

Abstract
BACKGROUND AND PURPOSE
Huntington's disease (HD) is a progressive neurodegenerative disorder characterized by a degeneration of striatal neurons. The possible role of COX and lipoxygenase (LOX) pathways has been well-documented in the pathology of several neurodegenerative disorders including HD. Licofelone is a competitive inhibitor of COX-1– and COX-2 and 5-LOX isoenzymes. Therefore, the present study was designed to investigate possible neuroinflammatory and apoptotic mechanisms in the neuroprotective effect of licofelone against 3-nitropropionic acid (3-NP)-induced HD-like symptoms in rats.
EXPERIMENTAL APPROACH
Rats were administered 3-NP (10 mg·kg−1day−1, i.p.) for 14 days. Licofelone (2.5, 5 and 10 mg·kg−1, p.o.) was given once a day, 1 h before 3-NP treatment for 14 days. Body weight and behavioural parameters (locomotor and rotarod activity) were assessed on the 1st, 5th, 10th and 15th day post-3-NP administration. Malondialdehyde, nitrite concentration, endogenous antioxidant enzymes (superoxide dismutase and catalase levels), mitochondrial enzyme complexes, pro-inflammatory compounds (TNF-α, IL-6, NF-κB), PGs (PGE2 and PGF2α) and caspase-3 activity were measured on day 15 in the striatum.
KEY RESULTS
Systemic 3-NP treatment significantly reduced body weight, locomotor activity, oxidative defence, mitochondrial enzyme complex activities and increased TNF-α, IL-6, caspase-3 activity, NF-κB and PGE2 and PGF2α levels in the striatum. Licofelone (2.5, 5 and 10 mg·kg−1) significantly attenuated the impairment in behavioural, biochemical and mitochondrial, pro-inflammatory and pro-apoptotic markers as compared with vehicle-treated group.
CONCLUSIONS AND IMPLICATIONS
The results demonstrate the involvement of pro-inflammatory compounds and the apoptotic cascade in the neuroprotective effect of licofelone against 3-NP-induced neurotoxicity.
Keywords: caspase-3, licofelone, mitochondria, NF-κB, 3-nitropropionic acid, oxidative stress, striatum, TNF-α
Introduction
Huntington's disease (HD) is a neurodegenerative disorder of complex neuropathology. It is characterized by ataxia, choreiform movements and dementia, and affects medium spiny neurons in the striatum (Gil and Rego, 2008). Recently, oxidative stress has been shown to be involved in the neurobiology of HD, although the cause of oxidative stress is uncertain (Brown et al., 1999). Recent reports show that oxidative stress activates the apoptotic and neuroinflammatory processes that trigger cell death in neurodegenerative diseases (Liot et al., 2009). In addition, results have been obtained that suggest excitotoxic events and mitochondrial dysfunction are involved in the pathophysiology of HD (Keller et al., 1998; Kumar and Kumar, 2009a). Apoptosis is a complex process involving activation of proteases of the caspase family, alterations in plasma membrane phospholipids, nuclear DNA condensation and fragmentation (Keller et al., 1998; Yang et al., 2004). Apoptosis has also been implicated in neurodegenerative diseases including HD and associated with mitochondrial dysfunction (Portera-Cailliau et al., 1995; Keller et al., 1998). Neuroinflammation is a key event involved in neuronal death and axonal degeneration (Mrak and Griffin, 2005). Prostaglandins are important mediators of the inflammatory process and a potent indicator of in vivo COX-mediated inflammatory processes (Mrak and Griffin, 2005). Clinical evidence also suggests that excess TNF-α is centrally involved in the pathogenesis of neurological diseases (Tobinick and Gross, 2008). A greater understanding of the apoptotic and neuroinflammatory cascades that mediate these effects, as well as their molecular interaction, can be used to elucidate the pathology of HD and aid the development of new drugs suitable to treat this and other neurodegenerative diseases. 3-Nitropropionic acid (3-NP) is a fungal toxin that produce HD-like symptoms both in animals and humans (Ludolph et al., 1991; Kumar and Kumar, 2009b). 3-NP irreversibly blocks the enzyme succinate dehydrogenase (SDH), decreases ATP levels and accelerates neuronal apoptosis (Coles et al., 1979; Tunez et al., 2006). 3-NP induces oxidative stress, neuroinflammation and excitotoxicity, all of which are associated with the pathogenesis of HD.
Licofelone ([2,2-dimethyl-6-(4-chloropheny-7-phenyl-2,3-dihydro-1H-pyrrazoline-5-yl] acetic acid), a competitive inhibitor of 5-lipoxygenase (5-LOX), COX-1 and COX-2, is currently in the clinical developmental phase for the treatment of osteoarthritis. Licofelone decreases significantly the production of pro-inflammatory mediators (leukotrienes and prostaglandins), which are involved in the pathophysiology of several neurological disorders (Singh et al., 2006). Both COX and 5-LOX inhibitors, alone or in combination, have been reported to have neuroprotective effects against several motor disorders (Bishnoi et al., 2005; 2006; 2007; Kalonia et al. 2009a,b,c;). However, their exact cellular mechanism is still not understood.
Therefore, the present study was designed to investigate the possible neuroinflammatory and apoptotic mechanisms in the neuroprotective effect of licofelone against 3-NP-induced HD-like symptoms in rats.
Methods
Animals
Male Wistar rats (300–350 g) bred in the Central Animal House of Panjab University, Chandigarh, were used. Animals were acclimatized to laboratory conditions prior to experimentation. The animals were kept in plastic cages (two to three in each cage) under hygienic conditions with a 12 h light and dark cycle with food and water ad libitum. All experiments were carried out between 09 h 00 min and 15h00 min. The protocol was approved by the Institution's Animal Ethics Committee and was carried out in accordance with the Indian National Science Academy Guidelines for use and care of animals.
Materials
3-NP (Sigma Chemicals, St. Louis, MO), licofelone animals (rats) from Central Animal House, Panjab University; sodium carboxymethyl cellulose (CMC) (Hi-Media, New Delhi, India), saline, BSA (Hi-Media, New Delhi); EGTA, elisa kit; actophotometer (IMCORP, Ambala, India); rotarod apparatus (Techno, Ambala, India); spectrophotometer (Norwalk, CT).
Drugs and treatment schedule
The following drugs were used in the present study. 3-NP was dissolved in saline (adjusted to pH 7.4) and administered i.p. in a volume of 0.5 mL 100 g−1 animal body weight. Licofelone was suspended in a 0.5% (w v−1) CMC solution and administered p.o., through a cannula, in a constant volume of 0.5 mL 100 g−1 body weight. Animals were randomly divided into six groups, consisting of 10–15 rats in each.
Group 1, vehicle-treated group (n = 15); group 2, vehicle + diet restriction-treated group (n = 10); group 3 received 3-NP (10 mg·kg−1, i.p.) for 14 days (n = 15); group 4 received licofelone (10 mg·kg−1 p.o.) alone (n = 12); groups 5 to 7 received licofelone (2.5, 5 and 10 mg·kg−1) + 3-NP (10 mg·kg−1, i.p.) for 14 days (n = 15 in each group).
Diet restriction protocol
All animals received food and water ad libitum. One week before the beginning of the experiment, food intake was recorded in order to know the average daily consumption of each rat. In the restricted diet-treated groups, animals received 15 g of commercial diet, which corresponded to 50% of the average amount of diet consumed by the animals in the previous week.
Measurement of body weight
Animal body weight was recorded on the first and last days of experiment; % change in body weight was calculated:
Behavioural assessments
Assessment of gross behavioural activity (locomotor activity)
The locomotor activity was monitored using an actophotometer. The motor activity was detected by infrared beams above the floor of the testing area. Animals were placed individually in the activity chamber for a 3 min acclimatization period before the actual activity tasks were started. Each animal was observed over a period of 5 min, and activity was expressed as counts 5 min−1 (Kumar et al., 2007).
Rotarod activity
The motor co-ordination and grip performance of the animals were evaluated using the rotarod apparatus. The rats were exposed to a prior training session to acclimatize them to rotarod performance. Rats were placed on a rotating rod with a diameter of 7 cm (speed, 25 r.p.m.). The cut-off time was 180 s, and each rat performed three separate trials after a 5 min gap. The average time of the fall was recorded (Kumar and Kumar, 2009a).
Dissection and homogenization
On the 15th day, animals were randomly divided into two groups, one for biochemical estimations and the other for mitochondrial enzyme complex estimations immediately after the behavioural assessments. The brains were dissected out. The striatum was separated and placed on ice. A 10% (w v−1) tissue homogenate was prepared in 0.1 M phosphate buffer (pH 7.4). The homogenate was centrifuged at 10 000×g for 15 min. Aliquots of the supernatant was separated and used for biochemical estimations.
Measurement of oxidative stress parameters
Measurement of lipid peroxidation
The quantitative measurement of lipid peroxidation in the brain striatum was performed according to the method of Wills (1966). The amount of malondialdehyde (MDA), a measure of lipid peroxidation, was measured by reaction with thiobarbituric acid at 532 nm using a Perkin Elmer lambda 20 spectrophotometer. The values were calculated using the molar extinction coefficient of the chromophore (1.56 × 105 M−1 cm−1) and expressed as a percentage of the vehicle-treated group (the protein concentration was 0.876 mg·mL−1).
Estimation of nitrite
The accumulation of nitrite in the striatum supernatant, an indicator of the production of NO, was determined by a colorimetric assay with Greiss reagent [0.1% N-(1-naphthyl) ethylenediame dihydrochloride, 1% sulphanilamide and 2.5% phosphoric acid] as described by Green et al. (1982). Equal volumes of supernatant and Greiss reagent were mixed, and this mixture was incubated for 10 min at room temperature in the dark. Absorbance at 540 nm was measured with a Perkin Elmer lambda 20 spectrophotometer. The concentration of nitrite in the supernatant was determined from a sodium nitrite standard curve and expressed as a percentage of the vehicle-treated group (the protein concentration was 0.876 mg·mL−1).
Catalase estimation
Catalase activity was assayed by the method of Luck (1971), in which the breakdown of hydrogen peroxide (H2O2) is measured at 240 nm. Briefly, the assay mixture consisted of 12.5 mM H2O2 in phosphate buffer (50 mM of pH 7.0) and 0.05 mL of supernatant from the striatum tissue homogenate (10%), and the change in absorbance was recorded at 240 nm. The results are expressed as mM of H2O2 decomposed mg−1 protein min−1 (the protein concentration was 0.876 mg·mL−1).
Superoxide dismutase (SOD) activity
SOD activity was assayed according to the method of Kono (1978), in which the inhibition of the reduction of nitrazobluetetrazolium (NBT) by SOD is measured at 560 nm using a spectrophotometer. Briefly, the reaction was initiated by the addition of 20 mM of hydroxylamine hydrochloride to the mixture containing 96 mM of NBT and 0.1 mL of the sample (striatum homogenate). The results are expressed as U mg−1 protein (the protein concentration was 0.876 mg·mL−1).
Protein estimation
The protein concentration was measured by the Biuret method using BSA as a standard (Gornall et al., 1949).
Mitochondrial complex estimation
Isolation of rat brain mitochondria
Rat brain mitochondria were isolated by the method of Berman and Hastings (1999). The brain striatum was homogenized in isolation buffer with EGTA (215 mM mannitol, 75 mM sucrose, 0.1% BSA, 20 mM hepes, 1 mM EGTA, pH 7.2). The homogenate was centrifuged at 13 000×g for 5 min at 4°C. The pellet was resuspended in isolation buffer with EGTA and spun again at 13 000×g for 5 min. The resulting supernatant was transferred to new tubes and topped off with isolation buffer containing EGTA and spun again at 13 000×g for 10 min. The pellet containing pure mitochondria was resuspended in isolation buffer without EGTA (the protein concentration was 0.676 mg·mL−1).
Complex I (NADH dehydrogenase activity)
Complex I was measured spectrophotometrically by the method of King and Howard (1967). The method involves the catalytic oxidation of NADH to NAD+ with subsequent reduction of cytochrome C. The reaction mixture contained 0.2 M glycyl glycine buffer, pH 8.5, 6 mM NADH in 2 mM glycyl glycine buffer and 10.5 mM cytochrome C. The reaction was initiated by the addition of a requisite amount of solubilized mitochondrial sample. The absorbance change at 550 nm was followed for 2 min (the protein concentration was 0.676 mg·mL−1).
Complex II (SDH activity)
SDH was measured spectrophotometrically according to the method of King (1967). The method involves the oxidation of succinate by an artificial electron acceptor, potassium ferricyanide. The reaction mixture contained 0.2 M phosphate buffer pH 7.8, 1% BSA, 0.6 M succinic acid and 0.03 M potassium ferricyanide. The reaction was initiated by addition of the striatum mitochondrial sample, and the absorbance change at 420 nm was followed for 2 min (the protein concentration was 0.676 mg·mL−1).
Complex IV (cytochrome oxidase assay)
Cytochrome oxidase activity was assayed in brain mitochondria according to the method of Sottocasa et al. (1967). The assay mixture contained 0.3 mM reduced cytochrome C in 75 mM phosphate buffer. The reaction was initiated by addition of the solubilized striatum mitochondrial sample, and absorbance change at 550 nm was followed for 2 min (the protein concentration is 0.676 mg·mL−1).
MTT assay
The 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl-H-tetrazolium bromide (MTT) assay is based on the reduction of MTT by hydrogenase activity in functionally intact mitochondria. The MTT reduction rate was used to assess reducing activity of the mitochondrial samples in isolated striatum mitochondria by the method of Liu et al. (1997). Briefly, 100 µL striatum mitochondrial samples were incubated with 10 µL MTT for 3 h at 37°C. The blue formazan crystals were solubilized with dimethyl sulphoxide and measured by an elisa reader with a 580 nm filter (Model 680 Microplate Reader, Bio-Rad, Japan) (the protein concentration was 0.676 mg·mL−1).
Preparation of lysates from tissue
(i)Cytoplasmic fraction collection
Striatum was weighed and cut into small pieces using a clean razor blade followed by wash with 5 mL of cold 1X PBS–PMSF. The striatal pieces were placed in a clean homogenizer, 5 µL of 1 M DTT and 500 µL of 10% detergent solution was added to 4.495 mL of ice-cold 1X hypotonic buffer g−1 tissue and then homogenized. The homogenate was incubated with ice for 15 to 30 min (whole-cell lysate), centrifuged for 10 min at 5590× g at 4°C, and the supernatant (cytoplasmic fraction; concentration, 1.1 mg·mL−1) was transferred to a 15 mL tube and stored at 4°C. The pellet was the nuclear fraction and concentration was 1 mg·mL−1.
(ii)Nuclear fraction collection
The nuclear pellet (concentration 1 mg·mL−1) was resuspended in 500 µL nuclear lysis buffer by pipetting up and down. The suspension was vigorously vortexed and incubated at 4°C for 30 min on a rocking platform. The suspension was centrifuged at 10956× g for 10 min at 4°C in a microcentrifuge. The supernatant (nuclear fraction; concentration is 0.91 mg·mL−1) was transferred into a pre-chilled microcentrifuge tube and stored at −80°C until further use. The protein concentration of the nuclear extract was 1 mg·mL−1.
Estimation of TNF-α and IL-6 in striatum
The quantifications of TNF-α and IL-6 were done by rat TNF-α and IL-6 immunoassay kit (R&D Systems, Minneapolis, MN). The Quantikine rat TNF-α and IL-6 immunoassay is a 4.5 h solid phase elisa designed to measure rat TNF-α and IL-6 levels. It is a solid-phase sandwich elisa using a microtitre plate reader. Concentrations of TNF-α were calculated from the standard curves. The protein concentration of the nuclear extract was 1 mg·mL−1.
Caspase-3 colorimetric assay in striatum
Caspase-3, also known as CPP-32, Yama or Apopain, is an intracellular cysteine protease that exists as a pro-enzyme, becoming activated during the cascade of events associated with apoptosis. The enzymatic reaction for caspase activity was carried out as using an Imgenex (San Diego, CA) caspase-3 colorimetric kit. The protein concentration of the nuclear extract was 1 mg·mL−1.
Quantification of NF-κB p65 subunit in striatum
The NF-κB/p65 Activ elisa (Imgenex) kit was used to measure NF-κB-free p65 in the nuclear lysate of the rat striatum. The nuclear levels of p65 may correlate positively with the activation of NF-κB pathway. The anti-p65 antibody-coated plate captures free p65, and the amount of bound p65 was detected by adding a second anti-p65 antibody followed by alkaline phosphatase (AKP)-conjugated secondary antibody using colorimetric detection in an elisa plate reader at 405 nm. The protein concentration of the nuclear extract was 1 mg·mL−1.
Estimation of PGE2 levels in striatum
The quantitative determination of PGE2 levels was done by using a PGE2 assay kit (R&D Systems). Briefly, the assay is based on the competitive binding technique in which PGE2 present in a sample competes with a fixed amount of horseradish peroxidase (HRP)-labelled PGE2 for sites on a mouse monoclonal antibody. During the incubation, the mouse monoclonal antibody becomes bound to the goat anti-mouse antibody coated onto the microplate. Following a wash to remove excess conjugate and unbound sample, a substrate solution is added to the wells to determine the bound enzyme activity. The colour development is stopped, and the absorbance is read at 450 nm. The intensity of the colour is inversely proportional to the concentration of PGE2 in the sample. The protein concentration of the nuclear extract was 1 mg·mL−1.
Estimation of PGF2α levels in striatum
The quantitative determination of PGF2α levels was done by using the PGF2α EIA kit (Cayman Chemical Co., Ann Arbor, MI, USA). The assay is carried out as per the procedure given by the manufacturer. The absorbance of the colour development due to the enzymatic reaction is read at 412 nm. The intensity of the colour is inversely proportional to the free amount of PGF2α present in the sample during incubation. The protein concentration of the nuclear extract was 1 mg·mL−1.
Statistical analysis
The data were analysed by one-way and two-way anova, followed by Tukey's test and two-way anova followed by Fisher's least significant difference test, respectively (Sigmastat 2.0 version, Cranes Software International Ltd., Bangalore, India). All the values are expressed as mean ± SEM. In all the tests, the criterion for statistical significance was P < 0.05.
Results
Effect of licofelone on body weight, locomotor activity and motor coordination in 3-NP-treated rats
Systemic treatment with 3-NP (10 mg·kg−1) significantly reduced body weight (Figure 1) and impaired locomotor (Figure 2) and motor co-ordination (rotarod) (Figure 3) on the 15th day as compared with the vehicle-treated group. The diet restriction followed by vehicle treatment did not significantly affect body weight, locomotor and rotarod activities of the animals as compared with vehicle alone-treated groups. Furthermore, licofelone (2.5, 5 and 10 mg·kg−1, p.o.) treatment attenuated the reduction in body weight, impairment in locomotor and motor co-ordination (rotarod) as compared with 3-NP-treated rats (P < 0.05). Licofelone (10 mg·kg−1, p.o.) per se did not show any significant effect on the behavioural parameters as compared with vehicle-treated group.
Figure 1.
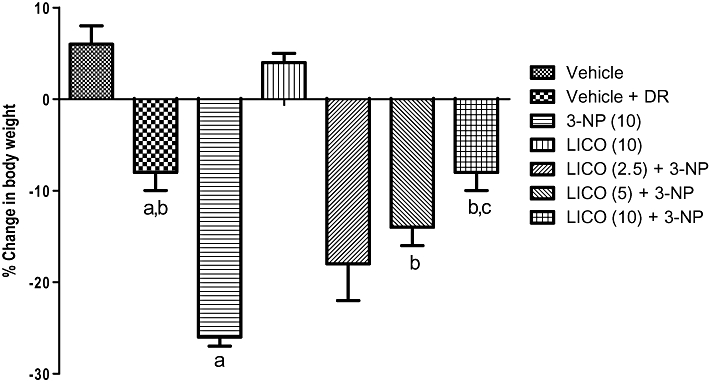
Effect of licofelone (LICO) on body weight in 3-NP-treated rats. Group 1, vehicle (n = 15); groups 2, vehicle + DR (n = 10); group 3, 3-NP (10 mg·kg−1) (n = 15); group 4, LICO (10 mg·kg−1) (n = 12); group 5, LICO (2.5 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15); group 6, LICO (5 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15); group 7, LICO (10 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15). aP < 0.05 versus vehicle-treated group, bP < 0.05 versus 3-NP, cP < 0.05 versus [LICO (2.5) + 3-NP]-treated group (one-way anova followed by Tukey's test). Data presented are the percentage of the vehicle-treated group (mean ± SEM). DR, diet restriction.
Figure 2.
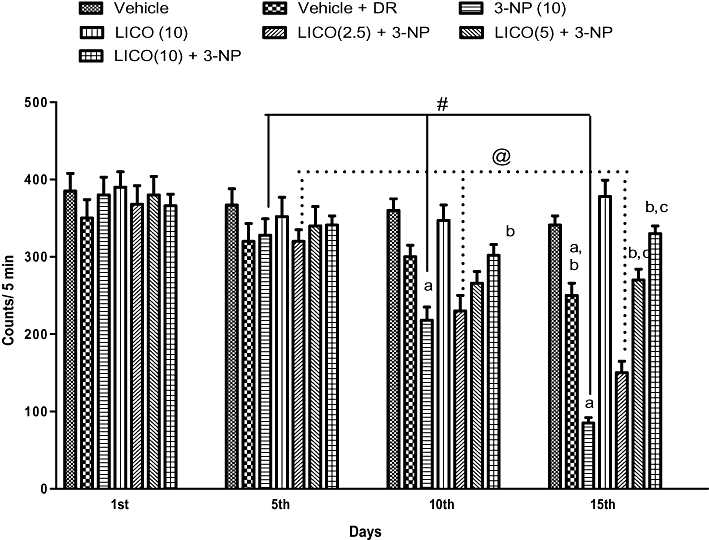
Effect of licofelone (LICO) on locomotor activity in 3-NP-treated rats. Group 1, vehicle (n = 15); group 2, vehicle + DR (n = 10); group 3, 3-NP (10 mg·kg−1) (n = 15); group 4, LICO (10 mg·kg−1) (n = 12); group 5, LICO (2.5 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15); group 6, LICO (5 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15); group 7, LICO (10 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15). aP < 0.05 versus vehicle-treated group, bP < 0.05 versus 3-NP, #P < 0.05 versus 3-NP treatment on 5th, 10th and 15th day, @P < 0.05 versus [LICO(2.5) + 3-NP] treated group on 5th, 10th and 15th days, cP < 0.05 versus [LICO(2.5) + 3-NP] treated group (two-way anova followed by Fisher's least significant difference test). Data presented are the percentage of the vehicle-treated group (mean ± SEM). DR, diet restriction.
Figure 3.
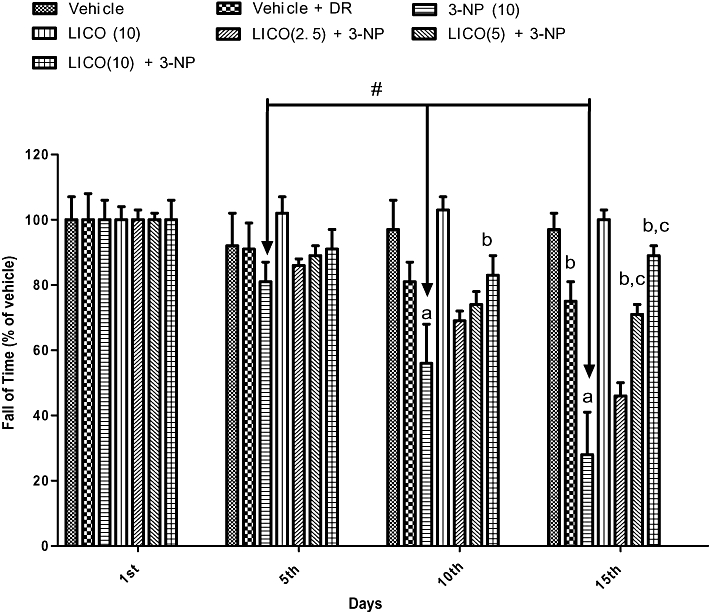
Effect of licofelone (LICO) on rotarod activity in 3-NP-treated rats. Group 1, vehicle (n = 15); group 2, vehicle + DR (n = 10); group 3, 3-NP (10 mg·kg−1) (n = 15); group 4, LICO (10 mg·kg−1) (n = 12); group 5, LICO (2.5 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15); group 6, LICO (5 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15); group 7, LICO (10 mg·kg−1) + 3-NP (10 mg·kg−1) (n = 15). aP < 0.05 versus vehicle-treated group, bP < 0.05 versus 3-NP, #P < 0.05 versus 3-NP treatment on 5th, 10th and 15th days. cP < 0.05 versus [LICO (2.5) + 3-NP] treated group (two-way anova followed by Fisher's least significant difference test). Data presented are the percentage of the vehicle-treated group (mean ± SEM). DR, diet restriction.
Effect of licofelone on lipid peroxidation, nitrite, SOD and catalase in striatum after 3-NP treatment
Systemic administration of 3-NP significantly increased lipid peroxidation, nitrite concentration and depleted SOD and catalase enzyme activity in the striatum as compared with the vehicle-treated group. Licofelone (2.5, 5 and 10 mg·kg−1, p.o.) significantly attenuated lipid peroxidation, nitrite concentration and restored endogenous antioxidant enzyme (SOD and catalase) activities as compared with the 3-NP-treated animals (P < 0.05) (Table 1). Furthermore, licofelone (10 mg·kg−1, p.o.) per se did not have a significant effect on lipid peroxidation, nitrite concentration, SOD and catalase enzyme activity as compared with the vehicle-treated group (Table 1).
Table 1.
Effect of licofelone on 3-NP treatment-induced biochemical changes in the striatum
| Treatment (mg·kg−1) | MDA, nmol mg−1 protein (% of vehicle) | Nitrite level, µmol mg−1 protein (% of vehicle) | SOD, U mg−1 protein (% of vehicle) | Catalase, µmol of H2O2 decomposed min−1 mg−1 protein (% of vehicle) |
|---|---|---|---|---|
| Vehicle | 100 ± 5.6 | 100 ± 6.3 | 100 ± 8.9 | 100 ± 6.6 |
| 3-NP (10) | 253 ± 5.5a | 195 ± 3.4a | 49 ± 4.0a | 55 ± 4.0a |
| LICO (10) | 93 ± 9.2 | 95 ± 5.7 | 101 ± 5.8 | 101 ± 6.9 |
| LICO (2.5) + 3-NP | 213 ± 3.3b | 177 ± 4.3 | 62 ± 4.3 | 64 ± 4.5 |
| LICO (5) + 3-NP | 173 ± 6.5b,c | 145 ± 6.8b | 78 ± 4.4b | 85 ± 7.5b |
| LICO (10) + 3-NP | 113 ± 10.3b,c,d | 114 ± 6.0b,c | 97 ± 6.8b,c | 94 ± 4.2b |
Values expressed as % of vehicle-treated group.
P < 0.05 versus vehicle
P < 0.05 versus 3-NP
P < 0.05 versus [LICO (2.5) + 3-NP]
P < 0.05 versus [LICO (5) + 3-NP].
Effect of licofelone on striatum mitochondrial enzyme complex levels in 3-NP-treated rats
Systemic 3-NP (10 mg·kg−1) administration significantly impaired all the mitochondrial enzyme complexes (I, II and IV) and MTT (mitochondrial redox) activity as compared with the vehicle-treated group. Licofelone (2.5, 5 and 10 mg·kg−1, p.o.) treatment significantly restored mitochondrial enzyme complex (I, II and IV) and MTT activities as compared to the 3-NP-treated group (P < 0.05) (Figure 4). Licofelone (10 mg·kg−1, p.o.) per se did not have a significant effect on mitochondrial enzyme complexes as compared with the vehicle-treated group.
Figure 4.
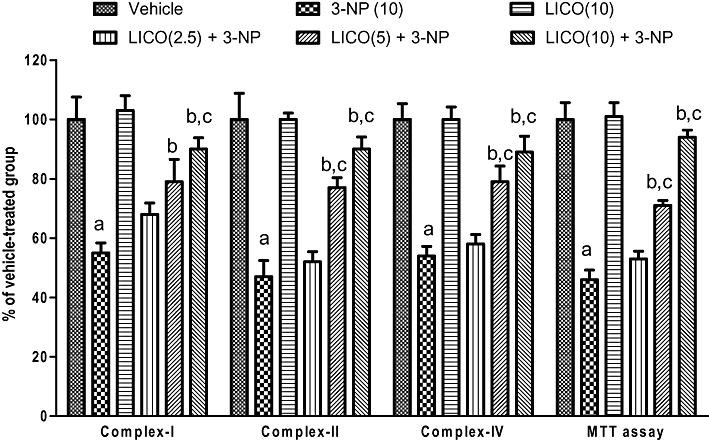
Effect of licofelone (LICO) on mitochondrial complexes I, II, IV and MTT assay in striatum in 3-NP-treated rats. aP < 0.05 versus vehicle-treated group, bP < 0.05 versus 3-NP, cP < 0.05 versus [LICO(2.5) + 3-NP] treated group (one-way anova followed by Tukey's test). Data presented are the percentage of the vehicle-treated group (mean ± SEM).
Effect of licofelone on NF-κB p65, TNF-α, IL-6 and caspase-3 activities in 3-NP-treated rats
Systemic 3-NP (10 mg·kg−1) treatment for 14 days significantly increased the levels of pro-inflammatory markers (NF-κB, p65, TNF-α, IL-6) as well as apoptosis (caspase-3) (Figure 5) as compared with vehicle-treated group. Licofelone (5 and 10 mg·kg−1) pretreatment significantly attenuated the levels of NF- κB, p65, TNF-α, IL-6 and caspase-3 levels in the striatum as compared with 3-NP-treated group (Figure 5). The lower dose of licofelone (2.5 mg·kg−1) failed to modify the increased levels of these neuroinflammatory markers (NF-κB, p65, TNF-α, IL-6) as well as (caspase-3) activity in the striatum induced by 3-NP treatment.
Figure 5.
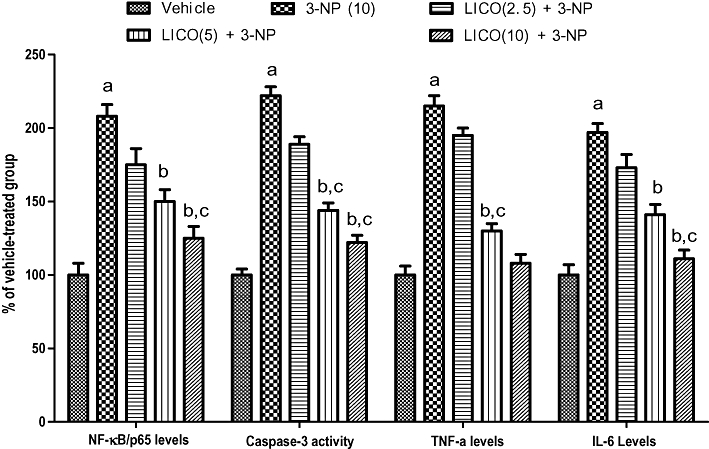
Effect of licofelone (LICO) on NF-κB/p65, IL-6, caspase-3 and TNF-α in striatum in 3-NP-treated rats. aP < 0.05 versus vehicle-treated group, bP < 0.05 versus 3-NP, cP < 0.05 versus [LICO(2.5) + 3-NP] treated group (one-way anova followed by Tukey's test). Data presented are the percentage of the vehicle-treated group (mean ± SEM).
Effect of licofelone on PGE2 and PGF2α activities in 3-NP-treated rats
Systemic 3-NP (10 mg·kg−1) treatment for 14 days significantly increased the levels of pro-inflammatory mediators (PGE2 and PGF2α) levels (Figure 6) as compared with vehicle-treated group. Licofelone (10 mg·kg−1) pretreatment significantly attenuated the levels of PGE2 and PGF2α in the striatum as compared with the 3-NP-treated group (Figure 6).
Figure 6.
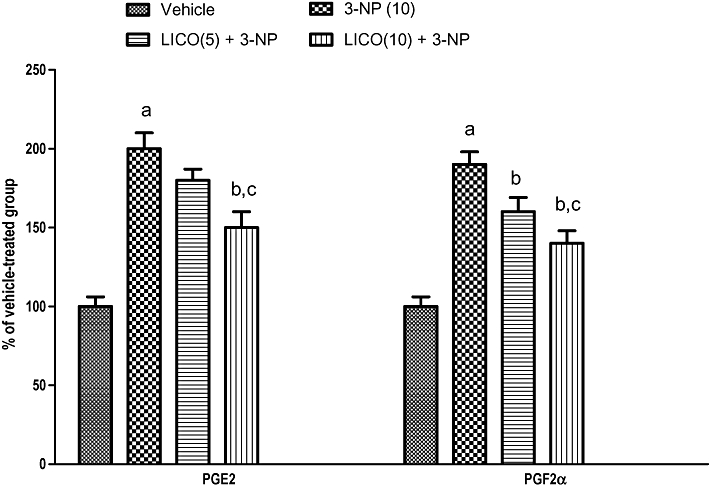
Effect of licofelone (LICO) on PGE2 and PGF2α in striatum after 3-NP treatment. aP < 0.05 versus vehicle-treated group, bP < 0.05 versus 3-NP, cP < 0.05 versus [LICO(2.5) + 3-NP] treated group (one-way anova followed by Tukey's test). Data presented are the percentage of the vehicle-treated group (mean ± SEM).
Discussion
Clinical use of COX inhibitors has been limited due to their side effects, mainly on the cardiovascular system (Fosslien, 2005). Researchers are now focusing on the alteration of another new class of anti-inflammatory drugs named dual COX/LOX inhibitors, and in particular dual COX/5-LOX inhibitors may represent an excellent substitute (Bishnoi et al., 2005; 2006; 2007; Kalonia et al., 2009a,b,c;). Based on the above reports, the present study is an attempt to explore the neuroprotective mechanism of licofelone (dual COX/5-LOX inhibitors) against 3-NP-induced behavioural, biochemical and cellular alterations in rats.
Systemic administration of 3-NP in rats and non-human primates produces homogeneous blockade of complex II within the brain (Kumar and Kumar, 2009a,b,c,d,e;), leading to preferential excitotoxic striatal degeneration associated with behavioural abnormalities that are highly reminiscent of HD (Brouillet et al., 1999; 1993; Bizat et al., 2003). The 3-NP experiment model mimics both the hyperkinetic and hypokinetic symptoms of HD, depending upon the duration of administration (Borlongan et al., 1997). In the present study, systemic administration of 3-NP for 14 days significantly reduced locomotor activity (hypokinetic movements), body weight and motor in coordination (grip strength performance) in rats, suggesting that the effects of 3-NP most probably mimic either the juvenile onset or late stages of HD-like behaviour. The diet restriction with vehicle group also showed alterations in the locomotor and rotarod activity compared with the control group. However, these were less marked than those produced in the 3-NP-treated group. Earlier reports from our laboratory had confirmed that 3-NP caused impairment in motor and cognitive functions, oxidative defence and mitochondrial enzyme complex activities in rats (Kumar et al., 2006; 2007; Kumar and Kumar, 2008; 2009a,b,c,d,e;). Observations using the this mitochondrial toxin (3-NP) also support the so-called ‘indirect excitotoxic’ hypothesis for HD (Kumar et al., 2006; 2007; Kumar and Kumar, 2008). Hyperstimulation of NMDA receptors leading to a massive calcium influx activates calcium-dependent PLA2 cascades (Arzberger et al., 1997). Furthermore, PLA2 cleaves membrane phospholipids to yield arachidonic acid (AA), which is converted into PGs by COX (Hurley et al., 2002). Licofelone treatment significantly attenuated the alterations in body weight, locomotor and grip strength performances, suggesting its therapeutic potential against HD-like behaviour. These findings are comparable with those from an earlier report involving a combination of COX-2 and 5-LOX inhibitors, indicating efficacy of COX-2/5-LOX inhibitors in quinolinic acid induced HD-like conditions (Kalonia et al. 2009a,b,c;).
There is substantial evidence that oxidative damage significantly contributes to the pathogenesis of several neurodegenerative diseases including HD (Kumar et al., 2007; Kumar and Kumar, 2009a,b,c,d,e;). In the present study, 3-NP significantly increased oxidative damage as evidenced by the increase in MDA, nitrite and the depletion of endogenous antioxidant defence enzyme (SOD and catalase) activities in the striatum. Disruption of the mitochondrial enzyme complex activity is associated with reactive oxygen species (ROS). In the present study, 3-NP significantly impaired mitochondrial enzyme complex activities (complexes I, II, IV) and mitochondrial redox activity in the striatum. Licofelone pretreatment significantly ameliorated the levels of oxidative stress parameters and restored the mitochondrial enzyme complexes activities in the striatum, suggesting it has antioxidant-like properties in addition to its inhibitory effects on the actions of COX/LOX. Several studies have also highlighted the possibility that individual COX-2 and 5-LOX inhibitors improve the activities of mitochondrial enzyme complexes, and that this could be one of the possible reasons for its beneficial effects against neurotoxin-induced neuronal degeneration (Bishnoi et al., 2005; 2006; 2007; Kalonia et al., 2009a,b,c;). Previous reports from our laboratory have also documented the therapeutic potential of COX-2/5-LOX inhibitors in HD-like conditions (Kalonia et al., 2009a; 2010;). Evidence has been obtained indicating that the expression of LOX is enhanced during a stimulus-evoked neurodegeneration (Bishnoi et al., 2007; Kalonia et al., 2009a,b;). The 5-LOX translocation and subsequent production of LTs increase in neurodegenerative processes (Qu et al., 2000), and an 5-LOX inhibitor can reduce oedema as well as ameliorate the traumatic and excitotoxic events associated with brain injury (Arai et al., 2001). Previous reports have also demonstrated that 5-LOX pathways are activated during excitotoxic brain injury and that licofelone has a protective effect in animal models of this condition (Kulkarni and Singh, 2007; 2008;).
Caspase-3 is considered to be a key protease responsible for many of the biological and morphological features of apoptosis (Tunez et al., 2006). Bizat and coworkers examined the involvement of death proteases of the caspase family and Ca2+-activated neutral protease calpains in 3-NP toxicity in rats (Bizat et al., 2003). In the present study, systemic 3-NP treatment for 14 days significantly increased caspase-3 activity, indicating the activation of apoptosis in the striatum. Licofelone pretreatment significantly suppressed caspase-3 activity suggesting it has an anti-apoptotic effect.
Members of the NF-κB family of transcription factors are activated within the CNS in the pathological settings of apoptosis and neurological diseases (Meffert and Baltimore, 2005). A variety of stimuli are known to activate NF-κB, which includes cytokines (e.g. TNF and IL-1) and oxidative stress (Meffert and Baltimore, 2005), but evidence for the involvement of NF-κB p65, TNF-α and IL-6 in the pathogenesis of 3-NP-induced neurotoxicity is lacking. In the present study 3-NP was found to significantly increase these neuroinflammatory markers (NF-κB, p65, TNF-α) and activate the apoptosis pathways (caspase-3) in the striatum. Furthermore, prostaglandin (PGE2 and PGF2α) levels were also found to be increased by 3-NP in the striatum. This study is the first of its kind to report an increased level of prostaglandins, which corresponds to increased COX activity, in the brains of 3-NP-treated rats. These findings highlight the involvement of COX early in the disease, which provides a reasonable and prudent target for therapeutics. These results further showed that there is an association between the behavioural and cellular alterations involved in the pathogenesis of 3-NP-induced HD-like symptoms in animals. Licofelone treatment significantly attenuated the levels of caspase-3, NF-κB, p65, TNF-α, IL-6 and prostaglandins in the striatum, indicating its action on these cellular cascades. Licofelone also had an inhibitory effect on COX-1, COX-2 and 5-LOX protein expression and directly decreased the production of neuroinflammatory markers by inhibiting the production of the PGs and LTs. Previous reports have also shown that licofelone has a protective effect against pain in animal models (Singh et al., 2005a,b; 2006;). In another study, Vidal and coworkers reported that the inhibitory effect of licofelone on 5-LOX expression was associated with the attenuation of the activation of NF-κB observed in vascular atheroma in rabbits (Vidal et al., 2007).
In summary, in the present study licofelone (dual 5-LOX/COX-2) was shown to have a neuroprotective effect against 3-NP-induced HD-like symptoms in rats. It was further demonstrated that the beneficial effects of licofelone are probably due to its ability to suppress various neuroinflammatory and apoptosis pathways. These findings indicate that licofelone could be used in the management of HD-like symptoms.
Acknowledgments
The authors gratefully acknowledged the financial support of the Indian Council of Medical Research (ICMR), New Delhi, in carrying out this work. The Senior Research Fellowship (Puneet Kumar) of the Indian Council of Medical Research (ICMR), New Delhi, is also gratefully acknowledged.
Glossary
Abbreviations
- 3-NP
3-nitropropionic acid
- BDNF
brain-derived neurotropic factor
- CMC
sodium carboxymethyl cellulose
- CREB
cAMP response element binding protein
- DMSO
dimethyl sulphoxide
- DTT
dithiothreitol
- HD
Huntington's disease
- LDH
lactate dehydrogenase
- LOX
lipoxygenase
- MDA
malondialdehyde
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole
- NBT
nitrazobluetetrazolium
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SOD
superoxide dismutase
Conflict of interest
All authors do not have conflict of interest.
References
- Arai K, Nishiyama N, Matsuki N, Ikegaya Y. Neuroprotective effects of lipoxygenase inhibitors against ischemic injury in rat hippocampal slice cultures. Brain Res. 2001;904:167–172. doi: 10.1016/s0006-8993(01)02491-x. [DOI] [PubMed] [Google Scholar]
- Arzberger T, Krampfl K, Leimgruber S, Weindl A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington's disease – an in situ hybridization study. J Neuropathol Exp Neurol. 1997;56:440–454. doi: 10.1097/00005072-199704000-00013. [DOI] [PubMed] [Google Scholar]
- Berman SB, Hastings TG. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J Neurochem. 1999;73:1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x. [DOI] [PubMed] [Google Scholar]
- Bishnoi M, Patil CS, Kumar A, Kulkarni SK. Protective effects of nimesulide (COX Inhibitor), AKBA (5-LOX Inhibitor), and their combination in aging-associated abnormalities in mice. Methods Find Exp Clin Pharmacol. 2005;27:465–470. doi: 10.1358/mf.2005.27.7.920929. [DOI] [PubMed] [Google Scholar]
- Bishnoi M, Patil CS, Kumar A, Kulkarni SK. Potentiation of antinociceptive effect of NSAIDs by a specific lipooxygenase inhibitor, acetyl 11-keto-beta boswellic acid. Indian J Exp Biol. 2006;44:128–132. [PubMed] [Google Scholar]
- Bishnoi M, Patil CS, Kumar A, Kulkarni SK. Co-administration of acetyl-11-keto-beta-boswellic acid, a specific 5-lipoxygenase inhibitor, potentiates the protective effect of COX-2 inhibitors in kainic acid-induced neurotoxicity in mice. Pharmacology. 2007;79:34–41. doi: 10.1159/000097627. [DOI] [PubMed] [Google Scholar]
- Bizat N, Hermel JM, Humbert S, Jacquard C, Creminon C, Escartin C, et al. In vivo calpain/caspase cross-talk during 3-nitropropionic acid-induced striatal degeneration: implication of a calpain-mediated cleavage of active caspase-3. J Biol Chem. 2003;278:43245–43253. doi: 10.1074/jbc.M305057200. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Koutouzis TK, Sanberg PR. 3-Nitropropionic acid animal model and Huntington's disease. Neurosci Biobehav Rev. 1997;21:289–293. doi: 10.1016/s0149-7634(96)00027-9. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, et al. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Condé F, Beal MF, Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- Browne SE, Ferrante RJ, Beal MF. Oxidative stress in Huntington's disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CJ, Edmondson DE, Singer TP. Inactivation of succinate dehydrogenase by 3-nitropropionate. J Biol Chem. 1979;254:5161–5167. [PubMed] [Google Scholar]
- Fosslien E. Cardiovascular complications of non-steroidal anti-inflammatory drugs. Ann Clin Lab Sci. 2005;35:347–385. [PubMed] [Google Scholar]
- Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- Gornall AG, Bardawill CJ, David MM. Determination of serum proteins by means of the biuret reaction. J Biol Chem. 1949;177:751–766. [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Ann Biochem. 1982;126:131–138. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- Hurley SD, Olschowka JA, O'Banion MK. Cyclooxygenase inhibition as a strategy to ameliorate brain injury. J Neurotrauma. 2002;19:1–15. doi: 10.1089/089771502753460196. [DOI] [PubMed] [Google Scholar]
- Kalonia H, Kumar P, Kumar A, Nehru B. Effect of caffeic acid and rofecoxib and their combination against intrastriatal quinolinic acid induced oxidative damage, mitochondrial and histological alterations in rats. Inflammopharmacology. 2009a;17:211–219. doi: 10.1007/s10787-009-0012-1. [DOI] [PubMed] [Google Scholar]
- Kalonia H, Kumar P, Nehru B, Kumar A. Neuroprotective effect of MK-801 against intra-striatal quinolinic acid induced behavioral, oxidative stress and cellular alterations in rats. Indian J Exp Biol. 2009b;47:880–892. [PubMed] [Google Scholar]
- Kalonia H, Kumar P, Kumar A, Nehru B. Effects of caffeic acid, rofecoxib, and their combination against quinolinic acid-induced behavioral alterations and disruption in glutathione redox status. Neurosci Bull. 2009c;25:343–352. doi: 10.1007/s12264-009-0513-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalonia H, Kumar P, Kumar A, Nehru B. Protective effect of rofecoxib and nimesulide against intra-striatal quinolinic acid-induced behavioral, oxidative stress and mitochondrial dysfunctions in rats. Neurotoxicology. 2010;31:195–203. doi: 10.1016/j.neuro.2009.12.008. [DOI] [PubMed] [Google Scholar]
- Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Bruce-Keller AJ, Mattson MP. Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci. 1998;18:4439–4450. doi: 10.1523/JNEUROSCI.18-12-04439.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TE. Preparation of succinate dehydrogenase and reconstitution of succinate oxidase. Methods Enzymol. 1967;10:322–331. [Google Scholar]
- King TE, Howard RL. Preparations and properties of soluble NADH dehydrogenases from cardiac muscle. Methods Enzymol. 1967;10:275–284. [Google Scholar]
- Kono Y. Generation of superoxide radical during auto-oxidation of hydroxylamine and an assay of superoxide dismutase. Arch Biochem Biophys. 1978;186:189–195. doi: 10.1016/0003-9861(78)90479-4. [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Singh VP. Licofelone – a novel analgesic and anti-inflammatory agent. Curr Top Med Chem. 2007;7:251–263. doi: 10.2174/156802607779941305. [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Singh VP. Licofelone: the answer to unmet needs in osteoarthritis therapy? Curr Rheumatol Rep. 2008;10:43–48. doi: 10.1007/s11926-008-0008-7. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A. Prolonged pretreatment with carvedilol prevents 3-nitropropionic acid-induced behavioral alterations and oxidative stress in rats. Pharmacol Rep. 2008;60:706–715. [PubMed] [Google Scholar]
- Kumar P, Kumar A. Effect of lycopene and epigallocatechin-3-gallate against 3-nitropropionic acid induced cognitive dysfunction and glutathione depletion in rat: a novel nitric oxide mechanism. Food Chem Toxicol. 2009a;47:2522–2530. doi: 10.1016/j.fct.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A. Possible neuroprotective effect of Withania somnifera root extract against 3-nitropropionic acid-induced behavioral, biochemical, and mitochondrial dysfunction in an animal model of Huntington's disease. J Med Food. 2009b;12:591–600. doi: 10.1089/jmf.2008.0028. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A. Protective effect of rivastigmine against 3-nitropropionic acid-induced Huntington's disease like symptoms: possible behavioural, biochemical and cellular alterations. Eur J Pharmacol. 2009c;615:91–101. doi: 10.1016/j.ejphar.2009.04.058. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A. Possible role of sertraline against 3-nitropropionic acid induced behavioral, oxidative stress and mitochondrial dysfunctions in rat brain. Prog Neuropsychopharmacol Biol Psychiatry. 2009d;33:100–108. doi: 10.1016/j.pnpbp.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A. Neuroprotective effect of cyclosporine and FK506 against 3-nitropropionic acid induced cognitive dysfunction and glutathione redox in rat: possible role of nitric oxide. Neurosci Res. 2009e;63:302–314. doi: 10.1016/j.neures.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Kumar P, Padi SS, Naidu PS, Kumar A. Effect of resveratrol on 3-nitropropionic acid-induced biochemical and behavioural changes: possible neuroprotective mechanisms. Behavioural Pharmacology. 2006;17:485–492. doi: 10.1097/00008877-200609000-00014. [DOI] [PubMed] [Google Scholar]
- Kumar P, Padi SS, Naidu PS, Kumar A. Cyclooxygenase inhibition attenuates 3-nitropropionic acid-induced neurotoxicity in rats: possible antioxidant mechanisms. Fundam Clin Pharmacol. 2007;21:297–306. doi: 10.1111/j.1472-8206.2007.00485.x. [DOI] [PubMed] [Google Scholar]
- Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy Wetzel E. Complex-II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ. 2009;16:899–909. doi: 10.1038/cdd.2009.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Peterson DA, Kimura H, Schubert D. Mechanisms of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolinium bromide (MTT) reduction. J Neurochem. 1997;69:581–593. doi: 10.1046/j.1471-4159.1997.69020581.x. [DOI] [PubMed] [Google Scholar]
- Luck H. Catalase. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. New York: Academic Press; 1971. pp. 885–893. [Google Scholar]
- Ludolph AC, He F, Spencer PS, Hammerstad J, Sabri M. 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci. 1991;18:492–498. doi: 10.1017/s0317167100032212. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Baltimore D. Physiological functions for brain NF-κβ. Trends Neurosci. 2005;28:37–43. doi: 10.1016/j.tins.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Potential inflammatory biomarkers in Alzheimer's disease. J Alzheimers Dis. 2005;8:369–375. doi: 10.3233/jad-2005-8406. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu T, Uz T, Manev H. Inflammatory 5-LOX mRNA and protein are increased in brain of aging rats. Neurobiol Aging. 2000;21:647–652. doi: 10.1016/s0197-4580(00)00167-6. [DOI] [PubMed] [Google Scholar]
- Singh VP, Patil CS, Kulkarni SK. Effect of licofelone against mechanical hyperalgesia and cold allodynia in the rat model of incisional pain. Pharmacol Rep. 2005a;57:380–384. [PubMed] [Google Scholar]
- Singh VP, Patil CS, Kulkarni SK. Effect of licofelone against NSAIDs-induced gastrointestinal ulceration and inflammation. Indian J Exp Biol. 2005b;43:247–253. [PubMed] [Google Scholar]
- Singh VP, Patil CS, Kulkarni SK. Anti-inflammatory effect of licofelone against various inflammatory challenges. Fundam Clin Pharmacol. 2006;20:65–71. doi: 10.1111/j.1472-8206.2005.00387.x. [DOI] [PubMed] [Google Scholar]
- Sottocasa GL, Kuylenstierna B, Ernster L, Bergstrand A. An electron-transport system associated with the outer membrane of liver mitochondria. A biochemical and morphological study. J Cell Biol. 1967;32:415–438. doi: 10.1083/jcb.32.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer's disease following perispinal etanercept administration. J Neuroinflammation. 2008;5:2. doi: 10.1186/1742-2094-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunez I, Collado JA, Medina FJ, Pena J, Del CMM, Jimena I, et al. 17 β-Estradiol may affect vulnerability of striatum in a 3-nitropropionic acid-induced experimental model of Huntington's disease in ovariectomized rats. Neurochem Int. 2006;48:367–373. doi: 10.1016/j.neuint.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Vidal C, Gomez-Hernandez A, Sanchez-Galan E, Gonzalez A, Ortega L, Gomez-Gerique JA, et al. Licofelone, a balanced inhibitor of cyclooxygenase and 5-lipoxygenase, reduces inflammation in a rabbit model of atherosclerosis. J Pharmacol Exp Ther. 2007;320:108–116. doi: 10.1124/jpet.106.110361. [DOI] [PubMed] [Google Scholar]
- Wills ED. Mechanisms of lipid peroxide formation in animal tissues. Biochem J. 1966;99:667–676. doi: 10.1042/bj0990667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Sugama S, Mischak RP, Kiaei M, Bizat N, Brouillet E, et al. A novel systemically active caspase inhibitor attenuates the toxicities of MPTP, malonate, and 3NP in vivo. Neurobiol Dis. 2004;17:250–259. doi: 10.1016/j.nbd.2004.07.021. [DOI] [PubMed] [Google Scholar]