

Abstract
BACKGROUND AND PURPOSE
The primary use of local anaesthetics is to prevent or relieve pain by reversibly preventing action potential propagation through the inhibition of voltage-gated sodium channels. The tetrodotoxin-sensitive voltage-gated sodium channel subtype Nav1.7, abundantly expressed in pain-sensing neurons, plays a crucial role in perception and transmission of painful stimuli and in inherited chronic pain syndromes. Understanding the interaction of lidocaine with Nav1.7 channels could provide valuable insight into the drug's action in alleviating pain in distinct patient populations. The aim of this study was to determine how lidocaine interacts with multiple inactivated conformations of Nav1.7 channels.
EXPERIMENTAL APPROACH
We investigated the interactions of lidocaine with wild-type Nav1.7 channels and a paroxysmal extreme pain disorder mutation (I1461T) that destabilizes fast inactivation. Whole cell patch clamp recordings were used to examine the activity of channels expressed in human embryonic kidney 293 cells.
KEY RESULTS
Depolarizing pulses that increased slow inactivation of Nav1.7 channels also reduced lidocaine inhibition. Lidocaine enhanced recovery of Nav1.7 channels from prolonged depolarizing pulses by decreasing slow inactivation. A paroxysmal extreme pain disorder mutation that destabilizes fast inactivation of Nav1.7 channels decreased lidocaine inhibition.
CONCLUSIONS AND IMPLICATIONS
Lidocaine decreased the transition of Nav1.7 channels to the slow inactivated state. The fast inactivation gate (domain III–IV linker) is important for potentiating the interaction of lidocaine with the Nav1.7 channel.
Keywords: Nav1.7, voltage-gated sodium channel, neuropathic pain, inactivation, channelopathies, local anaesthetics
Introduction
Voltage-gated sodium channels (VGSCs) are the primary target of local anaesthetics such as lidocaine (Taylor, 1959; Hille, 1966; 1977;). Local anaesthetics preferentially bind to open and/or inactivated VGSCs (Hille, 1977; Yeh, 1978). A common use of local anaesthetics is for treating pain by reversibly preventing action potential initiation and/or propagation by inhibiting VGSCs. Unfortunately, local anaesthetics are not isoform-specific and thus, they can produce clinically relevant side effects. The Nav1.7 channel subtype (channel nomenclature follows Alexander et al., 2009) is expressed at high levels in rat and human dorsal root ganglia and sympathetic ganglia (Black et al., 1996; Sangameswaran et al., 1997; Toledo-Aral et al., 1997) and responds to small, slow depolarizations thereby contributing to action potential generation (Cummins et al., 1998). Many of the neurons expressing Nav1.7 channels are nociceptive neurons (Djouhri et al., 2003). Although not used clinically, a relatively selective class of Nav1.7 channel inhibitors has been characterized (Williams et al., 2007) and could be very effect at treating various pain states. Nonetheless, establishing a detailed pharmacological and molecular basis for the interaction of local anaesthetics with Nav1.7 channels is important for improving pain therapeutics.
The interaction of local anaesthetics with specific gating configurations of VGSCs is controversial. Residues in the S6 segments whose side chains are positioned towards the inner portion of the pore (Fozzard and Hanck, 1996; Marban et al., 1998; Catterall, 2000) are proposed to interact with local anaesthetics (Ragsdale et al., 1994; Wright et al., 1998; Wang et al., 2000; Yarov-Yarovoy et al., 2001; 2002;). Point mutations of residues located in the S6 segments can reduce interaction of local anaesthetics to the inactivated state of VGSCs (Ragsdale et al., 1994; Wright et al., 1998; Li et al., 1999; Nau et al., 1999; 2003; Sheets et al., 2007). This provides structural evidence for a molecular site within VGSCs targeted by local anaesthetics. However, the conformation-dependent stabilization of lidocaine interaction continues to be explored.
Human cardiac VGSCs, lacking fast inactivation, display reduced sensitivity to lidocaine (Bennett et al., 1995) indicating fast inactivation is important for lidocaine interaction. Experiments using cardiac and skeletal muscle VGSCs in tissue or heterologous expression systems indicated the open-state of the channel was not necessary for interaction with lidocaine (Bean et al., 1983; Bennett et al., 1995; Balser et al., 1996b), but, in fact, lidocaine served to enhance VGSC inactivation. It has also been shown that fast inactivation can recover while lidocaine interaction remains (Vedantham and Cannon, 1999) creating controversy over the importance of the fast inactivated state in lidocaine inhibition of VGSCs.
Alternative theories for block suggest local anaesthetics interact with a slow inactivated state. Lidocaine block of rat Nav1.4 channels in Xenopus oocytes was similar for both fast and slow inactivated states (Balser et al., 1996a). Additional studies, with rat Nav1.4 channels, proposed that lidocaine interactions enhanced transition to a slow inactivated state (Chen et al., 2000; Ong et al., 2000; Fukuda et al., 2005). These studies implicate increased slow inactivation in lidocaine inhibition. However, our findings indicate that this is not the case for the neuronal Nav1.7 channel isoform.
Several inherited gain-of-function Nav1.7 channel mutations cause severe chronic pain disorders including paroxysmal extreme pain disorder (PEPD) and inherited erythermalgia (Dib-Hajj et al., 2007). Conversely, loss-of-function mutations in Nav1.7 channels have been identified in patients diagnosed with a congenital insensitivity to pain (Cox et al., 2006). Because Nav1.7 channels play a crucial role in the perception of pain, understanding the interaction of lidocaine with these channels is likely to yield useful pharmacological insights into its use as a treatment for pain. The aim of this study was to determine how lidocaine affected inactivated state transitions in Nav1.7 channels. Our findings indicate that lidocaine decreased the transition of Nav1.7 channels to a slow inactivated state.
Methods
cDNA vectors
The VGSC subtype human Nav1.7 was previously cloned and constructed into a modified pcDNA 3.1 (−) vector (Klugbauer et al., 1995). The Nav1.7-I1461T PEPD mutant construct was prepared as previously described (Jarecki et al., 2008). Nav1.7 channel constructs were co-transfected with the vectors pCD8-IRES-hβ1 and pEGFP-IRES-hβ2 (Lossin et al., 2002; 2003;), which contain the human β1 and β2 sodium channel subunits.
Transient transfection of human embryonic kidney 293 (HEK293) cells
Transient transfections were used for all experiments. HEK293 cells were grown under standard tissue culture conditions (5% CO2; 37°C) in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Transfections of human wild-type (WT) Nav1.7, or human mutant Nav1.7 (1461T), along with human β1 and β2 subunits, were performed using the calcium phosphate precipitation technique. The calcium phosphate–DNA mixture was added to naïve HEK293 cells on a 100 × 20 mm culture dish. The mixture was left on the cells in culture for 3–6 h, after which time the cells were washed with fresh DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Experiments testing for the expression of CD8 using Dynabeads (Dynal, Brown Deer, WI, USA) in enhanced green fluorescent protein (EGFP) positive cells indicate that ∼89% of HEK293 cells successfully transfected with the β2 subunit construct also were transfected with the β1 subunit construct; therefore, cells were subsequently selected based on EGFP fluorescence.
Solutions
For application during voltage-clamp experiments, lidocaine hydrochloride (Sigma Aldrich Co., St. Louis, MO, USA) was dissolved in standard bathing solution to give a stock solution of 100 mM. Subsequent dilutions were performed in standard bathing solution to give concentrations of (in mM): 0.1, 0.3, 1, 3, 10 and 30. The composition of the extracellular recording solution was (in mM): 140 NaCl, 1 MgCl2, 3 KCl, 1 CaCl2 and 10 HEPES, pH 7.3 (adjusted with NaOH). The composition of the standard pipette solution was as follows (in mM): 140 CsF, 10 NaCl, 1.1 EGTA and 10 HEPES, pH 7.3 with CsOH. Fluoride was used in the intracellular solutions as it substantially enhances the ability to perform the extended voltage-clamp protocols required for investigating lidocaine's interaction with slow inactivation. It should be noted that fluoride can shift the voltage-dependence of activation and steady-state inactivation by about −15 to −20 mV (Jarecki et al., 2008); data were not corrected for this. Although intracellular fluoride can also reduce the magnitude of persistent sodium currents associated with the I1461T mutation, in general we have found that the relative impact of the I1461T mutation on Nav1.7 properties is preserved in fluoride (Jarecki et al., 2008), especially for the peak transient currents, which are the focus of the current study. Lidocaine hydrochloride was diluted in the extracellular recording solution before being externally applied to the bath solution.
Whole cell patch-clamp recordings
Whole cell patch-clamp recordings were conducted at room temperature (∼21°C) using a HEKA EPC-10 double amplifier. Data were acquired on a Windows-based Pentium IV computer using the Pulse program (v 8.65, HEKA Electronic, Lambrecht/Pfalz, Germany). Fire-polished electrodes were fabricated from 1.7 mm VMR Scientific (West Chester, PA, USA) capillary glass using a Sutter P-97 puller (Novato, CA, USA). Isolated cells for whole cell electrophysiology were chosen by expression of EGFP. Recording electrodes had a typical resistance of 0.9–1.3 MΩ. The EPC10 amplifier's offset potential was zeroed with the electrode almost touching the cell of interest. Series resistance errors were compensated to be less than 3 mV and the capacitance artefact was cancelled using computer-controlled circuitry of the patch-clamp amplifier. Linear leak subtraction, based on resistance estimates from four to five hyperpolarizing pulses applied before the depolarizing test potential, was used for all voltage-clamp recordings.
Recordings were always started 3 min after establishing the whole cell configuration. This allows for equilibration of the pipette solution with the intracellular compartment and allows channels to stabilize in the resting configuration at the imposed holding membrane potential (typically −120 mV). Membrane currents were filtered at 5 kHz and sampled at 20 kHz. Cells having a leak current of more than 10% of the peak sodium current were not used for analysis. Voltage protocols are described within the Results. When the perfusion system (described below) was not used, compounds were added to the bath compartment by first withdrawing 25 µl l of bathing solution, then adding 25 µl of 10-fold concentrated compound and mixing 10–15 times with a 25 µl pipettor.
Perfusion system
We obtained concentration–response curves for lidocaine inhibition of Nav1.7 channel current using a SF-77B Perfusion Fast-Step system (Warner Instruments, LLC, Hamden, CT, USA) to deliver different concentrations of lidocaine focally onto the transfected HEK293 cell in whole cell configuration. During these experiments, we placed coverslips containing transfected HEK293 cells into a RC-26 Open Diamond Bath perfusion chamber (Warner Instruments, LLC, Hamden, CT, USA). The coverslips were affixed to the chamber with a small amount of Vaseline to prevent movement of the coverslip during recordings. This chamber allows a constant laminar flow of bath solution across the cells that was perfused at approximately 1 mL·min−1 using gravity flow and removed using a Masterflex C/L Single Channel Variable- Speed peristaltic pump (Cole-Parmer Instrument Company, Vernon Hills, IL, USA). The drug delivery 3-barrel tube was placed near the transfected HEK293 cell, and its positioning was calibrated before each experiment. The position of the drug delivery tube was controlled manually by a stepper motor with a response time of approximately 20 ms. Concentrations of lidocaine were applied in random order and were allowed to interact with the cell 30 s before recording. All solutions were kept at room temperature (∼21°C).
Data analysis
Voltage-clamp experimental data were analysed using the Pulsefit (v 8.65, HEKA Electronic) and Origin (OriginLab Corp., Northhampton, MA, USA) software programs. Slope factors of activation and steady-state inactivation curves were calculated using a Boltzmann function: 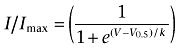 where I = measured current, Imax = maximum current, V = command voltage, V0.5 = voltage at which the normalized current value is 0.5, and k = slope factor describing the steepness of the relationship. An offset value was added to the Boltzmann equation when fitting to some slow inactivation profiles to address incomplete inactivation. The equation used for the one-site binding curve and two-site binding curves for concentration–inhibition relationship for lidocaine and the Nav channels were as follows:
where I = measured current, Imax = maximum current, V = command voltage, V0.5 = voltage at which the normalized current value is 0.5, and k = slope factor describing the steepness of the relationship. An offset value was added to the Boltzmann equation when fitting to some slow inactivation profiles to address incomplete inactivation. The equation used for the one-site binding curve and two-site binding curves for concentration–inhibition relationship for lidocaine and the Nav channels were as follows:
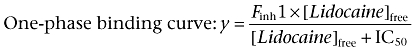 |
(1) |
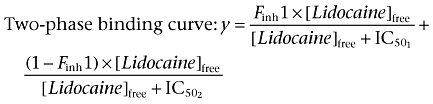 |
(2) |
Finh1 equals the fraction of current inhibited with the first component of lidocaine inhibition. Time constants for recovery from inactivation and onset of inactivation were estimated using first-, second- and third-order exponential fits. The first-order exponential growth equation used is as follows: 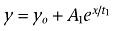 . The second- and third-order exponential growth equation used were as follows:
. The second- and third-order exponential growth equation used were as follows: 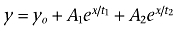 and
and 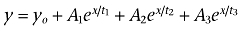 where for both equations yo = the initial value at time 0, A1 = weight factor (of total recovered) for fraction recovered with time constant t1, A2 = weight factor (of total recovered) for fraction recovered at t2, A3 = weight factor (of total recovered) for fraction recovered at t3, x = the growth constant, and t1 = time constant for recovery of fast inactivated channels, t2 and t3 = time constants for the recovery of potentially two distinct slow inactivated states of the channels. The order (i.e. first-order, second-order) of exponential fit was chosen after an increase in the order of the fit produced an extra parameter that: (i) was less than 5% of the total fit; and (ii) produced an estimated tau value outside the range of the fit. Fit precision was set at R2 > 0.90 for all fits. Results are presented as mean ± SEM, and error bars in the figures represent SEM. Statistical significance was set at P < 0.05.
where for both equations yo = the initial value at time 0, A1 = weight factor (of total recovered) for fraction recovered with time constant t1, A2 = weight factor (of total recovered) for fraction recovered at t2, A3 = weight factor (of total recovered) for fraction recovered at t3, x = the growth constant, and t1 = time constant for recovery of fast inactivated channels, t2 and t3 = time constants for the recovery of potentially two distinct slow inactivated states of the channels. The order (i.e. first-order, second-order) of exponential fit was chosen after an increase in the order of the fit produced an extra parameter that: (i) was less than 5% of the total fit; and (ii) produced an estimated tau value outside the range of the fit. Fit precision was set at R2 > 0.90 for all fits. Results are presented as mean ± SEM, and error bars in the figures represent SEM. Statistical significance was set at P < 0.05.
Results
Voltage dependence of fast and slow inactivation of Nav1.7 channels
The aim of this study was to determine how lidocaine interacts with fast and slow inactivated conformations of Nav1.7 channels. The voltage-dependence of steady-state fast inactivation Nav1.7 channels can be estimated with 200 ms depolarizing pre-pulses (Figure 1A) and under the recording conditions used for this study we estimate that half of the Nav1.7 channels are fast inactivated at −77.5 ± 1.4 mV (n = 11). The voltage-dependence of steady-state slow inactivation can be estimated using 10 s depolarizing pre-pulses followed by a 40 ms hyperpolarizing pulse to −120 mV to allow channels to recover from fast inactivation before the test pulse to determine channel availability. Under these conditions, slow inactivation is incomplete for Nav1.7 channels expressed in HEK293 cells and the voltage-dependence of slow inactivation is complex with possibly two distinct components (Figure 1B, n = 11).
Figure 1.
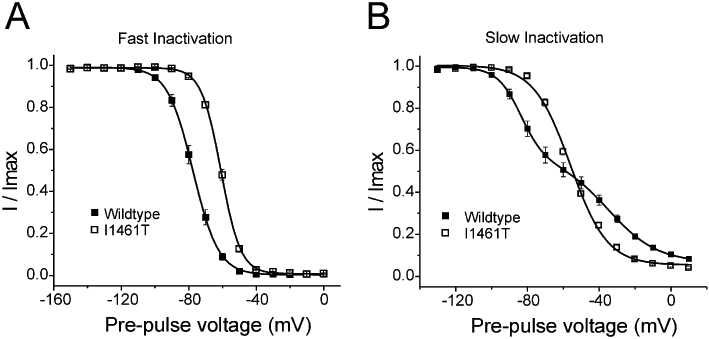
Voltage-dependence of wild type and I1461T Nav1.7 channels steady-state fast inactivation and slow inactivation. (A) The voltage-dependence of fast inactivation was determined using 200 ms pre-pulses to the potentials indicated followed by a test pulse to 0 mV to determine current availability for wild type and I1461T Nav1.7 channels. (B) The voltage-dependence of slow inactivation was determined using 10 s pre-pulses to the potentials indicated followed by a 40 ms pulse to −120 mV to allow channels to recover from fast inactivation and then a test pulse to 0 mV to determine current availability for wild type and I1461T Nav1.7 channels. Currents are normalized to the maximum current measured during the protocol (Imax).
Voltage and pulse duration determine concentration-response effects of lidocaine on Nav1.7 channels
We next established lidocaine concentration–response relationships for Nav1.7 channels pulsed to one of three potentials (−100, −50 and 0 mV) for 200 ms versus 10 s followed by a brief recovery pulse (Figure 2A). When pulsing for 200 ms, the concentration–response relationship was well fit with a one-site concentration–response model for all pulse potentials (Figure 2B–D). When pulsing to the various potentials for 10 s, the concentration–response relationship was best fit with a two-site binding model (Figure 2B–D, Table 1). This implied that the extension of pulse duration changed the Nav1.7 channels into a mixture of high and low affinity configurations, most likely fast and slow inactivated states.
Figure 2.
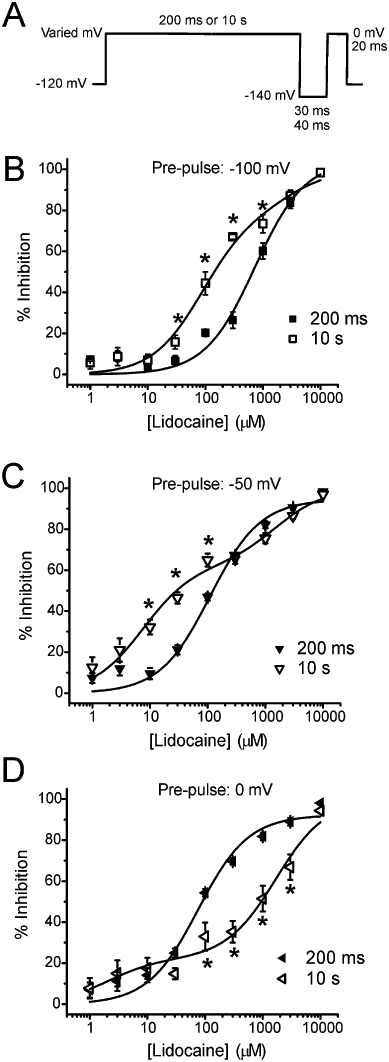
Concentration–response curves of lidocaine inhibition on Nav1.7 channels. (A) Protocol used for determining the concentration–response relationship of lidocaine inhibition on Nav1.7 channels. Pulse potential (A: −100 mV, B: −50 mV, C: 0 mV) is indicated at the top of each set of concentration–response curves. Concentration–response data resulting from pulsing channels for 200 ms were fit using a one-phase binding equation. Concentration–response data resulting from pulsing channels for 10 s were fit using a two-phase binding equation. The one- and two-phase binding equations are explained in Table 3. All fits had R2 >0.90. Data are presented as mean % decrease in current ± SEM. Statistical significance was determined using a Student's unpaired t-test (* = P < 0.05).
Table 1.
Summary of IC50 values established for lidocaine inhibition of Nav1.7 channel current at different potentials and pulse durations
| Pulse duration | ||||
|---|---|---|---|---|
| 200 ms | 10 s | |||
| Voltage (mV) | IC50 | IC50 1 | IC50 2 | Fraction IC50 1 |
| −100 mV | 727 ± 154 µM | 87.2 ± 14.6 µM | 2.8 ± 4.2 mM | 0.79 |
| −50 mV | 110 ± 20 µM | 7.5 ± 2.0 µM | 1.5 ± 0.6 mM | 0.62 |
| 0 mV | 76 ± 14 µM | 2.0 ± 1.8 µM | 1.7 ± 0.4 mM | 0.22 |
Values are mean IC50 values ± SEM estimated from the best fit line produced from a one-site or two-site binding equation. The average n value for all concentrations of lidocaine was 6 with no single concentration having an n < 4.
With more depolarized pulse potentials, lidocaine paradoxically showed more inhibition on Nav1.7 channels pulsed for 200 ms than those pulsed for 10 s (Figure 2C and D). The point of intersection for the separate concentration–response curves was dependent on pulse potential. For example, the point at which the 200 ms pulse to −50 mV begins producing more lidocaine inhibition than the 10 s pulse to −50 mV was approximately at 260 µM (Figure 2C). If the pulse is changed to 0 mV, this point of intersection shifts to approximately 25 µM (Figure 2D). Interestingly, the amplitude of the high-affinity (IC50∼ 2 µM) binding component is much smaller at 0 mV than at −50 mV (Figure 2, Table 1).
Lidocaine alters the recovery of Nav1.7 channels
We next tested the state dependence of lidocaine inhibition on Nav1.7 channels by evaluating recovery from inactivation. Cells expressing Nav1.7 channels were held at one of three different potentials (−100, −50 or 0 mV) for 10 s in the presence or absence of lidocaine (1 mM) followed by a recovery interval of variable duration to −140 mV before testing for active current with a 20 ms pulse to 0 mV (Figure 3A). The normalized amount of current measured was plotted versus the duration of the recovery time to determine time constants for recovery from inactivation or drug interaction. Recovery of control Nav1.7 channels always showed a two- or three-phase recovery profile dependent on pre-pulse potential (Figure 3B–D). Under these conditions tau 1 represented the time constant for recovery from fast inactivation and tau 2 and 3 represented time constants for recovery from slow inactivation (Table 2).
Figure 3.
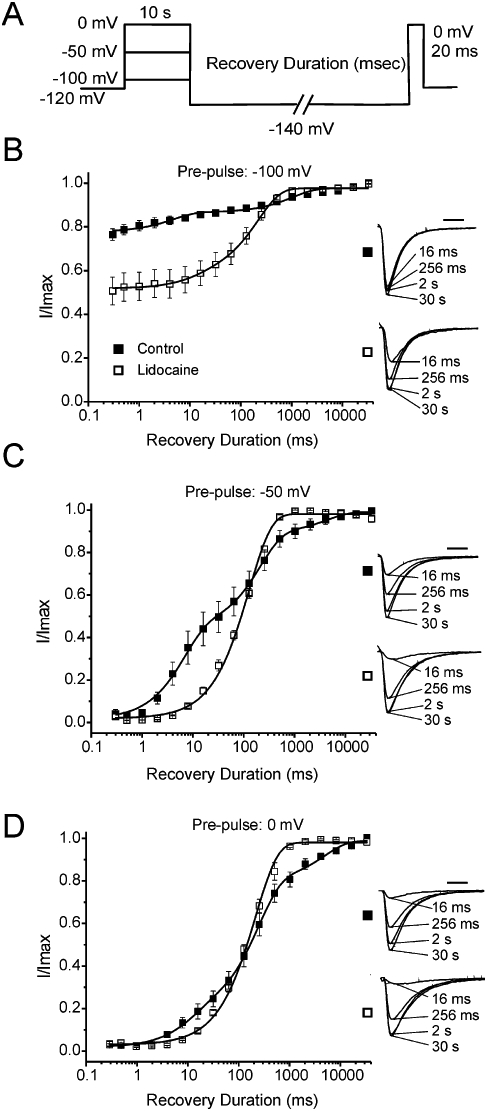
Recovery from prolonged inactivation and lidocaine inhibition for Nav1.7 channels. (A) Protocol used for examining recovery. (B) Recovery of control (n = 4) and lidocaine-treated (n = 5) Nav1.7 channels from a −100 mV pre-pulse. (C) Recovery profile of control (n = 4) and lidocaine-treated (n = 4) Nav1.7 channels from a −50 mV pre-pulse. (D) Recovery profile of control (n = 4) and lidocaine-treated (n = 3) Nav1.7 channels from a 0 mV pre-pulse. Insets: representative traces showing the magnitude of current recovered in the absence and presence of lidocaine (1 mM) for the specified pre-pulse voltage. All fits had R2 >0.90.
Table 2.
Estimated time constants for recovery of Nav1.7 channels from 10 s pulses in the absence and presence of lidocaine
| Pre-pulse voltage (mV) | No lidocaine | 1 mM lidocaine | |||||
|---|---|---|---|---|---|---|---|
| Tau 1 (ms) | A1 | Tau 2 (ms) | A2 | Tau 3 (s) | A3 | Tau (ms) | |
| −100 mV | 4.63 ± 1.6 | 0.14 | 1046 ± 300 | 0.11 | N/A | N/A | 220 ± 16 |
| −50 mV | 6.64 ± 0.9 | 0.44 | 207 ± 37 | 0.44 | 4.3 ± 3.0 | 0.10 | 129 ± 5.0 |
| 0 mV | 6.22 ± 4.3 | 0.21 | 245 ± 26 | 0.61 | 4.9 ± 1.4 | 0.18 | 189 ± 12 |
Values are mean ± SEM.
N/A, not applicable.
Recovery of Nav1.7 channels in the presence of lidocaine displayed a mono-phasic profile and was fit using a first-order exponential growth equation for all pre-pulse voltages tested (Table 2). Lidocaine altered the recovery of Nav1.7 channels from inactivation by inducing a channel-drug configuration that, at least initially, takes longer to recover than non-treated channels. However, at depolarized potentials (−50 and 0 mV) lidocaine paradoxically enhanced recovery of Nav1.7 channels at prolonged recovery durations, compared with control (Figure 3C and D). After recovery durations of approximately 140 ms for −50 mV pre-pulses and 75 ms for 0 mV pre-pulses, lidocaine-treated channels displayed greater recovery than control channels (Figure 3C and D). At shorter recovery durations, when control channels are recovering from a fast inactivated state, lidocaine slows recovery. These results suggest lidocaine stabilizes a drug-channel configuration in Nav1.7 channels that recovers slower than native fast inactivation but recovers faster than native slow inactivation.
The PEPD mutation I1461T decreases use-dependent inhibition by lidocaine
We further explored the relationship between inactivation states and lidocaine interaction by using a mutation (I1461T) in the putative isoleucine-phenylalanine-methionine-threonine inactivation gate. The I1461T mutation, implicated in PEPD (Fertleman et al., 2006), destabilizes transition to an inactivated state (Fertleman et al., 2006; Jarecki et al., 2008). We have previously shown that the I1461T mutation slows the rate of open-state fast inactivation and depolarizes the midpoint of the voltage-dependence of steady-state fast inactivation by ∼21 mV (Jarecki et al., 2008). We hypothesized the effects caused by the I1461T mutation might reduce the stability of lidocaine's interaction with Nav1.7 channels. Figure 1 compares the voltage-dependence of steady-state fast inactivation and slow inactivation of Nav1.7 WT and I1461T channels, under the recording conditions used in the present study.
Interestingly, we found that lidocaine created a similar shift in the midpoint of the voltage-dependence of fast inactivation for WT (−15.2 ± 1.9 mV; n = 5) and I1461T (−12.3 ± 0.5 mV; n = 7) channels. As lidocaine is known to exhibit use-dependent inhibition of sodium currents, we also examined the use-dependent effects of lidocaine on the Nav1.7 WT and I1461T channels by pulsing to −10 mV at a frequency of 5 Hz. Under control conditions, Nav1.7 WT and I1461T current amplitudes decreased by 20.5 ± 2.5% (n = 5) and 22.3 ± 2.0% (n = 7), respectively, when comparing current amplitudes evoked by the last pulse with those evoked by the first pulse, representing phasic inhibition. Application of lidocaine (1 mM) reduced the current amplitude elicited by the first pulse (tonic inhibition) and similarly produced phasic decreases for WT and I1461T channels (Figure 4A and B). These decreases were significant compared with time-dependent controls. Both the tonic and the phasic decreases in current amplitude for I1461T channels caused by lidocaine were significantly smaller than the decreases observed with the WT channel (Figure 4E).
Figure 4.
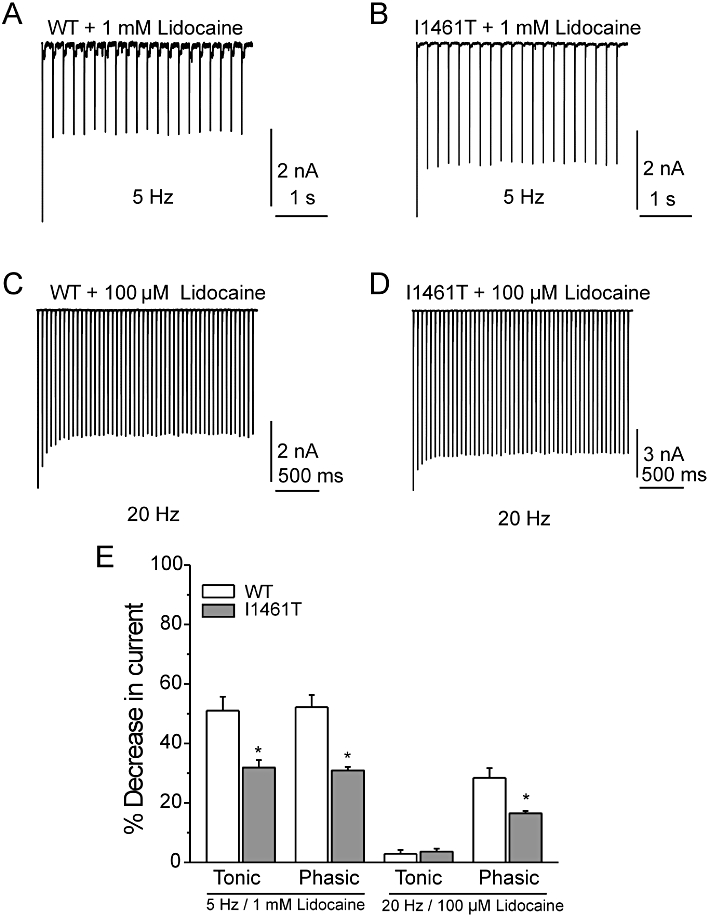
Use-dependent inhibition of Nav1.7 wild type (WT) and I1461T current by lidocaine. Representative traces of peak WT (A) and I1461T (B) currents during 5 Hz stimulation to −10 mV for 50 ms. Representative traces of peak WT (C) and I1461T (D) currents during 20 Hz stimulation to −10 mV for 10 ms. (E) Summary of tonic and phasic inhibition of WT and I1461T currents by either 1 mM lidocaine at 5 Hz stimulation or 100 µM lidocaine at 20 Hz stimuation. Tonic inhibition is presented as mean % decrease in current of the first pulse in lidocaine-treated and non-treated channels. Phasic inhibition is displayed as mean % decrease in current between the first and 20th pulse of lidocaine-treated channels. Statistical significance was determined using a Student's unpaired t-test (* = P < 0.05).
Because the phasic decreases in current stabilized after the second pulse for both WT and I1461T with 1 mM lidocaine, we lowered the concentration of lidocaine (100 µM) to reduce inhibition and re-examined phasic inhibition at stimulation frequencies of 5 and 20 Hz. Tonic inhibition by 100 µM lidocaine on WT and I1461T channels was less than 4% at 20 Hz (Figure 4E). Stimulating at 5 Hz created small amounts of phasic inhibition for both WT (7.0 ± 2.0 %) and I1461T (4.6 ± 0.4 %) channels and were not significantly different. By contrast, increasing the stimulation frequency to 20 Hz produced significantly higher phasic inhibition of WT currents (Figure 4C) compared with I1461T currents (Figure 4D). Overall (Figure 4E), these data indicated that the I1461T channels are less sensitive to tonic and phasic inhibition by lidocaine.
Lidocaine differentially alters the recovery of I1461T and WT channels from inactivation
Because lidocaine had similar effects on the voltage dependence of fast inactivation for I1461T and WT channels, we speculated that decreased use-dependent inhibition in I1461T channels was a result of altered recovery from inhibition. Interestingly, in the absence of lidocaine, I1461T channels pre-pulsed to −50 mV or 0 mV recovered more slowly from prolonged inactivation than WT channels (Figure 5A and B; Table 3). This significant slowing of recovery was likely to be due to increased transition of I1461T channels to slow inactivated states, which could result from the destabilized fast inactivation caused by the I1461T mutation (Featherstone et al., 1996). At −50 mV, lidocaine-treated I1461T channels displayed more recovery at short recovery durations (Figure 5C) compared with lidocaine-treated WT channels, consistent with our use-dependent data indicating lidocaine is less effective against I1461T channels. Paradoxically, at 0 mV lidocaine-treated I1461T channels displayed less recovery at long recovery durations (Figure 5D). One possibility for this is that the reduced stability of the lidocaine interaction with I1461T channels could allow for greater transition to slow inactivated states. However, we cannot rule out slow inactivation in I1461T channels that was independent of lidocaine interactions.
Figure 5.
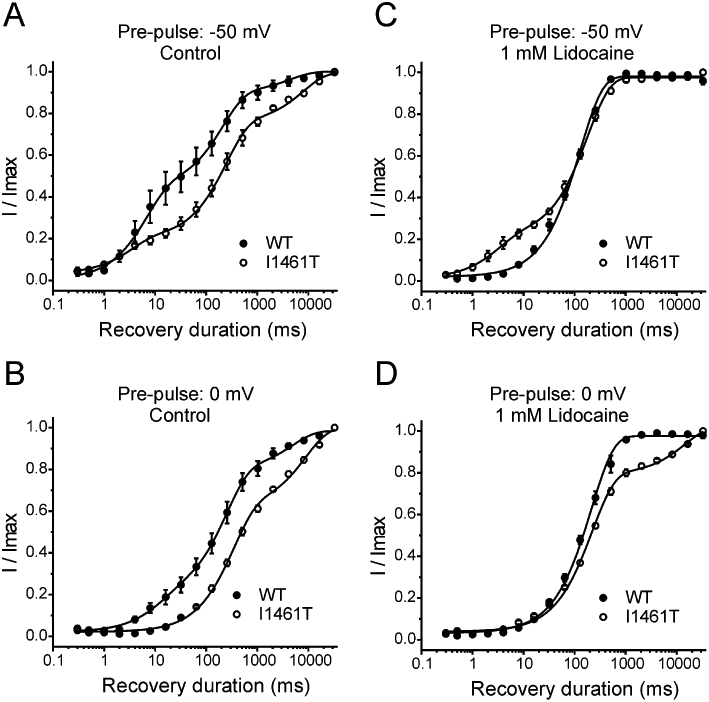
Recovery from long conditioning pulses and lidocaine inhibition for I1461T channels compared with wild-type (WT) channels. (A) Recovery of WT (n = 4) and I1461T (n = 3) channels from a 10 s pulse to −50 mV. (B) Recovery of WT (n = 4) and I1461T (n = 3) channels from a 10 s pulse to 0 mV. Recovery of lidocaine-treated WT (n = 4) and I1461T (n = 3) channels from a 10 s pulse to 50 mV (C) and 0 mV (D). All fits had R2 >0.90. WT plots are the same as in Figure 3.
Table 3.
Estimated time constants for recovery from prolonged inactivation and lidocaine inhibition for I1461T channels
| Pre-pulse voltage (mV) | No lidocaine | 1 mM lidocaine | |||||
|---|---|---|---|---|---|---|---|
| Tau 1 (ms) | A1 | Tau 2 (ms) | A2 | Tau 3 (s) | A3 | Tau (ms) | |
| −100 mV | 4.3 ± 1.7 | 0.11 | 1400 ± 400 | 0.08 | N/A | N/A | 220 ± 16 |
| −50 mV | 3.3 ± 0.7 | 0.19 | 243 ± 20 | 0.57 | 4.3 ± 3.0 | 0.10 | 129 ± 5.0 |
| 0 mV | N/A | N/A | 334 ± 24 | 0.60 | 4.9 ± 1.4 | 0.18 | 189 ± 12 |
Values are mean ± SEM.
N/A, not applicable.
When considering only I1461T channels, it is clear that lidocaine delayed recovery from inactivation for channels pulsed to −100 mV (Figure 6A). By contrast, with −50 mV and 0 mV conditioning pulses, lidocaine-treated I1461T channels recovered faster than non-treated I1461T channels (Figure 6B and C). These data indicate that lidocaine reduced or inhibited the transition of the I1461T channel to a slow inactivated state, which, in combination with decreased lidocaine stabilization, allows for more efficient recovery compared to non-treated I1461T channels (Figure 6C).
Figure 6.
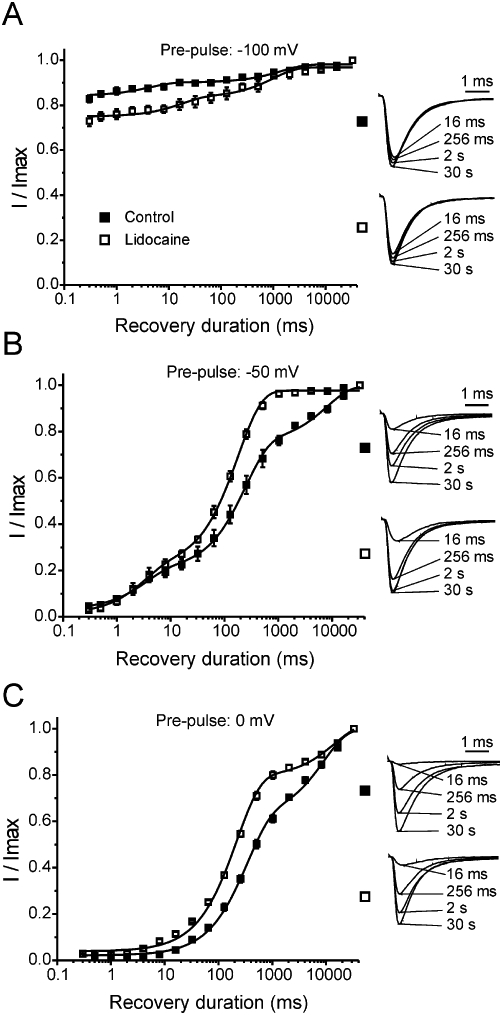
Recovery from prolonged inactivation and lidocaine inhibition for I1461T channels. (A) Recovery of control (n = 4) and lidocaine-treated (n = 3) I1461T channels from a −100 mV pre-pulse. (B) Recovery profile of control (n = 4) and lidocaine-treated (n = 3) I1461T channels from a −50 mV pre-pulse. (C) Recovery profile of control (n = 4) and lidocaine-treated (n = 3) I1461T channels from a 0 mV pre-pulse. Insets: representative traces showing the magnitude of current recovered in the absence and presence of lidocaine (1 mM) for the specified pre-pulse voltage. Under all conditions, the recovery profile was fit with a second-order exponential growth equation. All fits had R2 >0.90. Note, the data in (B) and (C) are the I1461T data from Figure 5 combined to better illustrate the effect of lidocaine on I1461T channel recovery.
The I1461T mutation increases slow inactivation
We next investigated the effects of lidocaine on the development of slow inactivation. To measure the time course for development of slow inactivation, we inserted a short recovery pulse between the conditioning pulse (0 mV for 1 ms to 10 s) and the test pulse (0 mV for 20 ms) to relieve fast inactivation. To assure that the duration of this pulse allowed sufficient relief from fast inactivation, we multiplied the time constants for Nav1.7 channel recovery (Table 2, tau 1) at −140 mV by five and used this value to set the recovery duration. Based on this calculation, for a holding potential of 0 mV the recovery duration was set at 40 ms. The fraction of slow inactivated channels was larger for I1461T channels compared with WT channels during long pulse durations (>2 s) to 0 mV (Figure 7A). Pulsing to 0 mV induced slow inactivation that developed with two distinct phases for WT channels (Figure 7B). Notably, lidocaine initially produced a decrease in current, but additional current reduction was minimal as pulse duration increased (Figure 7B). This ‘plateau’ phase in the time course for lidocaine inhibition took place at approximately the same duration where slow inactivation begins to occur under control (zero lidocaine) conditions. Interestingly, at pulse durations greater than 10 s, lidocaine-treated WT channels displayed greater current than control WT channels. Lidocaine also decreased I1461T current during short pulses to 0 mV compared with I1461T controls (Figure 7C). However, with long pulses (>2 s), I1461T channels generated larger current in the presence of lidocaine than in the absence of lidocaine (Figure 7C). By contrast this did not occur with WT channels until the pulse duration exceeded ∼10 sec.
Figure 7.
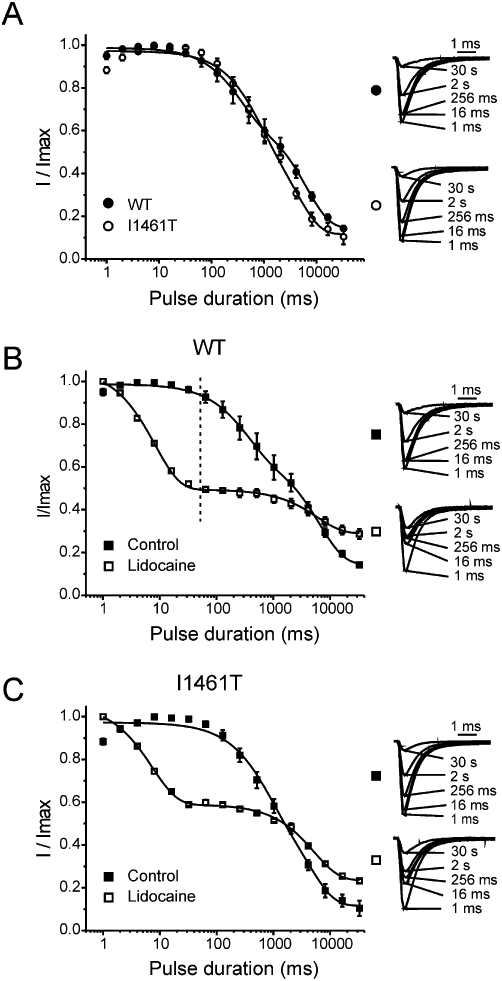
Development of slow inactivation and lidocaine inhibition for Nav1.7 channels and I1461T channels. (A) The development of slow inactivation for wild type (WT) (n = 7) and I1461T (n = 4) channels pulsed to 0 mV for increasing durations. Insets: representative traces showing the magnitude of slow inactivation for WT and I1461T channels. (B) Comparison of slow inactivation development (Control; n = 7) and lidocaine inhibition (Lidocaine; n = 6) for WT channels pulsed to 0 mV for increasing durations. (C) The development of slow inactivation (Control; n = 4) and lidocaine inhibition (Lidocaine; n = 4) for I1461T channels pulsed to 0 mV for increasing durations. In the case of control conditions, the onset profile fit better to a second-order exponential growth equation possibly because of the onset of two different forms of channel slow inactivation. Insets: representative traces showing the magnitude of slow inactivation under control conditions and lidocaine (1 mM) inhibition for the specified duration. All fits had R2 values >0.90.
Discussion and conclusions
Using WT and I1461T Nav1.7 channels expressed in HEK293 cells, we used multiple electrophysiological paradigms to investigate the effects of lidocaine during various voltage-dependent channel transitions. Our findings indicate lidocaine decreases channel transition to slow inactivation for Nav1.7 channels and the I1461 residue plays a role in stabilizing the interaction of lidocaine with the channel. We first observed that the concentration–response relationship for lidocaine inhibition relied on pulse duration. Short duration pulses (200 ms) produced a one-site binding profile while long duration pulses (10 s) produced a two-site binding profile (Figure 2). Surprisingly, at 0 mV, the concentration–response curves intersect, with higher lidocaine concentrations showing more inhibition on Nav1.7 channels pulsed for 200 ms than ones pulsed for 10 s (Figure 2D). This suggests that increasing the probability of the Nav1.7 transition to slow inactivation reduces channel interaction with lidocaine. The estimated IC50 values for the second component of lidocaine inhibition on Nav1.7 channels pulsed for 10 s are similar to IC50 values for resting Nav1.7 channels. Thus, we speculate that the pore configuration of Nav1.7 channels in a slow inactivated state may be similar to the pore configuration of resting channels, at least as it pertains to lidocaine binding. An alternative explanation for a biphasic concentration–response curve is that lidocaine might interact with closed fast inactivated and open fast inactivated channels differently. However, this is unlikely as the concentration–response response curves using 200 ms conditioning pulses were well fit with a one-site binding profile.
We next examined the effects of lidocaine on recovery of Nav1.7 channels from fast and slow inactivation. Interestingly, following 10 s depolarized conditioning pulses (−50 and 0 mV), the time course for recovery of current in the presence of lidocaine was faster than under control conditions. This enhancement of recovery by lidocaine suggests lidocaine is stabilizing Nav1.7 channels into a drug-channel confirmation that is energetically favourable and transition to a slow inactivated state is less favourable when lidocaine is bound compared to channels that are drug-free. In other words, it indicates that lidocaine hinders transition of Nav1.7 to a slow inactivated state.
For further investigation, we used a PEPD mutation (I1461T), which alters transition between the different inactivated configurations of Nav1.7 channels, as a tool to explore lidocaine inhibition. The I1461T mutation, while not in the theoretical local anaesthetic binding site, decreased use-dependent lidocaine inhibition. The I1461T mutation has no effect on activation of Nav1.7 channels (Fertleman et al., 2006; Jarecki et al., 2008) eliminating altered activation as the culprit of decreased lidocaine inhibition. The presence of a noticeable persistent component during the fast inactivation time course of I1461T currents indicated this mutation attenuates the ability of the Nav1.7 channel to inactivate completely (Fertleman et al., 2006; Jarecki et al., 2008). The anticonvulsant carbamazepine is usually at least partially effective at treating patients with PEPD (Fertleman et al., 2007) and evidence indicates that carbamazepine can inhibit persistent currents generated by I1461T channels (Fertleman et al., 2006). However, it should be noted that Fertleman et al. did not determine if WT and I1461T Nav1.7 channels exhibited differential sensitivity to carbamazepine. In the present study we used fluoride ions in the intracellular solution in order to obtain the prolonged recordings required to investigate interactions between lidocaine inhibition and slow inactivation. Fluoride reduces persistent currents associated with I1461T channels (Jarecki et al., 2008) and here we have focused on lidocaine's inhibition of peak transient currents. Therefore, we cannot address whether lidocaine is able to inhibit persistent currents generated by I1461T channels. However, although lidocaine created a roughly similar shift in the voltage-dependence of fast inactivation for WT and I1461T channels, we found that the I1461T PEPD mutation significantly decreased use-dependent inhibition by lidocaine. Although it has not been reported if patients with I1461T PEPD mutations respond to lidocaine or lidocaine analogues, our data suggest this would not be an effective treatment strategy.
We suspected the decreased use-dependent lidocaine inhibition of 1461T channels involved faster recovery from block. In contrast, I1461T channels recovered much slower than WT channels from long depolarizing pulses under control conditions, suggesting the I1461T mutation increases the probability of the slow inactivation transition. This is consistent with a previous study indicating that impairing fast inactivation of Nav1.4 channels enhances transition to the slow inactivated state (Featherstone et al., 1996). As with WT channels, lidocaine caused faster I1461T channel recovery from long depolarizing pulses, compared with control I1461T channels. This provided additional evidence that the interaction of lidocaine with Nav1.7 channels is hindering the transition to slow inactivation. The disparity in recovery between lidocaine-treated and control I1461T channels is more profound compared with WT channels. This is likely to be due to the innate recovery of I1461T channels being much slower. We conclude decreased stabilization of fast inactivation in I1461T channels allows faster recovery from lidocaine inhibition while not affecting lidocaine's hindrance of channel transition to the slow inactivated state.
For long depolarizing pulses, more I1461T channels changed to slow inactivation, compared with WT channels. This was consistent with slower recovery observed in I1461T channels. In the presence of lidocaine, WT and I1461T channels change more rapidly to a non-conducting state (likely a drug bound state) compared with the innate development of slow inactivation. However, after the fast decrease in current caused by lidocaine, inhibition decreased as pulse duration increased. This result is somewhat inconsistent with previous findings (Chevrier et al., 2004), which did not show a crossover in the profile for development of slow inactivation between control and lidocaine-treated Nav1.7 channels. However, in Chevrier et al. (2004) the conditions for studying Nav1.7 (i.e. expression system, slow inactivation protocol) were different. One possibility is that the interaction between fast and slow inactivated states is different for Nav1.7 channels expressed in Xenopus oocytes from those expressed in HEK293 cells. Although Chevrier et al. concluded that lidocaine enhanced entry into slow inactivation for Nav1.7 channels expressed in Xenopus oocytes, it is not clear if the ‘rapid recovery’ pulse used in their slow inactivation protocol was sufficient to allow for dissociation of lidocaine from fast inactivated channels in Xenopus oocytes.
Previous work in Nav1.4 channels has also indicated local anaesthetics induce slow inactivation (Balser et al. 1996b; Chen et al., 2000; Fukuda et al., 2005). Thus, it is possible that lidocaine is acting as a ‘catalyst’ in the development of slow inactivation for Nav1.7 channels and the decrease in lidocaine effect is simply a faster transition to slow inactivation. However, we feel this is very unlikely because: (i) non-treated WT and I1461T channels produced less current following long depolarizing pulses (Figure 7B and C); and (ii) lidocaine-treated WT and I1461T channels displayed faster recovery after long depolarizing pulses (Figures 3 and 6). Other work in Nav1.2 channels suggests etidocaine, another local anaesthetic, impedes channel entry into slow inactivation (Yarov-Yarovoy et al., 2002). Nav1.2 and Nav1.7 channels are ∼77% identical in their amino acid sequences, but Nav1.4 and Nav1.7 channels are only ∼61% identical. Thus, it is conceivable that lidocaine reduces entry into slow inactivation for Nav1.7 channels but enhances it in Nav1.4 channels.
Overall, the results from this study indicate the interaction of lidocaine with Nav1.7 channels creates a drug-channel conformation that is energetically favourable compared with fast inactivation and, if lidocaine is bound, the transition to a slow inactivated state is less probable. The two-site binding model used to fit the concentration–response curve for lidocaine inhibition of Nav1.7 channels (see Figure 2) was likely to be a result of lidocaine having different affinities for the fast inactivated configuration versus the slow inactivated configuration of the channel. To our surprise, lidocaine enhanced recovery of Nav1.7 channels from prolonged depolarization supporting our hypothesis that lidocaine hinders transition to slow inactivation. We showed also that lidocaine interaction with VGSCs was altered by the I1461T mutation, which: (i) destabilized transition to an inactivated state; (ii) was outside the theoretical local anaesthetic binding site of the channel; and (iii) caused greater transition to slow inactivation. With this increased transition to slow inactivation, the disparity in recovery between lidocaine and non-treated I1461T channels from prolonged depolarization was more pronounced, with lidocaine-treated channels showing faster recovery than in WT channels. More importantly, the fact that the I1461T mutation used in this study is implicated in the pain-causing disorder PEPD suggests local anaesthetic treatment may be less effective for patients expressing this mutation or in painful states in which fast inactivation of Nav1.7 channels is impaired or slow inactivation of Nav1.7 channels is enhanced.
Acknowledgments
This work was supported by NIH Grant NS053422 (T.R.C) and an Indiana Clinical and Translational Sciences Institute Career Development Award (PHS grant 5TL1RR025779) (B.W.J.). The authors thank Jay Z. Yeh for constructive suggestions on this manuscript and thank James O. Jackson II for his technical contribution to the study.
Glossary
Abbreviations
- EGFP
enhanced green fluorescent protein
- HEK293
human embryonic kidney 293
- PEPD
paroxysmal extreme pain disorder
- VGSC
voltage-gated sodium channels
- WT
wild-type
Conflict of interest
None of the authors have a conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser JR, Nuss HB, Orias DW, Johns DC, Marban E, Tomaselli GF, et al. Local anesthetics as effectors of allosteric gating. Lidocaine effects on inactivation-deficient rat skeletal muscle Na channels. J Clin Invest. 1996a;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser JR, Nuss HB, Romashko DN, Marban E, Tomaselli GF. Functional consequences of lidocaine binding to slow inactivated sodium channels. J Gen Physiol. 1996b;107:643–658. doi: 10.1085/jgp.107.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, Cohen CJ, Tsien RW. Lidocaine block of cardiac sodium channels. J Gen Physiol. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Valenzuela C, Chen LQ, Kallen RG. On the molecular nature of the lidocaine receptor of cardiac Na+ channels. Modification of block by alterations in the alpha-subunit III-IV interdomain. Circ Res. 1995;77:584–592. doi: 10.1161/01.res.77.3.584. [DOI] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, et al. Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res Mol Brain Res. 1996;43:117–131. doi: 10.1016/s0169-328x(96)00163-5. [DOI] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Chen Z, Ong BH, Kambouris NG, Marban E, Tomaselli GF, Balser JR. Lidocaine induces a slow inactivated state in rat skeletal muscle sodium channels. J Physiol. 2000;524:37–49. doi: 10.1111/j.1469-7793.2000.t01-1-00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier P, Vijayaragavan K, Chahine M. Differential modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br J Pharmacol. 2004;142:576–584. doi: 10.1038/sj.bjp.0705796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18:9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Na v 1.7 and human pain disorders. Trends Neurosci. 2007;30:555–563. doi: 10.1016/j.tins.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Djouhri L, Newton R, Levinson SR, Berry CM, Carruthers B, Lawson SN. Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel alpha subunit protein. J Physiol. 2003;546:565–576. doi: 10.1113/jphysiol.2002.026559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg-Olofsson O, Elmslie FV, et al. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome) Neurology. 2007;69:586–595. doi: 10.1212/01.wnl.0000268065.16865.5f. [DOI] [PubMed] [Google Scholar]
- Fozzard HA, Hanck DA. Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiol Rev. 1996;76:887–926. doi: 10.1152/physrev.1996.76.3.887. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Nakajima T, Viswanathan PC, Balser JR. Compound-specific Na+ channel pore conformational changes induced by local anaesthetics. J Physiol. 2005;564:21–31. doi: 10.1113/jphysiol.2004.081646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Common mode of action of three agents that decrease the transient change in sodium permeability in nerves. Nature. 1966;210:1220–1222. doi: 10.1038/2101220a0. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki BW, Sheets PL, Jackson JO, II, Cummins TR. Paroxymsal Extreme Pain Disorder mutations within the D3/S4-S5 linker of Nav1.7 cause moderate destabilization of fast-inactivation. J Physiol. 2008;586:4137–4153. doi: 10.1113/jphysiol.2008.154906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14:1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Galue A, Meadows L, Ragsdale DS. A molecular basis for the different local anesthetic affinities of resting versus open and inactivated states of the sodium channel. Mol Pharmacol. 1999;55:134–141. doi: 10.1124/mol.55.1.134. [DOI] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/s0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- Lossin C, Rhodes TH, Desai RR, Vanoye CG, Wang D, Carniciu S, et al. Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J Neurosci. 2003;23:11289–11295. doi: 10.1523/JNEUROSCI.23-36-11289.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. J Physiol. 1998;508:647–657. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau C, Wang SY, Strichartz GR, Wang GK. Point mutations at N434 in D1-S6 of mu1 Na(+) channels modulate binding affinity and stereoselectivity of local anesthetic enantiomers. Mol Pharmacol. 1999;56:404–413. doi: 10.1124/mol.56.2.404. [DOI] [PubMed] [Google Scholar]
- Nau C, Wang SY, Wang GK. Point mutations at L1280 in Nav1.4 channel D3-S6 modulate binding affinity and stereoselectivity of bupivacaine enantiomers. Mol Pharmacol. 2003;63:1398–1406. doi: 10.1124/mol.63.6.1398. [DOI] [PubMed] [Google Scholar]
- Ong BH, Tomaselli GF, Balser JR. A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J Gen Physiol. 2000;116:653–662. doi: 10.1085/jgp.116.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Sangameswaran L, Fish LM, Koch BD, Rabert DK, Delgado SG, Ilnicka M, et al. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem. 1997;272:14805–14809. doi: 10.1074/jbc.272.23.14805. [DOI] [PubMed] [Google Scholar]
- Sheets PL, Jackson JO, Waxman SG, Dib-Hajj SD, Cummins TR. A Nav1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol. 2007;581(Pt 3):1019–1031. doi: 10.1113/jphysiol.2006.127027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RE. Effect of procaine on electrical properties of squid axon membrane. Am J Physiol. 1959;196:1071–1078. doi: 10.1152/ajplegacy.1959.196.5.1071. [DOI] [PubMed] [Google Scholar]
- Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci USA. 1997;94:1527–1532. doi: 10.1073/pnas.94.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantham V, Cannon SC. The position of the fast-inactivation gate during lidocaine block of voltage-gated Na+ channels. J Gen Physiol. 1999;113:7–16. doi: 10.1085/jgp.113.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Nau C, Wang GK. Residues in Na(+) channel D3-S6 segment modulate both batrachotoxin and local anesthetic affinities. Biophys J. 2000;79:1379–1387. doi: 10.1016/S0006-3495(00)76390-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BS, Felix JP, Priest BT, Brochu RM, Dai K, Hoyt SB, et al. Characterization of a new class of potent inhibitors of the voltage-gated sodium channel Nav1.7. Biochemistry. 2007;46:14693–14703. doi: 10.1021/bi7018207. [DOI] [PubMed] [Google Scholar]
- Wright SN, Wang SY, Wang GK. Lysine point mutations in Na+ channel D4-S6 reduce inactivated channel block by local anesthetics. Mol Pharmacol. 1998;54:733–739. [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Brown J, Sharp EM, Clare JJ, Scheuer T, Catterall WA. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J Biol Chem. 2001;276:20–27. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall WA. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J Biol Chem. 2002;277:35393–35401. doi: 10.1074/jbc.M206126200. [DOI] [PubMed] [Google Scholar]
- Yeh JZ. Sodium inactivation mechanism modulates QX-314 block of sodium channels in squid axons. Biophys J. 1978;24:569–574. doi: 10.1016/S0006-3495(78)85403-4. [DOI] [PMC free article] [PubMed] [Google Scholar]