

Abstract
Cordycepin has been shown to interfere with a myriad of molecular processes from RNA elongation to kinase activity, and prevents numerous inflammatory processes in animal models. Here we show in a mouse model of LPS-induced acute lung injury that cordycepin prevents airway neutrophilia via a robust blockade of expression of several inflammatory genes, including the adhesion molecule ICAM-1 and VCAM-1, the cytokine/chemokine MCP-1, MIP-1α, MIP-2 and KC, and the chemokine receptor CXCR2. Such a blockade appears to be related to a severe reduction in TNF-α expression. Interestingly, in an in vitro system of A549 epithelial cell inflammation, cordycepin effectively blocked LPS-induced, but not TNF-α-induced, VCAM-1 expression. Such effects correlated with a marked reduction in p65-NF-κB activation as assessed by its phosphorylation at serine-536 but without an apparent effect on its nuclear translocation. The effects of cordycepin on the expression of VCAM-1 and ICAM-1, and of NF-κB activation and nuclear translocation upon TNF-α stimulation resembled the effects achieved upon poly(ADP-ribose) polymerase (PARP) inhibition, suggesting that cordycepin may function as a PARP inhibitor. Indeed, cordycepin blocked H2O2-induced PARP activation in A549 cells. In a cell-free system, cordycepin inhibited PARP-1 activity at nanomolar concentrations. Similar to PARP inhibitors, cordycepin significantly induced killing of breast cancer susceptibility gene (BRCA1)-deficient MCF-7 cells, supporting its therapeutic use for the treatment of BRCA-deficient breast cancers. With added antiinflammatory characteristics, therapies that include cordycepin may prevent potential inflammation triggered by traditional chemotherapeutic drugs. Cordycepin, to the best of our knowledge, represents the first natural product possessing PARP inhibitory traits.
INTRODUCTION
Airway inflammation is a complex process that can be mediated by a variety of stimuli from bacterial infection to allergen exposure. Interference with the expression of adhesion and chemotactic molecules, such as intracellular adhesion molecule (ICAM)-1 and vascular adhesion molecule (VCAM)-1, in response to inflammatory factors, such as lipopolysaccharide (LPS) or tumor necrosis factor-α (TNF-α), prevents inflammatory cell adhesion and transendothelial migration of leukocytes. A number of drugs have been suggested to block inflammatory processes and they may exhibit therapeutic potential by impairing the establishment or progression of inflammatory diseases. The inflammatory response during acute lung injury (ALI) that is triggered, for instance, by LPS, is characterized by the massive recruitment of neutrophils (1) and the production of numerous cytokines and chemokines, including monocyte chemotactic protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, interleukin (IL)-8, and MIP-2, as well as the expression of adhesion molecules and other inflammatory factors (2). The expression of these genes is believed to be associated with the production of TNF-α, as interference with the function or expression of TNF-α compromises the expression of the aforementioned genes (3,4) during LPS-induced ALI.
Our laboratory has investigated the role of poly(ADP-ribose) polymerase-1 (PARP-1) in tissue injury and its implications in several pathological conditions, including asthma, ALI, and atherosclerosis (5–12). PARP-1 is emerging as a viable therapeutic target for the treatment of inflammatory disease (13–15). In addition to its effects on cell and tissue homeostasis through the direct utilization of nicotinamide adenine dinucleotide (NAD)+, PARP-1 is increasingly believed to contribute to inflammation by regulating the expression of a variety of inflammatory genes, including adhesion molecules ICAM-1 and VCAM-1, MCP-1, MIP-1α, and a number of other factors (reviewed in 16–18). This function is linked to the ability of PARP-1 to regulate signal transduction events that result in the activation of the nuclear factor (NF)-κB, extracellular signal-related kinase (ERK), and activator protein 1 (AP1) (19–22). NF-κB is a pleiotropic transcription factor that plays a critical role in regulating the expression of numerous inflammatory genes, including ICAM-1, VCAM-1, TNF-α, MCP-1 and MIP-1α (23). We and others have reported that PARP-1 expression is required for the efficient translocation of NF-κB to the nucleus in response to LPS (10,24,25). However, we recently reported that this requirement does not apply when the stimulus is TNF-α (26). Interestingly, whereas NF-κB nuclear translocation in TNF-α-treated smooth muscle cells is sufficient for the expression of the adhesion molecule VCAM-1, ICAM-1 expression showed a critical requirement for PARP-1.
PARP-1 also is emerging as a very viable target in therapies aimed at either blocking or reducing cancer burden, including that of breast cancer (27). It is noteworthy that breast cancer is the most common form of malignancy among women and a leading cause of cancer-related deaths worldwide. Deficiency in the breast cancer susceptibility gene BRCA1 contributes to familial breast tumorigenesis that has long been known to have a very poor prognosis (28). BRCA-deficient cancers can be selectively targeted by PARP inhibitors, as inhibition of the enzyme promotes cell death and sensitization to DNA- damaging agents (27,29).
Cordycepin, a natural compound and adenosine analogue derived from Cordyceps militaris, has been shown to harbor a great potential for therapeutic use against a number of human diseases including inflammatory disease and cancer (30). The purified drug has been shown to possess numerous biological activities such as induction of cell death, blockade of cell growth, inhibition of expression of a variety of inflammatory genes and reduction of cell migration. In a variety of animal models, cordycepin has been shown to block inflammation and reduce tumor formation (30). Accumulating evidence suggests that cordycepin may interfere with a number of molecular processes, resulting in either the inhibition or modulation of a number of genes pertinent to inflammation or carcinogenesis, including inhibition of substrate phosphorylation by key kinases (30) or direct interference with protein synthesis (31). Although a number of studies have addressed the mechanism by which cordycepin interferes with such molecular processes to achieve its therapeutic potential, its mode of action appears multifaceted and remains poorly understood.
In the present study, we investigated whether cordycepin blocks ALI- associated inflammatory responses and expression of related genes by testing whether such effects were related to PARP inhibition. We also examined whether the potential PARP inhibition trait promotes the killing of BRCA1- deficient breast cancer cells.
MATERIALS AND METHODS
Animals
Mice (C57BL/6) were bred in a specific pathogen-free facility at the Louisiana State University Health Sciences Center (New Orleans, LA, USA) and allowed unlimited access to sterilized chow and water. Maintenance and experimental protocols were approved by the institution’s Animal Care and Use Committee. Five-wk-old mice (n ≥ 5) received a single injection intraperitoneally (i.p.) of 2 mg/kg cordycepin (Sigma-Aldrich, Saint Louis, MO, USA) suspended in ethanol and diluted 1/10 in saline, or vehicle 1 h before intratracheal administration of 50 μg/mouse LPS (Axxora, San Diego, CA, USA) as described recently (9). The dose of 2 mg/kg of cordycepin was selected based on a study by Rottenberg et al. reporting that this dose efficiently protects mice against trypanosomiasis without causing any detectable toxicity including wasting and diarrhea (32). Mice were euthanized 6 or 24 h after LPS treatment for tissue collection or bronchoalveolar lavage (BAL), respectively, essentially as described (9).
Cell Culture, Immunoblot Analysis and Immunofluorescence
The lung epithelial (A549) and the breast cancer (MCF-7) cell lines and PARP-1−/− lung smooth muscle cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin and streptomycin. The isolation of PARP-1−/− lung smooth muscle cells was conducted using an enzymatic digestion protocol and is described in details in the Supplementary Methods. Prior to treatment, cells at 50%–80% confluence were starved overnight by incubation in DMEM with 0.5% FBS. Cells were treated with 1 μg/mL LPS (Axxora) or 10 ng/mL TNF-α (Roche Diagnostics Corp, Indianapolis, IN, USA) for the indicated times in the absence or presence of increasing concentrations of cordycepin (Sigma-Aldrich). Cells were collected, washed and lysed using RIPA-lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein contents were assessed using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA). Immunoblot analyses were conducted essentially as described (33). Briefly, samples containing 20 μg of protein were fractionated by SDS-PAGE on 4%–20% gradient gels, and the separated proteins were transferred to nitrocellulose membranes. Membranes were stained with Ponceau S to confirm equal loading and transfer of proteins among lanes. After blocking with 5% nonfat milk in PBS + 0.05% Tween 20, the membranes were probed with antibodies to VCAM-1, ICAM-1, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), p65 or p50 NF-κB, the phosphorylated form of p65 NF-κB at serine 536, actin (all purchased from Santa Cruz Biotechnology), or to poly(ADP-ribose) (Axxora). Immune complexes were detected with appropriate secondary antibodies and chemiluminescence reagents (Pierce, Rockford, IL, USA). In some experiments, cells were treated with 500 μmol/L H2O2 (Sigma-Aldrich) with or without cordycepin or the PARP inhibitor NU1025 (Santa Cruz Biotechnology) or TIQ-A (Sigma-Aldrich) before processing for immunofluorescence or immunoblot analysis. Immunofluorescence was conducted essentially as described (25). Briefly, cells were fixed for 20 min in PBS containing 4% formaldehyde, washed with PBS. After washing, cells were permeabilized for 5 min in PBS containing 0.05% Triton X-100. Cells were then incubated overnight at 4°C with 20 μg/mL of antibodies to p65 or p50 NF-κB or poly(ADP-ribose). After a series of washes, antibody-antigen complexes were detected with Alexa-Fluor 594 or 488-conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA) using a Leica DMRA2 fluorescence microscope (Leica, Buffalo Grove, IL, USA).
RT-PCR (Conventional and Real-Time)
For RT-PCR, total RNA was extracted from cells using the RNeasy Plus Micro Kit (QIAGEN, Valencia, CA, USA). One μg of total RNA was used as a template to make first-strand cDNA by random priming using reverse transcriptase III (Invitrogen). Oligonucleotide primers (Supplementary Table 1) to specifically amplify a fragment of murine VCAM-1, ICAM-1, MCP-1, MIP-1α, MIP-2, TNF-α, keratinocyte-derived chemokine (KC) or β-actin, or human VCAM-1 or β-actin were purchased from Integrated DNA Technologies (Coralville, IA, USA). The PCR conditions were 30 s at 95° C followed by 30 s at 60° C then 30 s at 72° C for 40 cycles. The comparative Ct method was used as a quantitation approach for cDNA prepared from the different experimental groups by comparing the Ct values of the samples of interest with a control (samples from non-treated mice). The Ct values of both the control and the samples of interest were normalized to the housekeeping gene β-actin. Primers for the β-actin gene were designed specifically to avoid amplifying intronless actin pseudogenes. The quality (single peak and single band) of amplicons generated to test for messages of the different inflammatory genes is displayed in Supplementary Figure S1. All samples were tested in triplicates.
siRNA Transfection, Cell Viability and LDH Release Assays
MCF-7 cells were transfected transiently with specific small interfering RNA (siRNAs) (sc29219) targeting BRCA1 (Santa Cruz Biotechnology) or the negative control siRNA NEG2 (SA Bioscience Inc., Frederick, MD, USA) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cell viability was assessed in 96-well plates (10,000 cells/well) using the (3-[4,5-Dimethylthiozol-2-yl]-2-5) diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich). Lactate dehydrogenase (LDH) assay was conducted as described previously (5). Briefly, cells cultured in 96-well plates (10,000 cells/well) were treated as described in the figures; media was collected from the different experimental groups and tested for LDH activity using the CytoTox-Fluor Cytotoxicity Assay kit (Promega, Madison WI, USA). All samples were tested in triplicates.
Poly(ADP-ribosyl)ation in vitro
Purified recombinant PARP-1 (Axxora) was incubated in a reaction mixture (total volume 25 μL) containing reaction buffer (100 mmol/L Tris-HCl [pH 7.4]), 1 mmol/L dithiothreitol [DTT], 10 mmol/L MgCl2), 10 mg protein extracts from PARP-1−/− cells, different doses of cordycepin or PARP inhibitors,1 μg sonicated salmon sperm DNA and 2 mmol/L NAD+ (Sigma-Aldrich) for 30 min at 37°C. The reaction was terminated by the addition of an equal volume of 2 × sodium dodecyl sulfate (SDS) sample buffer (Bio-Rad) and heating at 95°C for 5 min. Samples then were subjected to immunoblot analysis with antibodies to poly(ADP-ribose) (PAR) (Axxora).
STATISTICAL ANALYSIS
Data are presented as means ± S.E.M. of more than three separate experiments performed. Comparisons between multiple groups were performed with analysis of variance (ANOVA) with Bonferroni’s test. Statistical significance was considered significant when P < 0.05.
All supplementary materials are available online at www.molmed.org.
RESULTS
Cordycepin Prevents LPS-Induced Airway Neutrophilia in Mice
Figure 1A shows that intratracheal administration of LPS induced ALI with a massive infiltration of neutrophils into the lungs of C57BL/6 mice. Such infiltration of inflammatory cells was almost completely blocked in animals receiving a single i.p. injection of cordycepin (2 mg/kg) 1 h prior to LPS administration. These results, although novel in their relevance to airway neutrophilia, confirm the antiinflammatory effects of cordycepin. The effects of cordycepin on neutrophil recruitment to the lungs of LPS-exposed mice correlate with a general reduction in the expression of inflammatory factors, including the adhesion molecules ICAM-1 and VCAM-1, the cytokines/chemokines MCP-1, MIP-1α, MIP-2 and KC, and the chemokine receptor CXCR2 (Figure 1B) as assessed by real-time PCR analysis of lung tissues collected 6 h after treatment. LPS is known to mediate its effects through TNF-α (3,4); Figure 1C shows that cordycepin may modulate LPS-induced effects in our animal model by reducing the expression of TNF-α. This finding is consistent with a previous report showing that cordycepin reduces TNF-α expression in response to LPS in vitro (34).
Figure 1.
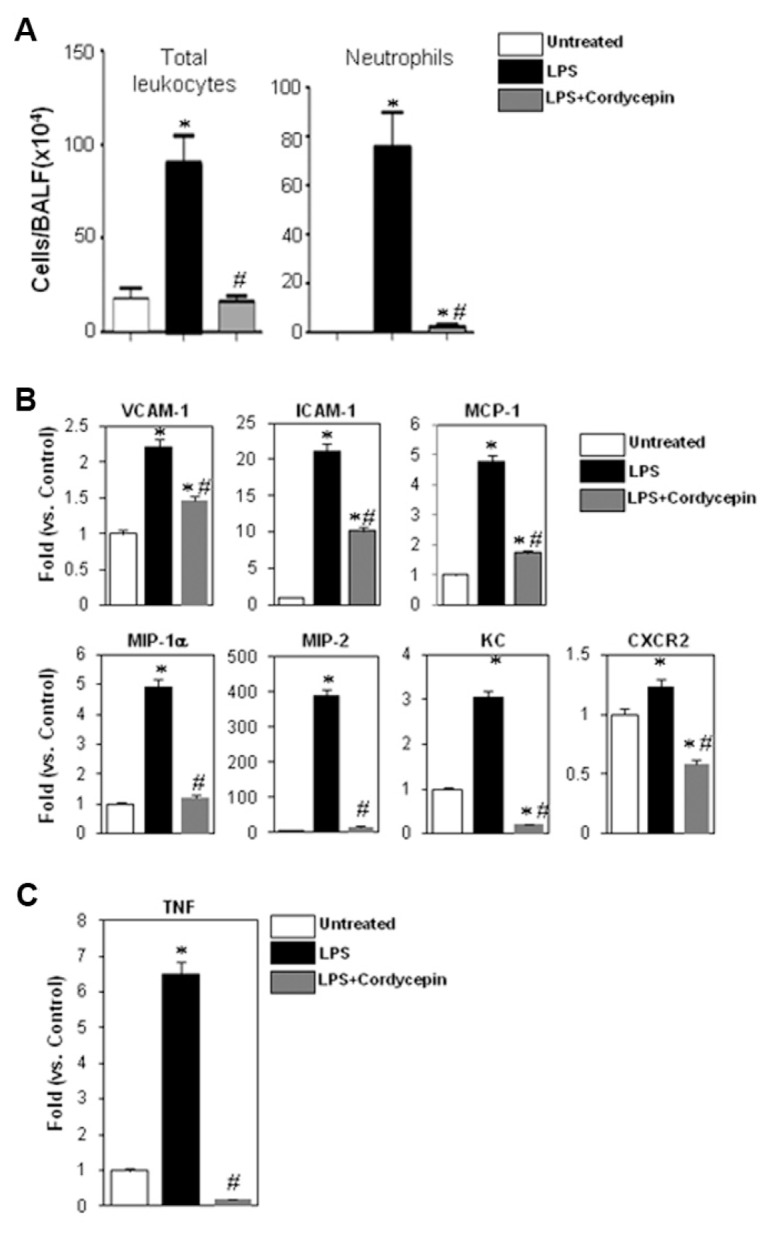
Effects of cordycepin on airway inflammatory cell recruitment in response to intratracheal administration of LPS in mice. C57BL/6 mice (n ≥ 5) were subjected to a single i.p. injection of 2 mg/kg cordycepin (suspended in ethanol and diluted 1/10 in saline) or vehicle 1 h before the intratracheal administration of LPS (50 μg/mouse) suspended in 50 μL saline. Either 6 or 24 h after treatment, mice were euthanized and subjected to either lung collection for RNA analysis or BAL, respectively. (A) BAL fluids were collected and centrifuged; cells were then differentially stained and visualized by light microscopy followed by a count of total cells and neutrophils. Data are given as the means ± SEM of values obtained from at least five mice per group. *Different from untreated control mice; P < 0.01. #Different from LPS-treated mice; P < 0.01. (B) Whole lungs were collected and subjected to RNA extraction followed by cDNA generation. Prepared cDNA was subjected to real-time PCR with primers for VCAM-1, ICAM-1, MCP-1, MIP-1α, MIP-2, KC or CXCR2; β-actin was amplified by its specific primers and used as an internal control and on which the attained values were normalized. (C) The cDNA from the different experimental groups described above was assessed for TNF-α by real-time PCR. *Different from untreated control mice; P < 0.01; #Different from LPS-treated mice; P < 0.01.
Cordycepin Effectively Blocks LPS-Induced, but not TNF-α-Induced, Expression of VCAM-1 in the Human Epithelial Cell Line A549
The results attained using animal ALI models are insufficient to provide a clear mechanistic explanation of how cordycepin blocks the expression of inflammatory genes, especially when TNF-α, for instance, acts as the primary mediator of responses to LPS exposure (3,4). Accordingly, we used an in vitro cell culture system and focused primarily on the effects of the drug on VCAM-1 expression. Figure 2A shows that cordycepin exerted a dose-dependent blockade of VCAM-1 expression in cells directly exposed to LPS. Conversely, the drug was not very effective in reducing the expression of VCAM-1 in A549 cells after TNF-α treatment; the modest reduction was observed only with 20 μmol/L cordycepin, the highest dose tested (Figure 2A). Interestingly however, cordycepin greatly reduced ICAM-1 expression in response to either LPS or TNF-α (see Figure 2A), clearly suggesting a differential effect of the drug on ICAM-1 and VCAM-1 in response to TNF-α but not LPS. Such differential effects were corroborated by examining the effects of the drug on VCAM-1 and ICAM-1 protein expression after a 12-h TNF-α treatment by immunoblot analysis (Figure 2B). The weak blockade of cordycepin on TNF-α-induced VCAM-1 expression occurred despite a clear reduction in p65 NF-κB activation as assessed by its phosphorylation status on serine 536 (Figure 2C). The overall levels of the two NF-κB subunits, p65 and p50, were not altered by cordycepin treatment (see Figure 2C; bottom panels). It is noteworthy that the effects of cordycepin on p65 NF-κB activation were not the result of a blockade of p65 or p50 nuclear translocation as assessed by immunofluorescence (Figure 2D).
Figure 2.

Cordycepin efficiently reduces VCAM-1 expression in response to LPS, but not TNF-α, in A549 cells, and such effects are associated with a reduction in p65 NF-κB phosphorylation but not its nuclear translocation. (A) A549 cells were treated for 6 h with 1 μg/mL LPS or 10 ng/mL TNF-α in the absence or presence of the indicated concentrations of cordycepin. Total RNA was subjected to RT-PCR analysis with primers to VCAM-1, ICAM-1 or β-actin. (B) A549 cells were treated with TNF-α for 12 h in the absence or presence of the indicated concentrations of cordycepin. Protein extracts were prepared and subjected to immunoblot analysis with antibodies to VCAM-1, ICAM-1 or GAPDH. (C) A549 cells were treated with TNF-α for 5 min in the absence or presence of the indicated concentrations of cordycepin. Protein extracts were prepared and subjected to immunoblot analysis with antibodies to p65 NF-κB, its serine-536 phosphorylated form, p50 NF-κB or GAPDH. (D) A549 cells were treated with TNF-α for 30 min in the absence or presence of the indicated concentrations of cordycepin. Cells were then fixed and subjected to immunofluorescence with antibodies to p65 or p50 NF-κB. Note that all cells were pre-treated with the indicated drug 30 min before treatment with either LPS or TNF-α in the continued presence of the drug.
Interestingly, the traits of cordycepin greatly resembled those observed upon PARP-1 inhibition; we recently reported that PARP-1 inhibition by gene knockout reduces ICAM-1 but not VCAM-1 in response to TNF-α despite the clear nuclear translocation of p65 NF-κB (26). Supplementary Figure 2 shows that pharmacological inhibition of PARP by NU1025 or TIQ-A also failed to reduce TNF-α-induced nuclear translocation of p65 NF-κB. This observation led us to examine whether cordycepin may reduce LPS-induced airway inflammation and expression of NF-κB-dependent genes, in part, by blocking the activity of PARP-1.
Cordycepin blocks H2O2-induced PARP activation in A549 cells and directly inhibits PARP-1 activity in vitro at very low concentrations. To test our hypothesis, we utilized H2O2 because of its ability to induce a robust activation of PARP as well as its relevance to inflammatory processes. The dose (500 μmol/L) of H2O2 used in this study induces an activation of the enzyme that is easily detectable in A549 cells as reported previously (5). Immunoblot analyses of protein extracts from H2O2-treated cells with antibodies to the poly(ADP-ribose) moieties of modified proteins revealed a prominent activation of PARP as evidenced by the presence of several bands (Figure 3A). Treatment with cordycepin severely reduced the generation of these bands, indicative of a modulation of PARP activity. These results were corroborated using immunofluorescence analysis with antibodies to poly(ADP-ribose) (Figure 3B). Treatment with H2O2 for 10 min induced a high level of PARP activity in A549 cells as most nuclei displayed strong staining; cordycepin prevented this activation. These results suggest that cordycepin may possess a PARP inhibitory trait. The inhibitory effect of cordycepin upon PARP-1 activity appeared to be partially reversible as removal of the drug allowed only a partial activation of PARP-1 by a second treatment of the cells with H2O2 (Figure 3C). It is noteworthy that these findings do not prove that cordycepin inhibits PARP-1 directly, as cordycepin is able to prevent the production of reactive oxygen species and, hence, the induction of DNA damage that is necessary for the activation PARP-1. Accordingly, we used a cell-free system with recombinant PARP-1 and sonicated DNA as a source for DNA damage as well as protein extracts from PARP-1−/− cells as substrates for poly(ADP-ribosyl)ation. Figure 3D shows that cordycepin inhibited PARP-1 at doses as low as 0.1 μmol/L, demonstrating its ability to function as a potent PARP-1 inhibitor. Taken together, these results strongly suggest that cordycepin may block LPS-induced airway inflammation and expression of NF-κB-dependent genes, in part, by blocking PARP-1 activity.
Figure 3.

Cordycepin reduces H2O2-induced PARP activation and inhibits in vitro PARP-1 activity with great potency. (A) A549 cells were treated with H2O2 for 10 min in the presence or absence of 10 or 20 μmol/L cordycepin. A portion of the H2O2-treated cells also was treated with the known PARP inhibitors NU1025 (100 μmol/L) or TIQ-A (2 μmol/L). Protein extracts were prepared and subjected to immunoblot analysis with antibodies to poly(ADP-ribose) (PAR) or GAPDH. (B) A549 cells were treated with H2O2 for 10 min in the presence or absence of 10 μmol/L cordycepin. Cells were then fixed and subjected to immunofluorescence with antibodies to poly(ADP-ribose). Note that the cytosolic staining is non-specific as it appears in all cells. (C) A549 cells were treated the same as in (B) after which they were washed with and incubated in complete medium for 1 h. Cells were then treated once more with H2O2 for 10 min in the absence of any additional cordycepin. Cells were then fixed and subjected to immunofluorescence with antibodies to poly(ADP-ribose). (D) Recombinant human PARP-1 was incubated for 5 min in an in vitro poly(ADP-ribosyl)ation reaction in the absence or presence of different concentrations of cordycepin or in the presence of NU1025 or TIQ-A (both PARP inhibitors) with NAD+, activated (sonicated) DNA, and protein extracts from PARP-1−/− smooth muscle cells. The reactions were terminated with sample buffer and subjected to immunoblot analysis with antibodies to poly(ADP-ribose) (PAR) or actin.
Cordycepin Induces Killing of BRCA-Deficient Breast Cancer Cells
Very recently, PARP inhibitors garnered substantial clinical interest owing to their promising efficacies in reducing tumor burdens in patients with breast cancer (27,29). Breast cancers that exhibit BRCA deficiency benefit the most from these inhibitors given the accumulation of DNA double-strand breaks and inefficient repair, leading to the specific killing of these cells. Given that our results show that cordycepin may harbor a very efficacious PARP inhibitory capability, we surmised that the drug might promote the killing of breast cancer cells in a BRCA deficiency-dependent manner. To test this hypothesis, we took advantage of the well-established cell line MCF-7. It is noteworthy that MCF-7 cells express functional wild-type BRCA1 (35). Initially, we tested the overall toxicity of cordycepin compared to that of the PARP inhibitor TIQ-A in MCF-7 cells by assessing the levels of LDH released into the culture medium. Figures 4A and B show that cordycepin and TIQ-A exhibit similar patterns of toxicity, albeit at different times of exposure. While high doses of the drugs (≥ 30 μmol/L), as expected, caused noticeable cell toxicities, low doses (1–15 μmol/L) of the drugs caused negligible toxicities. It is noteworthy that cordycepin caused no toxicity in normal cells such as primary smooth muscle cells (Supplementary Figure S3). Interestingly, BRCA1 deficiency achieved by gene knockdown using siRNA (Figure 4C) enhanced the cell-killing effects of both cordycepin and TIQ-A significantly (Figures 4D, E) especially at low doses of the drugs (15 μmol/L and 5 μmol/L, respectively). These results strongly suggest that cordycepin may act as a PARP inhibitor and promote the killing of cells that are deficient in BRCA1. Taken together, these results report for the first time that a natural compound isolated from a fungus can be utilized as a PARP inhibitor with promise for use in clinical settings.
Figure 4.

Cordycepin enhances killing of BRCA1-deficient MCF-7 cells in a manner similar to that achieved by the PARP inhibitor TIQ-A. MCF-7 cells were treated with different doses of cordycepin (A) or TIQ-A (B); subsequently, culture media were collected and assessed for LDH activity. Cytotoxicity is expressed as percent of LDH values detected in media of control-untreated cells. *Difference from control MCF-7 cells. (C) MCF-7 cells were transiently transfected with siRNA targeting BRCA1 or control siRNA. Cells, cultured in 96-well plates, were then treated with different doses of cordycepin for 24 h (D) or TIQ-A for 48 h (E), after which cell killing was assessed by MTT assay. *Difference from similarly treated control siRNA-transfected MCF-7 cells.
DISCUSSION
After the promising use of PARP inhibitors as therapies for the sensitization of cancers that are deficient in BRCA, the search for even better inhibitors continues. PARP inhibitors have a bright future in the fight against not only cancer but also against various inflammatory diseases (27,29). Our laboratory has been actively pursuing the search for mechanisms by which PARP-1 regulates inflammatory processes using both cell culture systems and animal models of diseases. Indeed, we have reported that PARP inhibition prevents the manifestation of asthma-related traits in an animal model of the disease (5–7), reduces atherogenesis in high fat diet–induced atherosclerosis using the ApoE−/− mouse model (10), and promotes the regression of already established atherosclerotic plaques (36). We associate such effects with the involvement of PARP-1 in these molecular processes, leading to the expression of inflammatory genes, primarily those regulated by NF-κB (5,9,25,26) and signal transducer and activator of transcription (STAT)-6 (37). The results of the present study provide evidence that cordycepin, a natural compound, inhibits PARP-1 with a rather potent efficacy. These findings shed new light on the mechanism behind the ability of cordycepin to modulate inflammation, inhibit the expression of a number of inflammatory genes and enhance the killing of cancer cells. Clearly, the ability of cordycepin to inhibit PARP could have an important clinical impact as it could be tested in the treatment of triple-negative breast cancers and reduce the inflammation associated with a number of chemotherapeutic strategies.
Cordycepin has been shown to exhibit a number of properties that block or reduce inflammation, angiogenesis, tumorigenesis, dislipidemia, microbial growth, aging and neurotoxicity (30). A number of molecular processes appear to be affected by cordycepin. Unfortunately, a large number of these studies have utilized high doses of the drug that would be rather difficult to translate into the clinic. In this study, we attempted to focus primarily on low micromolar concentrations to avoid overwhelming intracellular molecular processes, given the nucleotide-like structure of the drug. Although our finding relevant to the protective effects of cordycepin against LPS-induced ALI is novel, it was not completely unexpected given that many studies have demonstrated protective effects of the drug in other inflammatory disease models (30). According to our results, it appears that the ability of cordycepin to inhibit TNF-α expression in vivo is a primary cause of the reduction of the other inflammatory genes, especially given that most of the tested genes could be induced by TNF-α. This suggestion is further supported by our finding that cordycepin differentially affects the expression of adhesion molecules ICAM-1 and VCAM-1. It is important to note that these two genes, though tightly regulated by NF-κB, may also be differentially affected by other factors as well.
The remarkable aspect of these findings is that the differential effect of cordycepin on the expression of ICAM-1 and VCAM-1 directed our attention to the possibility that the drug may be acting as a PARP inhibitor. Our previous studies delineated certain aspects of the mechanism by which PARP-1 regulates the expression of ICAM-1 and VCAM-1 via the regulation of NF-κB activation (26). We showed that while p65 NF-κB nuclear translocation in TNF-α-treated cells is sufficient for the expression of VCAM-1, expression of ICAM-1 showed a critical requirement for PARP-1. Similar effects were observed when cordycepin was used in our cell culture system. The ability of the drug in blocking poly(ADP-ribose) formation upon H2O2 treatment provided initial evidence for the potential action of cordycepin as a PARP inhibitor. The PARP inhibitory action of cordycepin was fully confirmed using a cell-free system with purified recombinant PARP-1. How cordycepin inhibits PARP-1 is unclear, but because of its ability to inhibit the enzyme at very low concentrations, noncompetitive inhibition is likely, since competitive inhibitors such 3-aminobenzamide can only achieve full inhibition of PARP-1 at millimolar concentrations. However, to gain a precise mode of action and determine the IC50 of the drug, additional and precise experimentation is required.
The ability of cordycepin to inhibit PARP-1 suggests a promising use in the killing of BRCA-deficient breast cancer cells. It is noteworthy that a number of PARP inhibitors have already passed through a number of clinical trials and represent a promising strategy in the treatment of not only breast but also ovarian cancers (27,29,38). Cordycepin, to the best of our knowledge, represents the first natural product possessing PARP inhibitory traits that could function both as a promising drug for the treatment of breast and ovarian cancers in humans and as an initial structure to use for future modifications that may render it even more potent in inhibiting PARP-1 and killing cancer cells. These possibilities become even more significant given the antiinflammatory characteristics of cordycepin: if coadministered with traditional chemotherapeutic drugs, it could act to counteract any associated inflammatory processes triggered by such traditional therapies.
Supplemental Data
ACKNOWLEDGMENTS
This work was supported, in part, by grant HL072889 from the NIH and grant RSG-116608 from the American Cancer Society as well as funds from the Louisiana Cancer Research Consortium (New Orleans, LA) to H Boulares.
Footnotes
Online address: http://www.molmed.org
DISCLOSURES
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.Gonzalez PK, et al. Role of oxidant stress in the adult respiratory distress syndrome: evaluation of a novel antioxidant strategy in a porcine model of endotoxin-induced acute lung injury. Shock. 1996;6(Suppl 1):S23–6. [PubMed] [Google Scholar]
- 2.Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–7. doi: 10.2741/2853. [DOI] [PubMed] [Google Scholar]
- 3.Smith S, et al. The locus of tumor necrosis factor-alpha action in lung inflammation. Am J Respir Cell Mol Biol. 1998;19:881–91. doi: 10.1165/ajrcmb.19.6.3146. [DOI] [PubMed] [Google Scholar]
- 4.Koay MA, et al. Modulation of endotoxin-induced NF-kappa B activation in lung and liver through TNF type 1 and IL-1 receptors. Am J Physiol Lung Cell Mol Physiol. 2002;283:L1247–54. doi: 10.1152/ajplung.00036.2002. [DOI] [PubMed] [Google Scholar]
- 5.Boulares AH, et al. Gene knockout or pharmacological inhibition of poly(ADP-ribose) polymerase-1 prevents lung inflammation in a murine model of asthma. Am J Respir Cell Mol Biol. 2003;28:322–9. doi: 10.1165/rcmb.2001-0015OC. [DOI] [PubMed] [Google Scholar]
- 6.Oumouna M, et al. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol. 2006;177:6489–96. doi: 10.4049/jimmunol.177.9.6489. [DOI] [PubMed] [Google Scholar]
- 7.Naura AS, et al. Post-allergen challenge inhibition of poly(ADP-ribose) polymerase harbors therapeutic potential for treatment of allergic airway inflammation. Clin Exp Allergy. 2008;38:839–46. doi: 10.1111/j.1365-2222.2008.02943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naura AS, et al. Reciprocal regulation of iNOS and PARP-1 during allergen-induced eosinophilia. Eur Respir J. 2009;33:252–62. doi: 10.1183/09031936.00089008. [DOI] [PubMed] [Google Scholar]
- 9.Zerfaoui M, et al. Effects of PARP-1 deficiency on airway inflammatory cell recruitment in response to LPS or TNF: differential effects on CXCR2 ligands and Duffy Antigen Receptor for chemokines. J Leukoc Biol. 2009;86:1385–92. doi: 10.1189/jlb.0309183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oumouna-Benachour K, et al. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation. 2007;115:2442–50. doi: 10.1161/CIRCULATIONAHA.106.668756. [DOI] [PubMed] [Google Scholar]
- 11.Hans CP, et al. Protective effects of PARP-1 knockout on dyslipidemia-induced autonomic and vascular dysfunction in ApoE mice: effects on eNOS and oxidative stress. PLoS ONE. 2009;4:e7430. doi: 10.1371/journal.pone.0007430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hans CP, Zerfaoui M, Naura AS, Catling A, Boulares AH. Differential effects of PARP inhibition on vascular cell survival and ACAT-1 expression favouring atherosclerotic plaque stability. Cardiovasc Res. 2008;78:429–39. doi: 10.1093/cvr/cvn018. [DOI] [PubMed] [Google Scholar]
- 13.Pacher P, Szabo C. Role of poly(ADP- ribose) polymerase 1 (PARP-1) in cardiovascular diseases: the therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev. 2007;25:235–60. doi: 10.1111/j.1527-3466.2007.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pacher P, Szabo C. Role of the peroxyni-trite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giansanti V, Dona F, Tillhon M, Scovassi AI. PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol. 2010;80:1869–77. doi: 10.1016/j.bcp.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 16.Pieper AA, Verma A, Zhang J, Snyder SH. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci. 1999;20:171–81. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 17.Tentori L, Portarena I, Graziani G. Potential clinical applications of poly(ADP-ribose) polymerase (PARP) inhibitors. Pharmacol Res. 2002;45:73–85. doi: 10.1006/phrs.2001.0935. [DOI] [PubMed] [Google Scholar]
- 18.Cuzzocrea S. Shock, inflammation and PARP. Pharmacol Res. 2005;52:72–82. doi: 10.1016/j.phrs.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 19.Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol Life Sci. 2002;59:1534–53. doi: 10.1007/s00018-002-8527-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem. 1999;380:953–9. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- 21.Cohen-Armon M, et al. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell. 2007;25:297–308. doi: 10.1016/j.molcel.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Kraus WL, Lis JT. PARP goes transcription. Cell. 2003;113:677–83. doi: 10.1016/s0092-8674(03)00433-1. [DOI] [PubMed] [Google Scholar]
- 23.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 24.Oliver FJ, et al. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. Embo J. 1999;18:4446–54. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerfaoui M, et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1- mediated nuclear export of p65 NF-kappaB and retention upon TLR4 stimulation. J Immunol. 2010;185:1894–902. doi: 10.4049/jimmunol.1000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zerfaoui M, et al. Nuclear translocation of p65 NF-kappaB is sufficient for VCAM-1, but not ICAM-1, expression in TNF-stimulated smooth muscle cells: differential requirement for PARP-1 expression and interaction. Cell Signal. 2008;20:186–94. doi: 10.1016/j.cellsig.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frizzell KM, Kraus WL. PARP inhibitors and the treatment of breast cancer: beyond BRCA1/2? Breast Cancer Res. 2009;11:111. doi: 10.1186/bcr2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer. 2009;9(Suppl 2):S73–81. doi: 10.3816/CBC.2009.s.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blagden S, Gabra H. Promising molecular targets in ovarian cancer. Curr Opin Oncol. 2009;21:412–9. doi: 10.1097/CCO.0b013e32832eab1f. [DOI] [PubMed] [Google Scholar]
- 30.Ng TB, Wang HX. Pharmacological actions of Cordyceps, a prized folk medicine. J Pharm Pharmacol. 2005;57:1509–19. doi: 10.1211/jpp.57.12.0001. [DOI] [PubMed] [Google Scholar]
- 31.Wong YY, et al. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J Biol Chem. 2010;285:2610–21. doi: 10.1074/jbc.M109.071159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rottenberg ME, et al. Treatment of African trypanosomiasis with cordycepin and adenosine deaminase inhibitors in a mouse model. J Infect Dis. 2005;192:1658–65. doi: 10.1086/496896. [DOI] [PubMed] [Google Scholar]
- 33.Iyer S, et al. Induction of apoptosis in proliferating human endothelial cells by the tumor-specific antiangiogenesis agent combretastatin A-4. Cancer Res. 1998;58:4510–4. [PubMed] [Google Scholar]
- 34.Kim HG, et al. Cordycepin inhibits lipopolysaccharide-induced inflammation by the suppression of NF-kappaB through Akt and p38 inhibition in RAW 264.7 macrophage cells. Eur J Pharmacol. 2006;545:192–9. doi: 10.1016/j.ejphar.2006.06.047. [DOI] [PubMed] [Google Scholar]
- 35.Cousineau I, Belmaaza A. BRCA1 haploinsufficiency, but not heterozygosity for a BRCA1-truncating mutation, deregulates homologous recombination. Cell Cycle. 2007;6:962–71. doi: 10.4161/cc.6.8.4105. [DOI] [PubMed] [Google Scholar]
- 36.Hans CP, et al. Thieno[2,3-c]isoquinolin-5-one, a potent poly(ADP-ribose) polymerase inhibitor, promotes atherosclerotic plaque regression in high-fat diet-fed apolipoprotein E-deficient mice: effects on inflammatory markers and lipid content. J Pharmacol Exp Ther. 2009;329:150–8. doi: 10.1124/jpet.108.145938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datta R, et al. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy. 2011;66:853–61. doi: 10.1111/j.1398-9995.2011.02549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fong PC, et al. Inhibition of poly(ADP- ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.