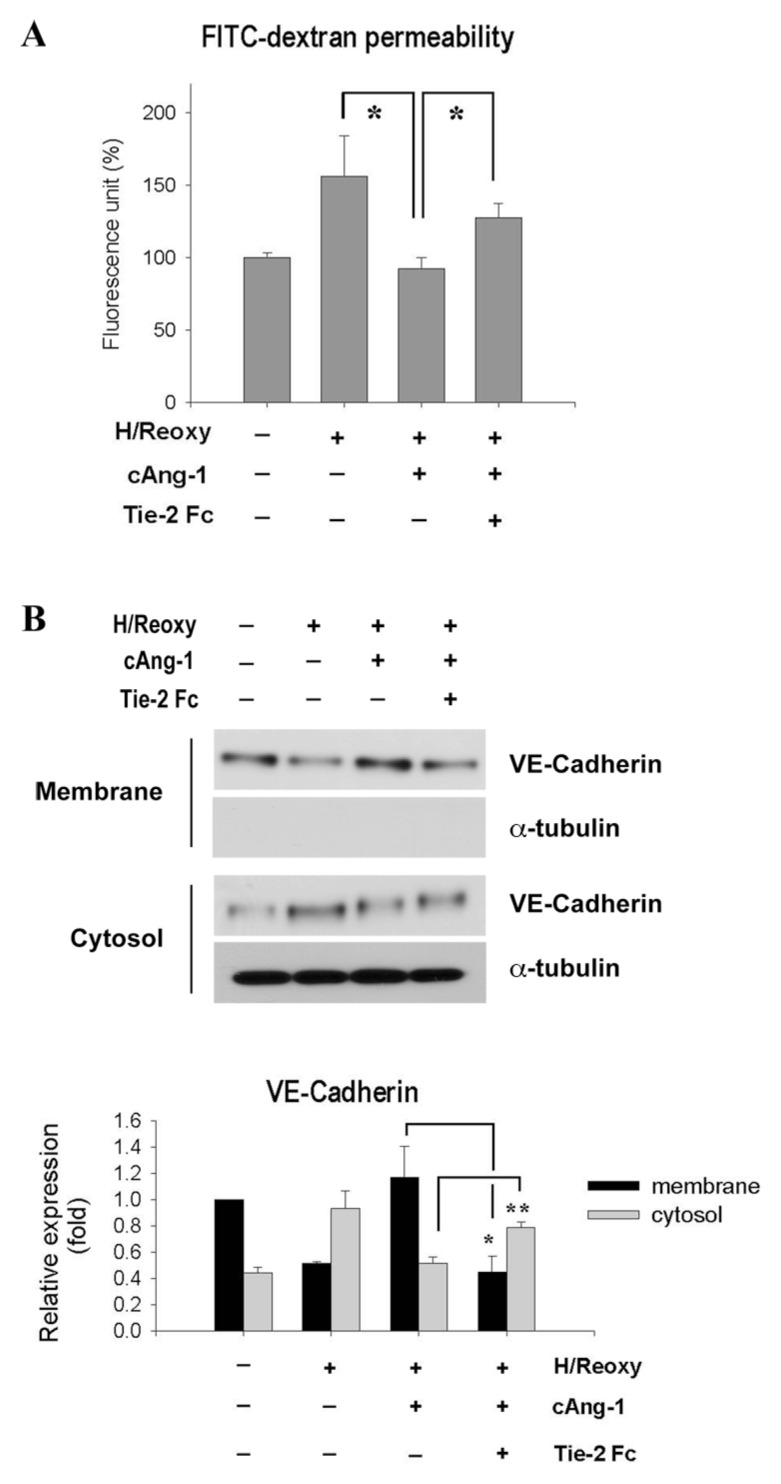
cAng-1 improved vascular integrity by regulating the membrane localization of VE-cadherin. (A) FITC-dextran permeability of endothelial monolayer. Endothelial cells seeded on transwell filters were incubated under hypoxic conditions for 16 h and then reoxygenated for 2 h (H/Reoxy). Tie-2Fc (10 μg/mL) was added 30 min before cAng-1 (200 ng/mL) treatment. n = 3, *P < 0.05. The effect of cAng-1 was blocked by Tie-2Fc. (B) H/Reoxy-induced shift of VE-cadherin from membrane to cytosol fraction, which was prevented by cAng-1 treatment. Distribution of VE-cadherin at the membrane and in the cytosol fraction is shown (top). The quantification graph of VE-cadherin immunoblots (bottom) is shown (n = 3; *P < 0.01; **P < 0.05; Tie-2Fc [10 μg/mL]; cAng-1 [200 ng/mL]).
Because VE-cadherin, an endothelial-specific adherens junctional protein, regulates endothelial permeability (21,22), we investigated whether cAng-1 affects VE-cadherin expression. The total amount of VE-cadherin was not changed either by H/Reoxy or by addition of cAng-1 (Supplementary Figure S2b). Therefore, we checked the cellular distribution of VE-cadherin (Figure 1B and Supplementary Figure S2c), because the integrity of endothelial cell–cell junctions depends on the cell membrane localization of VE-cadherin (20,21). VE-cadherin (red), under normoxic conditions, was found to be mainly located at the cellular membrane. After H/Reoxy, however, membrane VE- cadherin expression was markedly reduced, and its cytoplasmic localization correspondingly increased. Intriguingly, cAng-1 restored membrane VE-cadherin, which was blocked by Tie-2Fc (see Figure 1B and Supplementary Figure S2c), indicating that cAng-1 affects endothelial integrity by regulating VE-cadherin localization.
cAng-1 Maintained Membrane VE-Cadherin by SHP2- or PTPμ-Dependent Dephosphorylation of VE-Cadherin in Endothelial Cells
We next evaluated the effect of cAng-1 on the tyrosine phosphorylation status of VE-cadherin (pY-VE-cadherin), which has been suggested to be correlated with weakened cell adhesion and increased endothelial cell permeability (23). pY-VE-cadherin was not observed under normoxic conditions, whereas it was strongly upregulated after H/Reoxy in endothelial cells (Figure 2A). Importantly, cAng-1 significantly reduced pY-VE-cadherin, which was reversed by Tie-2Fc. Protein tyrosine phosphatases such as SHP2, VE-PTP/PTPβ and PTPμ were reported to regulate VE-cadherin phosphorylation (24). We knocked down these tyrosine phosphatases using their specific siRNA (Supplementary Figure S3a). The expression of each tyrosine phosphatase was remarkably and specifically blocked by specific siRNA, whereas it was not after transfection of control nonsilencing siRNA (siCon). We tested whether interaction between VE- cadherin and each phosphatase would be modulated by cAng-1 treatment (Figure 2B). We found that SHP2, PTPβ and PTPμ bound strongly to VE- cadherin, respectively, under normoxia and that this binding was significantly reduced under H/Reoxy conditions (see Figure 2B). Interestingly, cAng-1 remarkably restored the binding between SHP2 and VE- cadherin and also between PTPμ and VE-cadherin. Furthermore, we found that, even in the presence of cAng-1, inhibition of SHP2 or PTPμ phosphatase potentiated VE-cadherin phosphorylation at Tyr658, the phosphorylation of which is known to disrupt cell–cell barrier function (Figures 2C, D) (23). In parallel with Figure 2C, SHP2 inhibition reduced the membrane expression of VE-cadherin even in the presence of cAng-1 under H/Reoxy (Supplementary Figure S3b). The above findings suggest that cAng-1 stabilizes endothelial contacts by enhancing binding of phosphatases and VE-cadherin, resulting in dephosphorylation of VE-cadherin and maintenance of its membrane expression, thereby maintaining adherence junctions.
Figure 2.

cAng-1 reduces phosphorylation of VE-cadherin by interaction with phosphatases, such as SHP2 or PTPμ in HUVECs. (A) Immunoprecipitation with anti–VE-cadherin antibody followed by immunoblotting with phospho-tyrosine (P-Tyr) antibody. The same filter was reprobed with anti–VE-cadherin antibody, demonstrating equal loading. pY-VE-cadherin was increased after H/Reoxy. cAng-1 markedly reduced pY-VE-cadherin, which was reversed by Tie-2 Fc (n = 3, *P < 0.05; N.D., not determined). (B) Immunoprecipitates by anti-SHP2, anti-PTPβ or anti-PTPμ antibody were immunoblotted with anti–VE-cadherin antibody. Binding of VE-cadherin with phosphatase significantly decreased in H/Reoxy conditions. cAng-1 significantly restored the binding of VE-cadherin with SHP2 or PTPμ but negligibly with PTPβ (n = 3, *P < 0.05 versus Nor; **P < 0.05 versus Hy; N.D., not determined). (C, D) Inhibition of phosphatase using SHP2 siRNA or PTPμ siRNA remarkably increased pVE-cadherin (Tyr-658) levels (C, D, left) resulting in the increased ratio of phospho-VE-cadherin to VE-cadherin (C, D, right) (n = 3, *P < 0.05).
cAng-1 Restored Ultrastructural Vascular Integrity in the IR Myocardium
To determine whether our in vitro findings were valid in vivo, we examined mouse IR tissues at 24 h after surgery by electron microscopy (Figure 3A). Sections of sham heart showed normal mitochondria and myofilament architecture (see Figure 3A, a) as well as an intact and uniformly thick endothelial layer (see Figure 3A, b). In contrast, the peri-infarct myocardial area of the IR/Ad-β-gal group showed the disrupted endothelial linings of microvessels with red blood cell leakage (arrows) (see Figure 3A, c–e) as well as vacuoles, which are indicators of endothelial damage (arrowhead) (see Figure 3A, f ) (25). In the IR/Ad-cAng-1 group, however, pathological changes were significantly improved (see Figure 3A, g and h). Even in the peri-infarct zone, microvessels maintained an intact endothelial lining around damaged cardiomyocytes (see Figure 3A, g). Moreover, in the immunoprecipitation and Western blot analysis using myocardial protein, the binding between SHP2 and VE-cadherin was reduced in the IR/Ad-β-gal group, but was remarkably enhanced in the IR/Ad-cAng-1 group (Figure 3B). Binding of PTPμ/VE-cadherin was also remarkably increased in the IR/Ad-cAng-1 group (see Figure 3B).
Figure 3.

cAng-1 restored ultrastructural vascular integrity in the IR myocardium. (A) Transmission electron micorsscope (TEM) analysis of IR tissues at 24 h after surgery. (a, b) Sham group. (c–f ) IR/Ad-β-gal group. (e, f ) Enlarged view of the area indicated as rectangles from (d ). Arrows, vessel breakdown; arrowhead, vacuole in endothelium. (g, h) IR/Ad-cAng-1 tissue. Arrows, cardiomyocyte damage. Breakdown of vascular integrity and endothelial/cardiomyocyte damages are frequently observed in the IR myocardium, but cAng-1 treatment prevented such damages. Images are representative ones from each group. RBC, red blood cells. Scale bar = 2 μm. (B) Immunoprecipitation of cardiac protein with anti–VE-cadherin antibody followed by immunoblotting with anti-SHP2 or anti-PTPμ antibody. Binding of SHP2/VE-cadherin or PTPμ/VE-cadherin was notably decreased in the IR myocardium, which was remarkably restored by Ad-cAng-1 treatment (n = 3). WCL, whole cell lysate.
cAng-1 Protected Cardiomyocytes from H/Reoxy-Induced Death through Integrin-β1
Apoptosis was observed in hearts subjected to either continuous ischemia or ischemia followed by reperfusion (4). We evaluated the effect of cAng-1 on cardiomyocyte survival under H/Reoxy conditions (Supplementary Figure S4). Rat neonatal cardiomyocytes showed the reduced-viability under H/Reoxy conditions. Surprisingly, cAng-1 markedly protected cardiomyocytes from H/Reoxy-induced death (Supplementary Figure S4a), although cardiomyocytes did not express Tie2 (Supplementary Figure S4b).
It was recently reported that nonendothelial cells including cardiomyocytes and fibroblasts could bind to Ang-1 via integrin-β1 or -β3 (13). However, this previous study only suggested the possibility of binding between Ang-1 and integrins-β1 or -β3 under normal/resting conditions. But, it has not been tested which of those integrins is a real counter partner of Ang-1 or how the downstream signaling pathways of Ang-1/integrin axis affect survival of cardiomyocytes under IR conditions. Thus, we focused on integrin-β1 and -β3 and examined which has a role in Ang-1-mediated cardiomyocyte protection (Figure 4). Blocking antibody against integrin-β1 completely abolished the protective effect of cAng-1 on cardiomyocytes, whereas either integrin-β3 or isotype IgG blocking did not show remarkable change (see Figure 4). In Figures 4C and D, apoptotic cell numbers were markedly increased after H/Reoxy but were significantly reduced by cAng-1 treatment in the TUNEL assay. Caspase-3+ cells (red, arrowheads) indicate early apoptotic cells, and caspase-3+/TUNEL+ cells (arrows) are late apoptotic cells. In agreement with earlier results, an integrin-β1 blockade significantly abolished the protective effect of cAng-1 (n = 4, P < 0.01) (see Figure 4D).
Figure 4.

cAng-1 promoted cardiomyocyte survival under H/Reoxy conditions via integrin-β1. Each experiment showed rat neonatal cardiomyocyte viability after 16 h of hypoxia and 2 h of reoxygenation (H/Reoxy). IgG, anti–integrin-β1 and anti–integrin-β3 (50 μg/mL) were added 30 min before cAng-1 (200 ng/mL). Blocking antibody against integrin-β1 completely abolished the protective effect of cAng-1 on cardiomyocytes after H/Reoxy. (A) Lactate dehydrogenase (LDH) analysis (n = 3, *P < 0.05; P = NS, no significance). (B) PI–fluorescence-activated cell sorter (FACS) analysis (n = 3, *P < 0.05). (C) Immunofluorescence staining for cleaved caspase-3 and TUNEL. Arrows, caspase-3+/TUNEL+ cells; arrowheads, caspase-3+ cells. Scale bar = 40 μm. (D) TUNEL+ cells were counted in 10 different microscopic fields in at least 3 different culture plates (*P < 0.01).
cAng-1 Suppressed Cascade of Caspase-9 and Caspase-3 through ERK Activation in Cardiomyocytes
In Figure 5A, caspase-3 protein, a key element of the apoptotic pathway, was increased under H/Reoxy, which was significantly reduced by cAng-1 treatment. Interestingly, integrin-β1 blockade completely reversed the effect of cAng-1, whereas integrin-β3 blocking did not. We further confirmed that cAng-1 increased antiapoptotic Bcl-XL and reduced caspase-9, an initiator protease that activates caspase-3 (Figure 5B). Integrin-β1 blockade again completely reversed the effect of cAng-1. Exposure of cardiomyocytes to H/Reoxy + cAng-1 dramatically increased the phosphorylation of ERK, which was blocked by the integrin-β1 neutralizing antibody (Figure 5C). In contrast, total amount of ILK and the phosphorylation of Akt or GSK-3β were not remarkably affected by treatment of cAng-1 in H/Reoxy cardiomyocytes. To determine the roles of ERK in cAng-1-dependent alteration of caspase-9 protein, the ERK–mitogen-activated protein kinase (MAPK) pathway inhibitor PD98059 was applied to cardiomyocytes (Figure 5D). PD98059 reversed the caspase-9 activation, whereas the phosphatidylinositol 3-kinase (PI3K)/Akt pathway inhibitor LY294002 did not. We also checked the counterpart of integrin-β1 (Supplementary Figure S5). When we treated cardiomyocytes with blockade of integrin-α5, one of the β-integrin counterparts, caspase-3, was activated, even in the presence of cAng-1.
Figure 5.

cAng-1 suppressed apoptotic machinery in cardiomyocytes under H/Reoxy by an integrin-β1/ERK–dependent manner. Neonatal cardiomyocytes were incubated for 16 h of hypoxia and 2 h of reoxygenation (H/Reoxy). (A, B) Representative immunoblot and quantitative graph of cleaved caspase-3, Bcl-XL and cleaved caspase-9 (n = 4, each). Integrin-β1 blockade completely reversed the protective effect of cAng-1. (C) Levels of phosphorylated Akt, GSK-3β(Ser9) and ERK. Integrin-β1 neutralizing remarkably blocked the phospho-ERK. (D) PD98059 (10 μmol/L) or LY294002 (10 μmol/L) was incubated for 30 min before exposure to cAng-1. Only PD98059, an inhibitor of the ERK-MAPK pathway, blocked caspase-9 suppression by cAng-1.
cAng-1 Inactivated Caspase-9 through Phosphorylation at Thr125 and Suppressed Apoptosis in the Infarcted Myocardium
Next, we confirmed the effects of cAng-1 on cardiomyocyte apoptosis in in vivo IR tissue (Figure 6A). The ERK-MAPK pathway is known to mediate cell protection from apoptosis by inactivating caspase-9 through phosphorylation at the Thr125 site (26). In the IR tissue treated with cAng-1, the immunoreactivity of the inactivated or phosphorylated caspase-9(Thr125) was significantly increased, indicating that activation of caspase-9 is inhibited by phosphorylation at Thr125 by ERK in response to cAng-1. In parallel to this finding, the immunoreactivity of cleaved caspase-3, the downstream effector of caspase-9, remarkably decreased by cAng-1. Moreover, we quantitated the amount of phospho- caspase-9(Thr125) and active caspase-3 by immunoblot analysis using heart protein (Figure 6B, C). IR injury significantly decreased caspase-9(Thr125) and increased active caspase-3, which was reversed by cAng-1 treatment.
Figure 6.

In vivo evidence of cAng-1 to prevent apoptosis of cardiomyocytes in IR myocardium and to restore loss of cardiac performance in a mouse IR injury model. (A) Immunohistochemical staining of phosphorylated caspase-9(Thr125) and caspase-3 (brown) in heart tissues at 4 wks after IR. IR reduced caspase-9(Thr125) and activated caspase-3, which was prevented by cAng-1 treatment. Scale bar = 100 μm (magnification 400×). Representative images were shown (n = 4). (B) Myocardial proteins at 4 wks after IR were immunoprecipitated (IP) with anti–caspase-9 followed by immunoblotting with caspase-9(Thr125) antibody. The same filter was reprobed with anti–caspase-9 demonstrating equal loading (n = 3), N.D., not determined. (C) Immunoblotting using cardiac protein for cleaved caspase-3. Protein level of active caspase-3 was reduced in the cAng-1 group (n = 3). (D–G) Cardiac function measured by M-mode echocardiography 4 wks after IR. cAng-1–treated mice showed small LV-ESD and LV-EDD but greater EF and FS (n = 4 per group). *P < 0.05. (H) Schematic model for cardioprotection by cAng-1 after myocardial IR injury.
Treatment of cAng-1 Reduced Infarct Size and Improved Cardiac Function under IR Injury
To determine the effects of cAng-1 on pathological cardiac remodeling after IR injury, we measured the fibrosis area by Masson’s trichrome staining. The infarct areas were markedly reduced in the IR/Ad-cAng-1 group (Supplementary Figure S6). We then evaluated cardiac functional parameters by echocardiography at 4 wks after IR (Figures 6D–G). The IR/Ad-β-gal group showed markedly increased LV-ESD and LV-EDD with the reduced EF and FS. Importantly, we observed significant improvements in cardiac functional parameters in the IR/Ad-cAng-1 group compared with the IR/Ad-β-gal group, that is, higher ejection fraction (31.4% ± 9.34% versus 48.05% ± 4.53%) and fractional shortening (17.3% ± 5.76% versus 27.9% ± 3.14%) and smaller LV-ESD (4.18 ± 0.46 versus 3.13 ± 0.18 mm) and LV-EDD (5.05 ± 0.22 versus 4.3 ± 0.99 mm) values than in the IR/Ad-β-gal group (P < 0.05), which suggests cAng-1 attenuates postinfarction left ventricular remodeling.
DISCUSSION
The major new findings of the present study are that the cardioprotective effect of cAng-1 is based on two distinct mechanisms. Although cAng-1 was initially identified as an endothelial-specific regulator, we demonstrated that its coordinated protective effects act not only on endothelial cells, but also on cardiomyocytes during myocardial IR injury (Figure 6H).
Dephosphorylation of VE-Cadherin by cAng-1 to Maintain Endothelial Integrity during IR Injury
Vascular endothelial leakage associated with IR injury promotes edema and subsequently aggravates cardiomyocyte death (2,3). Impaired endothelial integrity, found also in several pathological conditions such as inflammation, sepsis and diabetes, ultimately leads to organ dysfunction (27). Consequently, preventing vascular permeability induced by IR injury may minimize organ damage, including myocardial infarction.
VE-cadherin is the most prominent adhesion molecule in endothelial cells and is directly involved in maintaining endothelial integrity (21,22). Tyrosine phosphorylation of VE-cadherin or its catenin binding partners is generally regarded to regulate the integrity of adherence junctions. Phosphorylation of Tyr658 and Tyr731 of VE-cadherin results in the uncoupling of p120- or β-catenin, respectively, from the cytoplasmic tail of VE-cadherin, leading to weakened cell adhesion and thereby increasing endothelial permeability (23,24). In this study, cAng-1 significantly reduced tyrosine phosphorylation of VE-cadherin in endothelial cells through activating protein tyrosine phosphatases.
Endothelial permeability is also regulated by internalization of VE-cadherin. For example, vascular endothelial growth factor (VEGF)-induced phosphorylation of VE-cadherin at Ser665, not at tyrosine residues, promotes clathrin-dependent internalization of VE-cadherin (28). In our experiments, cAng-1 also inhibited the serine-phosphorylation of VE-cadherin during hypoxic/reoxygenation events (data not shown).
Regulatory Mechanism of cAng-1 to Dephosphorylate VE-Cadherin
Tyrosine phosphorylation of VE- cadherin is regulated by competing actions of kinases and phosphatases (24). SHP2, an SH2 domain–containing protein-tyrosine phosphatase, was reported to be associated with the VE- cadherin complex, and its reduction was observed in vascular leakage (29). PTPμ is exclusively expressed in the endothelium and directly bound to VE-cadherin. Overexpression of PTPμ decreased tyrosine phosphorylation of VE-cadherin and enhanced endothelium integrity (30). PTPβ/VE-PTP is another endothelium-specific phosphatase suggested to inhibit VE-cadherin-tyrosine phosphorylation (31).
In our study, under hypoxic/reoxygenation conditions, the binding between VE-cadherin and these phosphatases decreased, resulting in increased tyrosine phosphorylation of VE-cadherin, especially at the Tyr658 residue (see Figure 2). We hypothesized that PTPβ and PTPμ binding to VE- cadherin might be regulated by cAng-1 because both can bind directly to VE-cadherin (30,31). Unexpectedly, however, this was not the case for PTPβ (see Figure 2). The binding activity of SHP2 or PTPμ to VE-cadherin was indeed restored by cAng-1 under H/Reoxy conditions (see Figure 2). In addition, blocking of SHP2 or PTPμ inhibited the effect of cAng-1 on dephosphorylation of VE-cadherin (Figures 2C, D), implicating that cAng-1 regulates tyrosine phosphorylation of VE-cadherin through SHP2 and/or PTPμ. In one previous report, SHP2 was found to bind to β-catenin in a VE-cadherin complex, a process that could be influenced by thrombin (29). Therefore, further study will be required to elucidate how cAng-1 regulates the association of SHP2 with VE-cadherin leading to tyrosine dephosphorylation.
Direct Effect of cAng-1 on Cardiomyocyte Survival: Interaction between cAng-1 and β1-Integrin
It has been suggested that Ang-1 exerts its activity in nonendothelial cells through integrins. In a previous study, treatment with integrin-β1– and integrin-β3–blocking antibodies inhibited cell adhesion to Ang-1-coated surfaces, leading the authors to suggest that integrin-β1 and integrin-β3 are essential for Ang-1-mediated effects (12,13). However, this line of experiments was conducted under normoxic conditions and did not investigate the mechanism by which Ang-1 regulates cardiomyocyte protection through integrins under injury conditions. We showed that cAng-1 protects cardiomyocytes during IR injury by inhibiting apoptosis through integrin-β1– dependent caspase-3 reduction (Figures 4 and 5). Caspase inhibition prevents myocardial contractile protein degradation, reduces apoptosis and improves myocardial function (32).
After confirming cAng-1 and integrin interaction, we investigated possible signal transduction routes that are located downstream of integrin-β1 and that regulate cardiomyocyte viability. In a couple of previous reports, Ang-1 increases ILK, a key regulator of integrin signaling, and consequently reduces cardiac hypertrophy (33). Ang-1 inhibits cell apoptosis through PI3K activation (14,34). Akt, the downstream effector of PI3K, phosphorylates GSK-3β at Ser9 and prevents car-diomyocyte death (35). On the basis of these reports, we expected the PI3K/ILK/Akt/GSK-3β axis would be the active downstream pathway after Ang-1 stimulation. However, cAng-1 did not affect the expression or activity of ILK, Akt and GSK-3β in hypoxic/reoxygenated cardiomyocytes. In our investigation, the ERK pathway was associated with protection of cells and inhibition of caspase-9 activation (see Figure 5).
Caspase-9 plays a central role in the initiation of apoptosis by the mitochondrial pathway. Cardiomyocytes have abundant mitochondria, and the mitochondria-mediated intrinsic apoptosis pathway plays a crucial role in IR injury (4,36). The release of cytochrome C from mitochondria into cytosol leads to the activation of caspase-9. Once activated, caspase-9 cleaves and activates caspase-3 and downstream caspases (36). Recent reports indicate caspases are also substrates for kinases (26,37). To uncover how kinases such as ERK regulate the caspase-9/caspase-3 cascade in response to cAng-1, we evaluated the phosphorylation status of caspase 9. The ERK-MAPK pathway inhibits caspase-9 activity by direct phosphorylation at residue Thr125. Phosphorylation at Thr125 blocks caspase-9 processing and subsequent caspase-3 cleavage (26). In contrast, Akt signaling phosphorylates caspase-9 at Ser196 (26). We observed that cAng-1 remarkably increased phospho-caspase-9(Thr125) (Figure 6), suggesting that this novel action mechanism of cAng-1 sequentially inactivated caspase-9 and -3 and improved cardiac performance during IR injury.
Merits of Ang-1 as a Therapeutic Agent for Patients with Myocardial Infarction
Several angiogenic growth factor– based therapies have been attempted to treat ischemic cardiovascular disease. To date, VEGF gene therapy has proven promising to promote angiogenesis but has shown severe drawbacks, such as generation of immature and leaky vessels or angioma (38,39). In contrast to VEGF, Ang-1 therapy might have potential benefits for IR treatment because Ang-1 alone is insufficient to promote angiogenesis, unless combined with VEGF (40). In a cornea micropocket assay, Ang-1 alone did not promote neovascularization, although pellets containing only VEGF induced corneal angiogenesis. The combination of VEGF with Ang-1 (VEGF + Ang-1) produced thick vessels without vessel sprouting (40). This result suggested that Ang-1 does not supply a direct angiogenic signal but rather may potentiate the effects of other angiogenic factors and promote vascular network maturation. It has been reported that VEGF is elevated in hypoxia/reoxygenated rat myocardium (41) and in patients with acute coronary syndromes (42). Thus, when we administer cAng-1, it may synergistically act with VEGF to enhance neovascularization in ischemic myocardium, especially to produce nonleaky and mature vessels. Sufficient blood flow through increased formation of mature blood vessels might minimize cardiomyocyte loss and thereby stimulate functional recovery of the heart. This finding indicates the potential benefits of Ang-1 therapy for IR: improvement in endothelial integrity and direct protection of cardiomyocytes (Figure 6H).
Reperfusion injury damages cardiomyocytes and the endothelium, destroys endothelial integrity of the microvasculature and further aggravates myocardial damage. Here, we found that cAng-1 maintains the integrity of the endothelial lining by dephosphorylating VE- cadherin via an interaction with SHP2 or PTPμ phosphatase. Additionally, cAng-1 exerts a direct antiapoptotic effect on cardiomyocytes exposed to H/Reoxy stress through integrin-β1. Our findings suggest that recombinant Ang-1 (such as cAng-1) can protect the infarcted heart from endothelial leakage and myocardial damage in patients undergoing reperfusion therapy, if administered before reperfusion.
Supplemental Data
ACKNOWLEDGMENTS
This study was supported by a grant for the Innovative Research Institute for Cell Therapy (A062260) and a National Research Foundation grant funded by the Korea government (Ministry of Education, Science and Technology [MEST]) (2010-0020257). H-S Kim is a professor of Molecular Medicine and Biopharmaceuticals Sciences, Seoul National University, and is sponsored by the World Class University program from the Ministry of Education and Science, Korea.
S-W Lee performed project planning, experimental design and data interpretation and prepared the manuscript. J-Y Won, H-Y Lee, J-Y Lee and S-W Youn performed cell culture and experiments and analyzed data. H-J Lee, C-H Cho, H-J Cho, S Oh and I-H Chae provided the mouse myocardial ischemia (MI) model and data analysis. H-S Kim provided project planning, data interpretation and funding and prepared the manuscript.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Bijnens B, Sutherland GR. Myocardial oedema: a forgotten entity essential to the understanding of regional function after ischaemia or reperfusion injury. Heart. 2008;94:1117–9. doi: 10.1136/hrt.2007.135392. [DOI] [PubMed] [Google Scholar]
- 2.Kang HJ, Kim HS. G-CSF- and erythropoietin-based cell therapy is a promising strategy for angiomyogenesis in myocardial infarction. Expert Rev Cardiovasc Ther. 2008;6:703–13. doi: 10.1586/14779072.6.5.703. [DOI] [PubMed] [Google Scholar]
- 3.Lefer AM, Tsao PS, Lefer DJ, Ma XL. Role of endothelial dysfunction in the pathogenesis of reperfusion injury after myocardial ischemia. FASEB J. 1991;5:2029–34. doi: 10.1096/fasebj.5.7.2010056. [DOI] [PubMed] [Google Scholar]
- 4.Fliss H, Gattinger D. Apoptosis in ischemic and reperfused rat myocardium. Circ Res. 1996;79:949–56. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- 5.Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection: the “dark side” of reperfusion. Circulation. 2009;120:2105–112. doi: 10.1161/CIRCULATIONAHA.108.814640. [DOI] [PubMed] [Google Scholar]
- 6.Lee SW, Kim WJ, Jun HO, Choi YK, Kim KW. Angiopoietin-1 reduces vascular endothelial growth factor-induced brain endothelial permeability via upregulation of ZO-2. Int J Mol Med. 2009;23:279–84. [PubMed] [Google Scholar]
- 7.Suri C, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–80. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 8.Lee SW, et al. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat Med. 2003;9:900–6. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- 9.Yancopoulos GD, et al. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–8. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 10.Abdulmalek K, et al. Differential expression of Tie-2 receptors and angiopoietins in response to in vivo hypoxia in rats. Am J Physiol Lung Cell Mol Physiol. 2001;281:L582–90. doi: 10.1152/ajplung.2001.281.3.L582. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K, et al. Adenoviral-delivered angiopoietin-1 reduces the infarction and attenuates the progression of cardiac dysfunction in the rat model of acute myocardial infarction. Mol her. 2003;8:584–92. doi: 10.1016/s1525-0016(03)00230-2. [DOI] [PubMed] [Google Scholar]
- 12.Carlson TR, Feng Y, Maisonpierre PC, Mrksich M, Morla AO. Direct cell adhesion to the angiopoietins mediated by integrins. J Biol Chem. 2001;276:26516–25. doi: 10.1074/jbc.M100282200. [DOI] [PubMed] [Google Scholar]
- 13.Dallabrida SM, Ismail N, Oberle JR, Himes BE, Rupnick MA. Angiopoietin-1 promotes cardiac and skeletal myocyte survival through integrins. Circ. Res. 2005;96:e8–24. doi: 10.1161/01.RES.0000158285.57191.60. [DOI] [PubMed] [Google Scholar]
- 14.Cho CH, et al. Long-term and sustained COMP-Ang1 induces long-lasting vascular enlargement and enhanced blood flow. Circ Res. 2005;97:86–94. doi: 10.1161/01.RES.0000174093.64855.a6. [DOI] [PubMed] [Google Scholar]
- 15.Barone GW, Farley PC, Conerly JM, Flanagan TL, Kron IL. Morphological and functional techniques for assessing endothelial integrity: the use of Evans blue dye, silver stains, and endothelial derived relaxing factor. J Card Surg. 1989;4:140–8. doi: 10.1111/j.1540-8191.1989.tb00270.x. [DOI] [PubMed] [Google Scholar]
- 16.Corada M, et al. Vascular endothelial- cadherin is an important determinant of microvascular integrity in vivo. Proc Natl Acad Sci U S A. 1999;96:9815–20. doi: 10.1073/pnas.96.17.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williamson CL, Dabkowski ER, Dillmann WH, Hollander JM. Mitochondria protection from hypoxia/reoxygenation injury with mitochondria heat shock protein 70 overexpression. Am J Physiol Heart Circ Physiol. 2008;294:H249–56. doi: 10.1152/ajpheart.00775.2007. [DOI] [PubMed] [Google Scholar]
- 18.Mestril R, Chi SH, Sayen MR, O’Reilly K, Dillmann WH. Expression of inducible stress protein 70 in rat heart myogenic cells confers protection against simulated ischemia-induced injury. J Clin Invest. 1994;93:759–67. doi: 10.1172/JCI117030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee MJ, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–12. doi: 10.1016/s0092-8674(00)81661-x. [DOI] [PubMed] [Google Scholar]
- 20.Youn SW, et al. COMP-Ang1 stimulates HIF-1α-mediated SDF-1 overexpression and recovers ischemic injury through BM-derived progenitor cell recruitment. Blood. 2011;117:4376–86. doi: 10.1182/blood-2010-07-295964. [DOI] [PubMed] [Google Scholar]
- 21.Navaratna D, McGuire PG, Menicucci G, Das A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes. 2007;56:2380–7. doi: 10.2337/db06-1694. [DOI] [PubMed] [Google Scholar]
- 22.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009;19:8–15. doi: 10.1016/j.tcb.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–12. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 24.Young BA, et al. Protein tyrosine phosphatase activity regulates endothelial cell-cell interactions, the paracellular pathway, and capillary tube stability. Am J Physiol Lung Cell Mol Physiol. 2003;285:L63–75. doi: 10.1152/ajplung.00423.2002. [DOI] [PubMed] [Google Scholar]
- 25.Weis S, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–94. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allan LA, et al. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003;5:647–54. doi: 10.1038/ncb1005. [DOI] [PubMed] [Google Scholar]
- 27.Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–70. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- 28.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–34. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 29.Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem. 2000;275:5983–6. doi: 10.1074/jbc.275.8.5983. [DOI] [PubMed] [Google Scholar]
- 30.Sui XF, et al. Receptor protein tyrosine phosphatase micro regulates the paracellular pathway in human lung microvascular endothelia. Am J Pathol. 2005;166:1247–58. doi: 10.1016/s0002-9440(10)62343-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nawroth R, et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. 2002;21:4885–95. doi: 10.1093/emboj/cdf497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–81. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
- 33.Hannigan GE, Coles JG, Dedhar S. Integrin-linked kinase at the heart of cardiac contractility, repair, and disease. Circ Res. 2007;100:1408–14. doi: 10.1161/01.RES.0000265233.40455.62. [DOI] [PubMed] [Google Scholar]
- 34.Lee SW, et al. Angiopoietin-1 protects endothelial cells from hypoxia-induced apoptosis via inhibition of PTEN. Korean Circ J. 2009;39:57–65. [Google Scholar]
- 35.Song JQ, Teng X, Cai Y, Tang CS, Qi YF. Activation of Akt/GSK-3beta signaling pathway is involved in intermedin 1–53 protection against myocardial apoptosis induced by ischemia/reperfusion. Apoptosis. 2009;14:1061–9. doi: 10.1007/s10495-009-0382-2. [DOI] [PubMed] [Google Scholar]
- 36.Allan LA, Clarke PR. Apoptosis and autophagy: regulation of caspase-9 by phosphorylation. FEBS J. 2009;276:6063–73. doi: 10.1111/j.1742-4658.2009.07330.x. [DOI] [PubMed] [Google Scholar]
- 37.Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138:838–54. doi: 10.1016/j.cell.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Losordo DW, et al. Gene therapy for myocardial angiogenesis: initial clinical results with direct myocardial injection of phVEGF165 as sole therapy for myocardial ischemia. Circulation. 1998;98:2800–4. doi: 10.1161/01.cir.98.25.2800. [DOI] [PubMed] [Google Scholar]
- 39.Schwarz ER, et al. Evaluation of the effects of intramyocardial injection of DNA expressing vascular endothelial growth factor (VEGF) in a myocardial infarction model in the rat-angiogenesis and angioma formation. J Am Coll Cardiol. 2000;35:1323–30. doi: 10.1016/s0735-1097(00)00522-2. [DOI] [PubMed] [Google Scholar]
- 40.Asahara T, et al. Tie2 receptor ligands, angiopoietin-1 and angiopoietin-2, modulate VEGF-induced postnatal neovascularization. Circ Res. 1998;83:233–40. doi: 10.1161/01.res.83.3.233. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki H, Ray PS, Zhu L, Galang N, Maulik N. Oxidative stress due to hypoxia/reoxygenation induces angiogenic factor VEGF in adult rat myocardium: possible role of NFkappaB. Toxicology. 2000;155:27–35. doi: 10.1016/s0300-483x(00)00274-2. [DOI] [PubMed] [Google Scholar]
- 42.Lee KW, Lip GY, Blann AD. Plasma angiopoietin-1, angiopoietin-2, angiopoietin receptor tie-2, and vascular endothelial growth factor levels in acute coronary syndromes. Circulation. 2004;110:2355–60. doi: 10.1161/01.CIR.0000138112.90641.7F. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.