

Abstract
BACKGROUND AND PURPOSE
Systemic iron deficiency concomitant with macrophage iron retention is characteristic of iron-refractory anaemias associated with chronic disease. The systemic misdistribution of iron, which is further exacerbated by parenteral iron supplementation, is mainly attributable to iron retention exerted on resident macrophages by hepcidin-mediated down-regulation of the iron exporter ferroportin. We aimed at developing an experimental macrophage-based cell model that recapitulates pathophysiological features of iron misdistribution found in chronic disorders and use it as a screening platform for identifying agents with the potential for relocating the accumulated metal and restoring affected functions.
EXPERIMENTAL APPROACH
A RAW macrophage subline was selected as cell model of iron retention based on their capacity to take up polymeric iron or aged erythrocytes excessively, resulting in a demonstrable increase of cell labile iron pools and oxidative damage that are aggravated by hepcidin.
KEY RESULTS
This model provided a three-stage high throughput screening platform for identifying agents with the combined ability to: (i) scavenge cell iron and thereby rescue macrophage cells damaged by iron-overload; (ii) bypass the ferroportin blockade by conveying the scavenged iron to other iron-starved cells in co-culture via transferrin but (iii) without promoting utilization of the scavenged iron by intracellular pathogens. As test agents we used chelators in clinical practice and found the oral chelator deferiprone fulfilled essentially all of the three criteria.
CONCLUSIONS AND IMPLICATIONS
We provide a proof of principle for conservative iron relocation as complementary therapeutic approach for correcting the misdistribution of iron associated with chronic disease and exacerbated by parenteral iron supplementation.
Keywords: macrophages, iron, oxidative stress, infection, anaemia, chelators
Introduction
The phagocytosis of aged erythrocytes involved in the recycling of iron by the monocyte/macrophage system is a critical component of systemic iron metabolism (Recalcati et al., 2010). While it satisfies an estimated 90% of the body's daily iron management, its disruption may lead to impaired iron supply for de novo erythropoiesis and ultimately to anaemia of chronic disease (ACD): some linked to particular gene mutations (Finberg, 2009) and others associated with chronic conditions, mostly with inflammatory components, infections or malignancies (Weiss and Goodnough, 2005). ACDs are characterized by inadequate erythrocyte production in the setting of low plasma iron despite normal plasma iron-binding capacity and preserved or even increased macrophage iron stores in the bone marrow, liver and spleen (Agarwal and Prchal, 2009; Beaumont and Delaby, 2009; Theurl et al., 2009). Recent advances in understanding of the function of the circulating peptide hepcidin have elucidated the sequence of events responsible for this imbalance or misdistribution of iron (Kemma et al., 2009; Babitt and Lin, 2010). Summarized briefly, cytokine release due to inflammation/infection induces the hepatic production of hepcidin and its release into plasma. The proposed cell-surface receptor for hepcidin is the iron exporting protein ferroportin (found in macrophages, hepatocytes and duodenal enterocytes), which, upon hepcidin binding, undergoes internalization and degradation (Nemeth et al., 2004; Recalcati et al., 2010). Cytokines can also affect cell iron accumulation by affecting the transcriptional expression of transporters (Ludwiczek et al., 2003; Yang et al., 2002). At the systemic level, shutdown of ferroportin expression leads to obstruction of enteral iron absorption and impaired iron release from spleen macrophages. This also provides an explanation for the refractoriness to oral iron supplementation found in ACD and also in iron-refractory iron deficient anaemia (IRIDA) (Theurl et al., 2009). At the cellular level, ferroportin inactivation leads to intracellular entrapment of iron and its ensuing retention (Nemeth et al., 2004), which in turn increases the risk of infection by intracellular pathogens (Nairz et al., 2007; Paradkar et al., 2008). A point in case is asymptomatic malaria where impaired iron recycling caused by elevated hepcidin leads to macrophage iron retention (Nairz et al., 2007; Paradkar et al., 2008) and ensuing anaemia, but also to a higher predisposition to infection by bacteria (Schaible and Kaufman, 2004). A potentially exacerbating factor in the treatment of ACD is i.v. supplementation of polymeric iron forms that are primarily ingested and processed by macrophages. Although i.v. iron bypasses the block in enteral iron absorption, it can further increase the regional accumulation of stored or unprocessed iron, leading to potentially toxic macrophage iron accumulation and ensuing oxidative damage (Weiss and Goodnough, 2005; Agarwal and Prchal, 2009; Babitt and Lin, 2010).
A major therapeutic challenge in disorders associated with intracellular iron entrapment at the expense of the organism is how to circumvent the block in the iron egress pathway and render iron metabolically available for erythropoiesis, while avoiding its utilization by microorganisms. That might be theoretically achieved by hepcidin antagonists (Nemeth, 2010) or possibly by anti-hepcidin antibodies (Sasu et al., 2010) and by upstream factors that can down-modulate hepcidin production by the liver (Babitt et al., 2007; Kawabata et al., 2007; Yu et al., 2008; Babitt and Lin, 2010). An alternative/complementary approach for correcting disorders of iron misdistribution entails the use of pharmacological agents that can mobilize iron from sites of accumulation and convey it to physiological acceptors for reutilization (Breuer and Cabantchik, 2009; Kakhlon et al., 2010). Such a conservative mode of action can be expected from agents that can act as siderophores at pharmacologically attainable concentrations (Kakhlon et al., 2010). Siderophore-like agents chelate labile cell iron (LCI) and thereby reduce metal-evoked oxidative damage (Glickstein et al., 2006) but can also donate the complexed metal to extracellular acceptors (Evans et al., 1992; Sohn et al., 2008) and/or to cell metabolic machinery, such as that involved in iron-sulphur cluster biosynthesis (Kakhlon et al., 2008). Taking advantage of the fact that moderate doses of deferiprone (DFP) are not toxic to cells (Glickstein et al., 2006; Lescoat et al., 2007) and will cause no short-term systemic iron deficiency in patients with no iron overload (Vreugdenhil et al., 1990; Boddaert et al., 2007), we assessed here the concept of conservative relocation of iron in disease-simulating settings (i.e. inflammation/infection) and its feasibility for identifying agents with therapeutic potential. For that purpose we designed an analytical platform based on a mouse macrophage RAW264.7 subline (IS) that displays sensitivity to iron accumulated by erythrophagocytosis or exposure to polymeric sources of iron and becomes hypersensitive to iron when supplemented with the hormone hepcidin. This experimental model enabled us to assess the possibility of restoring iron-affected cell functions with chelating agents that are not detrimental to normal cells and might even differentially support growth of iron-deficient cells but not of cell-invading bacteria.
Methods
Materials
Calcein green (CALG; 3,3′-bis[N,N-bis(carboxymethyl) aminomethyl] fluorescein), calcein blue (CALB; methylumbelliferone-8-methyleneiminodiacetic acid) and their respective acetomethoxy (AM) precursors CALG-AM and CALB-AM and 5-(&6)-carboxy-2′,7′-dichlorofluorescein diacetate-acetoxymethyl ester (CDCHF-DA-AM) were from Molecular Probes (Invitrogen Corp., Carlsbad, CA, USA); salicyl isonicotinoyl hydrazone (SIH) was a gift from Dr. P. Ponka (Montreal) dihydrorhodamine 123 (DHR) from Biotium Inc. (Hayward, CA, USA). Ferric ammonium citrate (FAC), deoxycholate, sulphorhodamine B, propidium iodide (PI), Hoechst 33342, anti-protease mix, protein bicinchoninic acid (BCA) reagent, Hoechst 33342, bathophenanthroline sulphonate, caspase 3 = Ac-DEVD-AMC (read at Exc 355 nm:Em 460 nm/Exc/Em maxima are 380:460 nm) were from Sigma Chem. Co. (St. Louis, MO, USA). DFP (DFP = ferriprox™ = 1,2-dimethyl-3-hydroxypyridin-4-one) was from Apo Pharma Inc. (Toronto, Canada); deferrioxamine (DFO) and deferasirox (DFR; Exjade) from Novartis-Pharma (Basel, Switzerland). Hepcidin from Peptides International (Louisville, KY, USA) and the iron-saccharate polymer Venofer™ from Vifor International, St Gallen, Switzerland.
Cell culture
RAW264.7 mouse macrophages and human erythroleukaemia K562 cells were grown in 5% CO2 Dulbecco's modified Eagle's (DMEM) medium supplemented with 10% fetal calf serum, 4.5g·L−1 D-glucose, glutamine and antibiotics (Biological Industries, Kibbutz Bet Haemek, Israel). Cells were plated a day before experimentation onto 96 well plates, or onto microscopic slides glued onto to perforated 3 cm diameter tissue culture plates. The iron-sensitive (IS) subline was obtained by continual growth of the wild-type (WT) RAW264.7 cells at high density for >8 passages. The WT cell spontaneously transformed to a stable IS phenotype that is characterized by high susceptibility to polymeric iron sources.
Cell iron load
Cell iron loading comprised incubating cells for 6–18 h in the presence of FAC (100 µM) or the iron-saccharate i.v. formulation venofer (V; 500 µM Fe) followed by alternate incubations with 10 µM DFO in phosphate-buffered saline (PBS) for 10 min at room temperature (to remove traces of extracellular iron) and washings in PBS.
Erythrophagocytosis
Human erythrocytes [red blood cells (RBCs)] washed in PBS and opsonized by incubating 1 × 108 RBCs with rabbit anti-human RBC IgG (1:50) (Abcam, Cambridge, UK) for 20 min at 37°C, then washed twice in PBS and suspended in DMEM medium. The erythrocytes (1–5 × 107) were laid over to 1 × 106 a RAW264.7 cells in 6 well plates and incubated for 1 h at 37°C. Cell surface adsorbed RBCs were lysed by incubation for 5 min at 37°C with lysing solution (140 mM NH4Cl, 17 mM Tris, pH 7.6) followed by two washes with PBS.
Western blots
Ferritin Cells harvested by scraping and washing with PBS, were lysed in 0.15 mL of ice-cold PBS containing 0.5% Triton X-100 (Fluka, Buchs, Switzerland) and protease inhibitor mix and lysates clarified by centrifugation and analysed for protein (BCA assay) of which 50 µg were electrophoretically separated on 12% SDS-polyacrylamide gels. After transfer to nitrocellulose membrane and blocking with nonfat dry milk the blot was incubated overnight with rabbit anti-human ferritin (1:1000) (Sigma Chem. Co.), followed by washing, 1 h incubation with horseradish peroxidase-linked goat anti-rabbit antibody and chemiluminescence analysis.
Ferroportin Membrane fractions were prepared as described elsewhere (Germann, 1997) and protein concentrations determined by the Bradford assay (Bio-Rad, Hercules, CA, USA). Proteins mixed with sample buffer at room temperature for 30 min were separated on a 10% SDS-polyacrylamide gel, transfered to nitrocellulose membrane, blocked, blotted overnight at 4°C with affinity-purified rabbit anti-ferroportin (3 µg·mL−1, Alpha Diagnostics, San Antonio, TX, USA), washed, incubated for 1 h with horseradish peroxidase-linked goat anti-rabbit followed by chemiluminescence analysis.
Protein carbonyls (COs)
Protein COs (Reznick and Packer, 1994) were determined by incubating cell lysates (1 mL) for 1 h with dinitrophenylhydrazine (DNPH; from Sigma Chem. Co.) (2.5 mM in 4 mL 2N HCl), proteins precipitation with an equal volume of 20% (w·v−1) trichloroacetic acid, centrifugation (6500× g for 5 min), dispersion of the precipitate and washing with 4 mL of 10% (w·v−1) trichloroacetic acid and thrice with 4 mL of ethanol : ethyl acetate (1:1) and finally dissolution in 6 M guanidine–HCl. DNP-protein adducts were quantified by absorbance at 365 nm. Protein contents were estimated from control, unreacted cell lysates prepared in parallel, using as standard bovine serum albumin in guanidine–HCl and reading the absorbance at 280 nm.
ROS production
Cell ROS production was determined by incubating cells at 37°C with 10 µM CDCHF-DA-AM in HEPES-buffered saline (HBS) supplemented with 10 mM glucose. The conversion of the non-fluorescent 2-7 carboxy-dichlorodihydrofluorescein-diacetate (CDDHCF-DA) to the fluorescent 2-7-carboxy-dichlorofluorescein (CDCF) was measured on line at 37°C either in a fluorescence plate reader (Tecan-Safire, Neotec, Männedorf, Austria) (Exc: 488 nm; Em: 517 nm) or under the fluorescence microscope (Nikon TE 2000 microscope equipped with a thermostated stage and a Hamamatsu Orca-Era CCD camera) driven by a Volocity 4 operating system (Improvision, Coventry, UK) that was used for both image data acquisition and analysis (Glickstein et al., 1994; Sohn et al., 2008). At the indicated times, H2O2 (50 µM final) and/or chelator (50–100 µM DFP or SIH) were added.
Cytosolic/mitochondrial ROS formation was determined by incubating cells at 37°C with 50 µM DHR, which is oxidized intracellularly to rhodamine 123 and analysed fluorimetrically/microscopically as described for Method 1 (Exc. 488 nm; Em. 517 nm). At the indicated times, H2O2 (50 µM final), and/or chelator were added.
Cell protein
Cell protein was determined as described previously (Voigt, 2005). Cells (in culture plates) were treated for 1 h at 4°C with 15% TCA, washed three times in distilled water, reacted with sulphorhodamine B (0.4% w·v−1 in 1% acetic acid) for 30 min and washed three times with 1% acetic acid. The rotein-bound stain was extracted with 10 mM Tris-base and absorbance read at 492 nm.
Cell viability/metabolic activity
Cell viability/metabolic activity was determined as described previously (Nairz et al., 2009). RAW264.7 cells in 96 well culture plates were supplemented with Alamar Blue (AB) reagent (10% by volume) and the fluorescence read on a plate-reader after 1–4 h incubation at 37°C (Exc: 530–560 nm; Em: 590 nm). The percent dead cells were also determined by microscopic inspection with Trypan Blue (0.4% for 5 min) or by fluorescence microscopy following propidium iodide staining (10 µM for 30 min at room temperature) of dead/damaged cells and in conjunction with co-staining of viable cells by CAL-AM loading (0.5 µM for 10 min) and inspection of the accumulated fluorescence CAL (fluorescein settings).
Nucleic acid
Cells washed with PBS were reacted with 10 µM propidium iodide (PI) in DMEM-HEPES or HBS buffer, incubated for 30 min at room temperature in the dark, then supplemented with 10 µM Hoechst 33342 at 37°C for 30 min and visualized under a fluorescence microscope using a filter set for rhodamine and DAPI (Exc: 350 nm; Em: 461 nm). Staurosporine (0.5 µM) was used as a positive control to induce apoptosis.
Co-culture
RAW264.7 cells (preloaded with FAC 100 or by erythrophagocytisis) were co-cultured with iron-depleted K562 cells (by culturing overnight with 10 µM DFO) in medium with iron-depleted serum [prepared by acidification to pH 5.0, passage through a Chelex-100 column (BioRad Labs, Hercules, CA, USA) and neutralization to pH 7.4 with NaOH]. The K562 cells growing in suspension (unlike the substrate-attached RAW264.7 cells) were collected and transferred to a 96 well plate for staining with Hoechst 33342.
LCI
The CALB assay was used to determined LCI (Glickstein et al., 1994). Cells exposed to CALB-AM 5 µM at 37°C for 10 min in DMEM-HEPES were washed in HBS, pH 7.4 and subsequently incubated at 37°C in either HBS or DMEM-HEPES containing 0.5 mM probenecid (to minimize probe leakage). The fluorescence changes of CALB were monitored by epi-fluorescence microscopy as described earlier (Exc: 390 nm; Em: 430 nm). DFP (50 µM) was added to cells for the measurements of cytosolic LIP.
CALG-Fe loading via endocytosis
RAW264.7 cells were exposed to CALG-Fe (50 µM) in serum-free DMEM-HEPES medium for 30 min at 37°C and washed with HBS. The CALG fluorescence intensity was monitored by confocal microscopy with a FV-1000 confocal microscopy (Olympus, Tokyo, Japan) equipped with an IX81 inverted microscope, after attaining maximal recoverable fluorescence by adding the chelator SIH (50 µM).
Cell non-haeme iron
Iron was extracted from RAW264.7 cells by adding 0.4 mL of 3N HCl, 10% trichloroacetic acid (iron free) and 3% thioglycolic acid for 1 h at room temperature. The supernatant was diluted with one volume of distilled water and mixed with an equal volume of BPS reagent (bathophenanthroline sulphonate in 2 M Tris-base, 0.2%thioglycolic acid) for 15 min at room temperature and absorption was read at 535 nm. For protein determination, the precipitated protein was dissolved with 1 M NaOH and reacted with the Bradford reagent (Pierce Chemical Co., Rockford, IL, USA).
Salmonella infection
RAW264.7 (WT) cells were switched to antibiotic free medium (DMEM + 10% FCS + 2 mM L-glutamate) with/without 100 µM FAC or 500 µM V (Fe equivalents)18 h prior to infection, seeded (1 million cells per well) 5 h before infection with WT Salmonella enterica serovar thyphimurium strain ATCC 14028 at a multiplicity of infection of 5:1 (Nairz et al., 2009). After 1 h at 37°C the cells were washed three times in PBS containing 25 µg·mL−1 gentamycin and cultured with DMEM containing no other antibiotics but gentamycin. The infected cells were grown without or with 100 µM FAC or or 500 µM 100 µM or with DFP for 24 h, lysed with 0.5% deoxycholic acid and the lysates were plated under sterile conditions onto LB agar plates for colony assessment [colony-forming units (CFU)] following incubation at 37°C.
Statistical analysis
Regression and statistical analysis (paired t-test, anova or Bonferroni-variance test-parametric) were performed with the aid of Origin 8.1 program (OriginLab Corp., Northampton, MA, USA) or with the SPSS package (SPSS Inc., Chicago, IL, USA).
Results
Properties of a macrophage cell line susceptible to iron loading
The parental RAW264.7 mouse macrophage cell line (WT) has served as model for assessing numerous biological properties, especially those related to erythrophagocytosis (Aderem and Underhill, 1999). In typical culture conditions the WT cells can withstand exposure to large concentrations of iron supplemented in various chemical forms, showing no detectable impairments in growth rates or metabolic activities (Figure 1). However, when continuously grown in high density media (>8 passages), WT RAW cells apparently adapt to these growth restrictive conditions by spontaneously transforming into a subline that showed normal rate of growth under standard culture conditions, but underwent growth arrest when exposed to particular iron sources, as shown in Figure 1. Exposure to various FAC concentrations for 18 h caused an incremental impairment in the metabolic activity of the IS cells, as measured by their capacity to reduce the reagent AB. When hepcidin (1 µM) was added to the suspension of IS RAW cells, the FAC inhibitory effect was more pronounced, whereas in the same experimental conditions hepcidin had no effect on WT cells (Figure 1A). A similar pattern of results was obtained following an overnight exposure to polymeric iron-saccharose (V = Venofer™), an i.v. formula of iron supplementation (Figure 1B) or to erythrophagocytosis (Figure 1C). In both cases, IS cell growth was reduced following exposure to the macromolecular/ particulate iron source and this effect was further aggravated in the presence of supplemented hepcidin. We found that the rapidly-permeating, soluble complex iron : 8-hydroxyquinoline (1:1 stoichiometry) was equally toxic to WT and IS cells at concentrations > 5 µM (1 h exposure time), while iron-saturated holotransferrin at 4 mg·mL−1 (approximately 100 µM iron) had no effect on IS cell viability (data not shown). We tentatively attribute the enhanced cytotoxicity of the various ferric sources to an apparently impaired ability of IS cell to cope with iron taken up by endocytic routes and exacerbated by hepcidin down-modulation of the iron exporter ferroportin.
Figure 1.

Iron loading and erythrophagocytosis by RAW 264.7 macrophages: effects on wild-type (WT) and iron-sensitive (IS) cells. (A) Cells were incubated with ferric ammonium citrate (FAC) at the indicated concentrations in the absence or presence of 1 µM hepcidin and cell viability was determined after 24 h with the fluorescent redox probe Alamar Blue. Results are shown as means ± standard deviation (SD) (n = 5) of Alamar Blue's fluorescence for the indicated treatment relative to the respective untreated controls. (B) Cells were incubated for 6–8 h with Venofer (V; 500 µM Fe) in the absence or presence of 1 µM hepcidin (+Hep), and subsequently washed and cultured for 18 additional h and analysed as described in (A). Results shown are relative to the respective untreated controls. (C) Cells were incubated for 1 h with or without opsonized erythrocytes (+RBC) as described in the Methods section, and then cultured overnight with or without 1 µM hepcidin (+Hep). Cell viability was measured with Alamar Blue and is shown relative to respective untreated controls ± SD (*P < 0.05).
Uptake of iron salts and of red blood cells by WT and IS macrophages
The susceptibility of IS macrophages to polymeric iron complexes or aged RBCs implies that cell damage is incurred by acquisition of iron via endocytic routes. In preliminary studies with the fluid endocytosis marker sulphorhodamine, we observed that the two RAW variants showed similar patterns of fluorescent endosomes when exposed to growth medium but different ones when exposed to medium supplemented with iron complexes. The latter, which tend to adsorb onto membrane surfaces and trigger local endocytosis, can be visualized when incubating cells with complexes of calcein green-Fe(III) (CALG-Fe; 1:1 stoichiometry) as a fluorescent surrogate of polycarboxylic: Fe(III) salt uptake by cells or with calcein-V (CALG-V) as an example of polymeric iron that is used clinically in the supportive treatment of iron refractory anaemias. Whereas uptake of CALG alone by RAW cells is hardly detectable even after hours of incubation (not shown), the quenched CALG-Fe or CALG-V complexes or opsonized RBCs are taken up into the endosomal compartment (Figure 2). The appearance of fluorescent endosomes indicates that Fe is released from CALG-Fe complexes following their endocytosis. The Fe-quenched CALG is revealed with the permeant SIH, a chelator that scavenges CALG-bound Fe and thereby restores endosomal-associated fluorescence. We tentatively interpret the distinctive properties of IS cells in terms of significantly increased endocytic/phagocytic activity leading to excessive iron accumulation. It is not known whether the latter is also accompanied by ineffective handling of particulate iron forms and/or the ensuing cellular processing of the metal.
Figure 2.

Endocytic properties of wild-type (WT) and iron-sensitive (IS) lines of RAW 264.7 macrophages. WT and IS cells were incubated in culture conditions with growth medium supplemented with 50 µM of calcein green (CALG) given as either CALG-Fe (1:1 complexes) for 30 min or CALG-V (1:2 complexes) for 4 h or with opsonized erythrocytes for 1 h, washed and analysed by phase contrast, differential interference contrast (DIC) and fluorescence microscopy, as described in the Methods section. Following CALG-V, a subset of washed cell samples was treated with the permeant chelator salicyl isonicotinoyl hydrazone (SIH) (50 µM) for 10 min (in order to reveal all Fe quenched complexes). Image analysis was carried out on confocal fluorescence images obtained after loading with CALG-Fe (fluorescence superimposed on phase contrast) or CALG-V (fluorescence) and on DIC images obtained after erythrophagocytosis. Results of cell image analysis of n = 3 independent experiments yielded statistically different (P < 0.05) parameters (mean values ± standard deviation determined in three areas of interest (one area per single cell) between WT and IS cells incubated with CALG-Fe: # of endosomes/cell: 22 ± 7 versus 56 ± 5; endosomes mean fluorescence (rel units): 36 ± 3 versus 28 ± 5; cell mean fluorescence (rel units): 11 ± 2 versus 19 ± 1; cell total fluorescence (rel units): 277 ± 143 versus 1096 ± 50. The mean numbers of erythrophagocytosed red blood cells (RBCs) per macrophage (WT vs. IS) were 1.2 ± 0.6 and 3.6 ± 1.8.
Iron and oxidative stress parameters following endocytosis of iron salts and of red blood cells
The basal non-haeme iron content of WT and IS cells increased significantly (by 3- to 5-fold) following 18 hexposure to 100 µM Fe given as FAC or to 500 µM Fe given as V (a concentration attained during i.v. iron supplementation) and to even higher levels in the presence of hepcidin (Table 1).
Table 1.
Non-haeme iron content in RAW 264.7 WT and IS cells following iron loading
| Treatment | Haeme content | |
|---|---|---|
| Treatment | WT | IS |
| Basal (-FAC) | 0.49 ± 0.09 | 0.67 ± 0.05 |
| FAC | 1.61 ± 0.09* | 3.24 ± 0.11*,** |
| FAC + Hep | 1.84 ± 0.05 | 4.27 ± 0.7 |
| Basal (-V) | 0.43 ± 0.06 | 0.43 ± 0.04 |
| V | 3.82 ± 0.20* | 2.91 ± 0.49** |
| V + hep | 4.87 ± 0.10 | 3.66 ± 0.27 |
| Basal (-RBC) | 0.55 ± 0.04* | 0.66 ± 0.05 |
| RBC | 0.83 ± 0.02* | 1.22 ± 0.34*,** |
| RBC + hep | 0.96 ± 0.12 | 1.20 ± 0.23 |
WT and IS cells were incubated: (a) for 1 h with opsonized erythrocytes [red blood cells (RBCs)] and then cultured overnight without or with 1 µM hepcidin (+Hep) or (b) for overnight in regular culture medium, or in medium supplemented with either 100 µM ferric ammonium citrate (FAC) ± 1 µM hepcidin (FAC + Hep) or with Venofer (V; 500 µM Fe) ± 1 µM hepcidin (hep).
The total non-haeme iron content (nmol·µg−1 protein) in untreated cells (basal levels) and iron loaded cells was determined after extensive washing of the cells as described in the Methods section and is given as mean values ± standard deviation with * denoting significant difference at P < 0.05 from the respective control (indicated as basal) ** significant difference between iron sensitive (IS) and wild type (WT) for the equivalent treatment.
The increases in non-haeme iron induced by erythrophagocytosis and followed by overnight incubation, were also significantly higher in IS as compared with WT cells (P < 0.05, n = 3) (Table 1), but apparently less pronounced when compared with the increases induced by overnight exposure to iron complexes. A factor that contributed to this apparent difference is the substantial increase in protein mass per cell following erythrophagocytosis.
Since by measuring total cell-associated iron one cannot distinguish between internalized and surface adsorbed metal (despite extensive cell washing with strong and impermeant iron chelators such as DFO), we opted for examining microscopically the iron loading properties of WT and IS cells in terms of LCI levels by tracing labile iron with the fluorescent iron probe calcein blue (Figure 3A). Under unchallenged (or basal) growth conditions, LCI was barely detectable in both WT and IS cells, but following incubation with FAC (100 µM for 4 h) the levels rose significantly to 12 ± 4 for WT and 70 ± 6 U for IS. Both types of cells showed a similar capacity to respond to prolonged (24 h) iron loads by induction of H-ferritin protein levels and parallel repression of TfR levels (Figure 3B). Increased ferroportin levels, were observed in IS, but not in WT cells (Figure 3B). Furthermore, IS cells responded robustly to hepcidin exposure, as shown by decreased ferroportin and a further increase in H-ferritin protein levels (Figure 3B). In contrast, ferroportin protein levels in FAC-loaded WT cells were below Western blot detection levels and H-ferritin expression did not increase further in response to hepcidin (data not shown). Possibly, lower iron accumulation in WT cells compared with IS cells leads to a diminished requirement for ferroportin and hence lower ferroportin protein levels.
Figure 3.
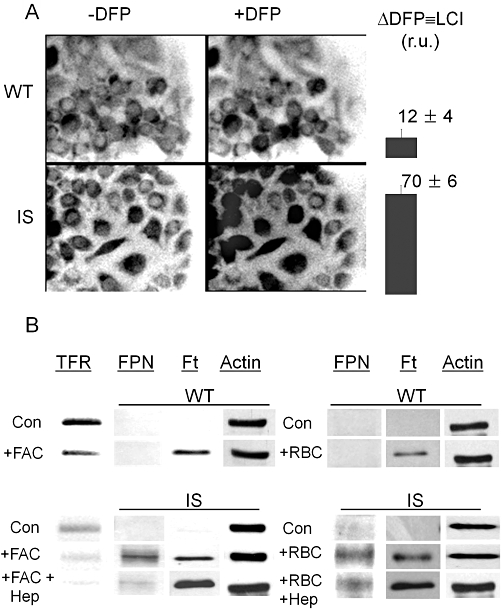
Iron loading and erythrophagocytosis by RAW 264.7 [wild type (WT) and iron-sensitive (IS)] macrophages: effects on labile iron pools, ferroportin, ferritin and transferrin receptor levels. WT and IS cells were incubated 4–18 h in regular culture medium supplemented with 100 µM ferric ammonium citrate (FAC). (A) The cytosolic labile cell iron (LCI) levels in 4 h iron-loaded WT and IS cells were assessed by fluorescence microscope imaging with the fluorescent intracellular iron probe Calcein blue as described in the Methods section. LCI is shown for one experiment in relative fluorescence units determined in five different fields of cells (mean ± standard error of the mean) as determined from the increase in fluorescence 15 min after addition of 100 µM deferiprone (DFP) (Δ DFP). The range of LCI in iron loaded cells (in relative fluorescence units r.u. obtained with the same microscope settings in three independent experiments) was 4–20 for WT cells and 62–95 for IS cells. (B) Ferroportin (FPN), transferrin receptor (TfR) and ferritin (Ft) levels in WT (top panel) and IS (bottom panel) cells were assessed by Western blotting after overnight incubation of cells without or with 100 µM of FAC (+FAC) or after 1 h with or without opsonized erythrocytes (+RBC), followed by overnight incubation with or without 1 µM Hepcidin (+Hep). The data displayed are representative of one of five experiments performed in two different laboratories, showing similar Western blot patterns.
The different LCI levels in WT and IS cells before and after iron loading could be predicted to result in a commensurate propensity to generate intracellular ROS, which we measured with dihydro-CDCF (Figure 4A), which oxidizes to fluorescent CDCF in a labile-iron dependent manner. The time-dependence conversion is highlighted for selected systems in the inset of the figure and the calculated mean rates of conversion obtained in 3 independent experiments are shown in the histogram of Figure 4A. The rate of fluorescence increase of CDCF elicited by 50 µM H2O2 was significantly (75%) higher in IS than in WT cells. Following overnight exposure to FAC there was a 2.0 ± 0.6-fold increase in cytosolic reactive oxygen intermediates (ROI) production in WT cells as compared with 2.9 ± 0.5-fold increase in IS cells. Thus, IS cells generate significantly higher levels of ROI than WT cells, which is further magnified after iron loading. The fact that the rapidly-penetrating, iron chelator DFP produced a swift and major inhibition of dihydro-CDCF oxidation, implicates iron in the measured cell ROI formation. Similar results were obtained with the probe DHR, which is converted by ROI into fluorescent rhodamine 123 (R) accumulating in mitochondria potentiometrically (Figure 4B). Again ROI production was significantly higher in IS cells (time dependence of formation shown illustratively in Figure 4B inset and calculated rates from three independent experiments in the main figure). Oxidative challenge evoked by H2O2 further increased ROI production whereas DFP partially inhibited it, possibly because of lower accessibility of the chelator to mitochondrial compartments.
Figure 4.
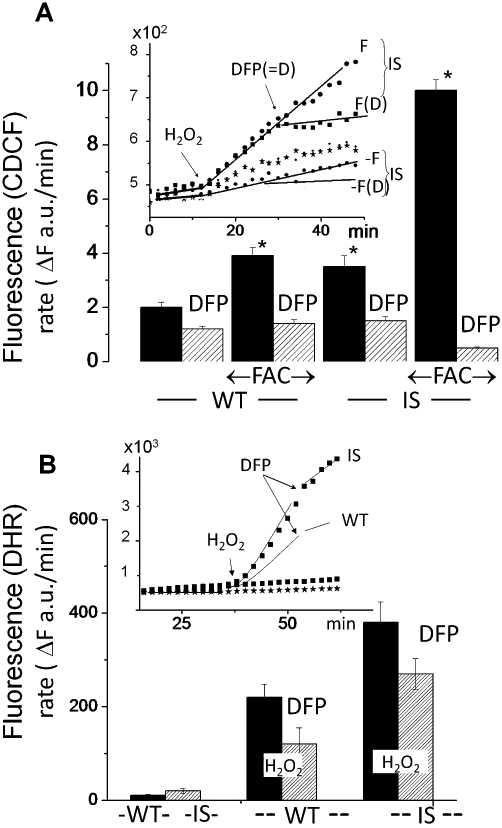
Iron loading and erythrophagocytosis by RAW 264.7 [wild type (WT) and iron-sensitive (IS)] macrophages: formation of reactive oxygen intermediates (ROI). WT and IS cells were preincubated overnight with/without 50 µM ferric ammonium citrate (FAC) (F) and subsequently loaded with dihydro-2-7-carboxy-dichlorofluorescein (dihydro-CDCF) [via the cell permeant precursor acetoxymethyl ester CDCF-acetomethoxy (CDCF-AM) (A) or with dihydrorhodamine 123 (B)]. The time-dependent changes in fluorescence intensity [(mean of triplicate values given in arbitrary fluorescence units (a.u.)] are shown in the insets of (A) and (B) illustratively for only the indicated treatments. The fluorescence traces shown in each graph were obtained concurrently at 37°C in a thermostated fluorescence plate reader. After a stable baseline cell fluorescence signal was detected, 50 µM H2O2 and 50 µM deferiprone (DFP) (= D) were added sequentially 20 min apart (indicated by arrows; the sets not exposed to H2O2 are denoted as -). Histograms in the main figure (A, B) show the rates of fluorescence increase (ΔF a.u.·min−1) calculated from the linear segments of the fluorescence profiles shown in the insets (shown illustratively for the indicated conditions).
The extent to which IS cells are affected by oxidation reactions due to exposure to iron sources was also assessed in terms of cell protein CO group formation (Table 2). When grown in standard culture conditions, the CO group content of IS cells was not significantly different (P < 0.05) from that of WT cells. However, following exposure to FAC or V, the protein CO levels rose differentially and significantly in IS cells and attained even higher levels in the presence of hepcidin. Thus, IS cells are endowed with an intrinsic ability to respond to iron overload by triggering the formation of ferritin and of ferroportin while remaining susceptible to hepcidin. These features recapitulate properties of RES cells in inflammation whereby cell iron retention follows erythrophagocytosis and/or endocytosis of polymeric i.v. iron supplements in the background of high hepcidin.
Table 2.
Protein carbonyl (CO) groups in RAW 264.7 wild type (WT) and iron sensitive (IS) cells following iron loading
| Cell CO groups (nmol·mg−1 protein ± SD) | |||||
|---|---|---|---|---|---|
| RAW264.7 cell type | Basal | FAC | FAC + Hep | V | V + Hep |
| WT | 0.57 ± 0.09 | 0.68 ± 0.2 | 0.71 ± 0.3 | 0.75 ± 0.11 | 0.84 ± 0.13** |
| IS | 0.62 ± 0.21 | 1.97 ± 0.31*,** | 2.57 ± 0.30*,** | 1.97 ± 0.33*,** | 2.86 ± 0.47*,** |
The CO group levels in basal growth conditions and following 48 exposure to 100 µM FAC or Venofer (V) (500 µM Fe) ± hepcidin (Hep) (1 µg·mL−1) were determined with the 2,4-dinitrophenyl-hydrazine assay as described in the Methods section.
The indicated values represent means of three equivalent experiments ± SD and significant differences (at P < 0.05) between IS and WT cells for a given treatment are indicated as * and between treatment and basal conditions for a given cell type as **.
Effect of chelators on RAW 264.7 macrophages
The capacity of iron chelators to either prevent or correct iron toxicity in IS cells was assessed by various methods. Cell metabolic activity or viability (Figure 5A) and % cell death (Figure 5B) were assessed following iron loading with 100 µM FAC for 4 h or 500 µM V (±1 µM hepcidin) for 24 h, or erythrophagocytosis (RBC) for 1 h, followed by washing and incubation for a further 24 h period in growth medium. In the experimental conditions used, iron loading resulting from exposure to either FAC or V (±hepcidin) or by erythrophagocytosis markedly reduced IS cell metabolism/viability as assessed with the redox indicator AB. Chelators assessed for potential reversal of metabolic inhibition were used either at pharmacological concentrations attained clinically (DFP at 100 µM and DFR at 50 µM) or for the poorly permeant DFO at 10-fold above clinical concentrations (100 µM). The cell metabolic activity/viability affected by either method of iron loading was largely (80–90%) restored by treatment with DFP, whereas with DFR or DFO the recovery was markedly lower. We obtained essentially similar results when cell proliferation was measured as total cell protein with the protein stain sulphorhodamine B (data not shown). The ability of DFP or DFR to reverse the toxic effects of iron loading of IS cell was also assessed in terms of preventing cell death. Cell death, manifested as a loss in plasma membrane integrity, was measured by the penetration of the otherwise impermeant DNA-binding dye propidium iodide vis a vis Hoechst 33342 (Figure 5B). Unlike WT cultures, which remained unaffected by iron loading (1% mean cell death index), cell death in IS cultures significantly increased following 4 h exposure to FAC or 24 h to V and was further increased by hepcidin. Importantly, DFP at 100 µM and DFR at 50 µM significantly reduced the iron-evoked cell death, the first by >75% (P < 0.01) and the second by <40% (P < 0.05).
Figure 5.
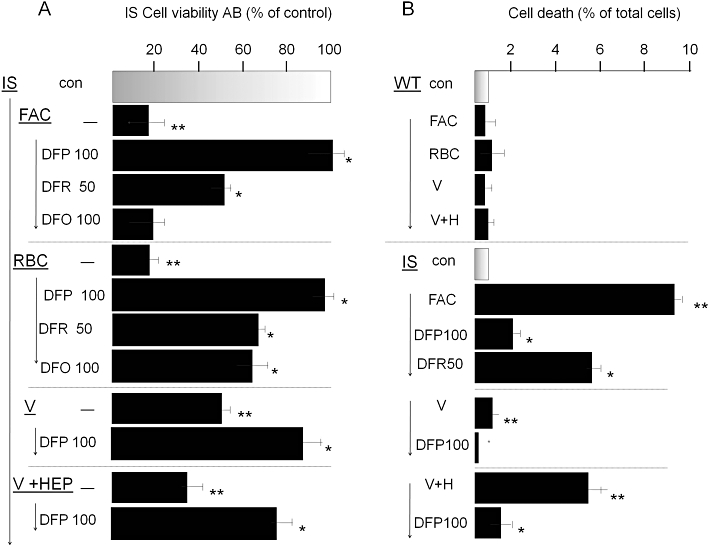
Reversal of iron toxicity in iron sensitive (IS) RAW 264.7 macrophages by chelators. IS cells were preincubated overnight with 100 µM ferric ammonium citrate (FAC), or for 1 h with opsonized erythrocytes [red blood cells (RBCs)] or 24 h with Venofer (500 µM), washed and cultured without or with added iron chelators deferiprone (DFP; 50 and 100 µM), deferasirox (DFR; 100 µM) or deferrioxamine (DFO; 100 µM). Following overnight incubation, they were assessed for cell viability with Alamar Blue (AB) (shown in A) and for maintenance of plasma membrane integrity (cell death) by the propidium iodide-Hoechst 33342 double stain described in the Methods section (shown in B). After exposure to Venofer, cells were cultured without and with hepcidin (‘V’ and ‘V + HEP’). Data are given as % of control cells (no FAC or RBC treatment) and shown as means ± standard deviation of three independent experiments. Staurosporin (0.5 µM) served as positive control and caused >90% cell death (not shown). The higher level of iron toxicity compared with that described in Figure 1 is associated with the use of lower initial cell seeding density in the cultures, necessitated by the 48 h duration of the experiment.
The cytoprotective effect of chelators from iron-driven oxidative damage is thought to be achieved by the neutralization of LCI. However, most of the accumulated cell iron is stored within ferritin, which undergoes degradation in response to continuous iron depletion, particularly after prolonged chelator treatment. (Konijn et al., 1999; De Domenico et al., 2009). We therefore assessed the relative efficacies of the chelators in extracting iron from cell iron stores by following the ferritin levels in IS cells preloaded with iron via FAC or erythrophagocytosis (Figure 6). In the experimental period of 24 h, DFR at 50 µM was relatively more efficacious than 100 µM DFP or DFO in triggering ferritin degradation in iron loaded IS cells. Thus the ability of chelators to protect cells from the toxic effect of accumulated iron is not directly related to their ability to reduce cellular ferritin levels.
Figure 6.
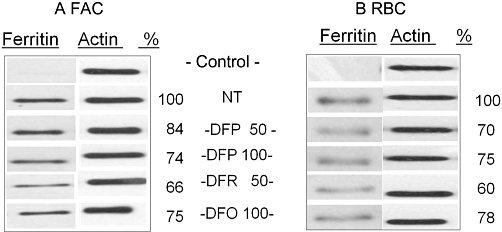
Effect of chelation on ferritin levels in iron-loaded RAW 264.7 (IS) macrophages. Cells were (A) incubated overnight with 100 µM FAC, or (B) for 1 h with opsonized erythrocytes [red blood cells (RBCs)], then washed and grown for 24 h in the presence of deferiprone (DFP), deferasirox (DFR) or deferrioxamine (DFO), given in micromolar. Ferritin levels in whole cell lysates were determined by Western blotting with antibodies against H-ferritin and blots were re-probed for actin. Relative values of ferritin levels (given as % of control) determined by densitometry are shown. Data are representative of one out of four independent experiments.
Chelator-mediated transfer of iron from iron-loaded to iron-depleted cells but not to intracellular bacteria
While all three chelators prevented to various extents the iron-mediated damage to iron-loaded cells, it was of interest to assess whether the chelated iron could be rendered available to other cells and yet withheld from engulfed bacteria. To this end, macrophage cells that had been iron-loaded via erythrophagocytosis (Figure 7A) or via exposure to V (Figure 7B) were co-cultured with iron-starved, growth-arrested K562 human erythroleukaemia cells for 18 h. K562 cells were brought to growth arrest by previous iron deprivation via DFO chelation and the co-culture with macrophages was performed in media supplemented with iron-depleted serum containing iron-free apo-transferrin in order to minimize iron supply from any sources other than the macrophages. The result of chelator-mediated transfer of iron was assessed by determination of the growth of both RAW macrophages and K562 cells (Figure 7). As shown, erythrophagocytosis blocked IS cell growth, but only DFP restored it to control levels (value of 1) whereas all others remained significantly different (P < 0.01) from 1. Conversely, in K562 cells that were deprived of iron, resumption of DNA synthesis s was observed only when the cells were co-cultured with iron preloaded RAW cells [either via erythrophagocytosis (A) or via V (B)] and only in the presence of 50 or 100 µM DFP. In contrast, incubation in the presence of 50 µM DFR or DFO led to K562 cell growth arrest and early cell death manifested as decreased DNA content, in line with previous observations (Sohn et al., 2008).
Figure 7.
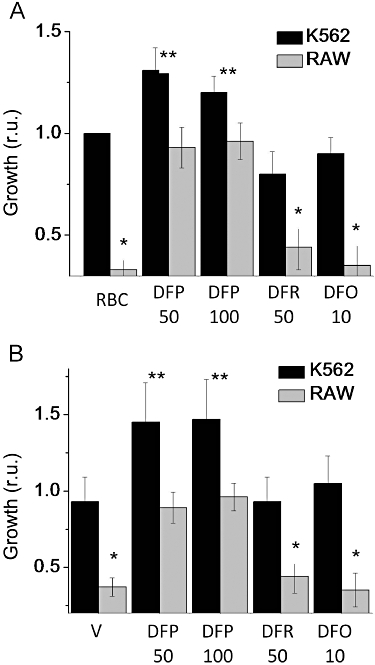
Chelator-mediated iron relocation from iron-loaded RAW 264.7 macrophages to iron-depleted K562 cells in co-culture. Iron-sensitive (IS) cells were iron-loaded either (A) by incubation for 1 h with opsonized erythrocytes to allow erythrophagocytosis, followed by washing of erythropcytes and incubation overnight in culture conditions followed by removal of non-internalized RBC by hypotonic lysis and washing, or (B) by overnight exposure to 500 µM Venofer (V) and washing. Iron-deprived K562 cells were added on top of the RAW 264.7 cells and the mixed cell culture was grown for 24 h in iron-free, apo-transferrin containing medium supplemented with the iron chelators deferiprone (DFP), deferasirox (DFR) or deferrioxamine (DFO) at the indicated concentrations (µM). The K562 cells, which grow in suspension, were carefully separated from the substrate-attached RAW 264.7 IS cells by aspiration and the relative numbers of K562 cells and of RAW cells were quantified in a fluorescence plate reader following staining with the DNA fluorescent stain Hoechst 33342. Data are expressed as mean growth (in fluorescence units) relative to the control ± standard deviation (n = 3). The asterisks denote statistically significant differences between treatment and the respective control (* for RAW cells and ** for K562 cells).
In order to test whether iron redistribution by DFP also increased iron availability to macrophage-ingested bacteria, we assessed its effect on Salmonella multiplication in RAW macrophages exposed to FAC or V (Table 3). Preloading of WT cells with FAC or V before bacterial ingestion caused a slight but significant increase in bacterial multiplication relative to control. Furthermore, in accordance with previous results (Nairz et al., 2009), the continued presence of iron, in the form of FAC or V during the post-ingestion period led to a further increase in bacterial multiplication within cells, being indicative of the requirement for iron for optimal intra-macrophage growth of these bacteria. However, incubation with DFP after bacterial ingestion did not increase bacterial multiplication in both control and FAC-or V-preloaded macrophages, indicating that in this experimental system the agent does not enhance iron availability to ingested bacteria.
Table 3.
Chelator-mediated iron relocation in iron-laden macrophages: effects on multiplication of ingested bacteria
| Colony forming units in treated relative to control cells | ||||
|---|---|---|---|---|
| Pretreatment | None | FAC | Venofer (V) | DFP |
| Control | 100 ± 20 | 156 ± 16xx | 439 ± 19xx | 131 ± 14 |
| FAC | 150 ± 17* | 236 ± 18*,xx | 559 ± 55**,xx | 108 ± 24 |
| V | 239 ± 65** | 389 ± 65**,xx | 862 ± 43**,xx | 288 ± 42 |
Wild-type cells, either untreated (control) or preloaded with 100 µM FAC or 500 µM V overnight (preincubation, FAC or V, respectively), were allowed to ingest bacteria (S. thyphimurium) (pretreatment) then washed and treated for 24 h in regular medium with no additions or with 100 µM ferric ammonium sulphate (FAC) or 500 µM Venofer (V) and/or 100 µM deferiprone (DFP) (post-treatment)and bacterial colony-forming units (CFUs) were quantified as described in the Methods section.
Data are given as % of control cells (no treatment) and values presented as mean ± standard deviation (n = 6). The statistical analysis (using SPSS, Bonferroni variance test-parameters) on sets of data obtained independently indicate significant differences (*P < 0.03; **P < 0.001) between pretreatment with FAC or V versus the respective control and (xxP < 0.001) between treatment with FAC or V versus none or versus DFP.
Discussion
Iron dyshomeostasis resulting in tissue iron retention, such as IRIDA (Finberg, 2009) or ACD (Weiss and Goodnough, 2005; Finberg, 2009; Babitt and Lin, 2010) are linked to the activity of the iron transporter ferroportin, a key player in the release of iron from erythrocyte-recycling macrophages and iron absorption in the gut (Theurl et al., 2009; Recalcati et al., 2010). This has led to experimental approaches aimed at targeting ferroportin (Nemeth, 2010), its principal regulator hepcidin (Sasu et al., 2010) and its associated signal-response machineries (Babitt et al., 2007; Yu et al., 2008; Mair and Weiss, 2009), but a possible interference with physiologically essential functions might limit their clinical use (Mair and Weiss, 2009).
We assessed an alternative or complementary therapeutic strategy for coping with disease-related macrophage iron accumulation in the setting of systemic iron deficiency, by applying agents with a potential for cell iron detoxification in concert with conservative redistribution of the scavenged metal (Breuer and Cabantchik, 2009; Kakhlon et al., 2010). We reasoned that a potential candidate should, at the minimum, be devoid of side effects that exacerbate the iron deficiency per se and, optimally, even reduce it. Agents with the requisite therapeutic properties are expected to attain a conservative redistribution of the metal by scavenging excess iron from overloaded cells and conveying the chelated metal to circulating apo-Tf (Sohn et al., 2008) and/or to other cells for reutilization (Breuer and Cabantchik, 2009; Kakhlon et al., 2010). Such an approach might be of therapeutic value in patients with iron restrictive anaemia as well as in those frequently treated with combinations of haematopoietic stimulating agents and iron supplements (Macdougall, 2010). As i.v. iron formulations are composed of polymeric micro-particulate iron forms, they are ingested and processed mainly within resident macrophages in liver and spleen (Mair and Weiss, 2009).
In search of models for exploring iron redistribution we isolated a subline of murine macrophage RAW 264.7 cells (IS) with an increased iron sensitivity. The IS cells appear to spontaneously over-accumulate iron under normal culture conditions, as indicated by elevated total cell iron, increased basal reactive O- intermediate levels in the cytosol and mitochondria, induction of ferritin and ferroportin synthesis. Furthermore, a preliminary gene expression microarray screen showed that the most significant difference in basal mRNA levels between IS and WT cells is a fourfold decrease in transferrin receptor 1 expression (Y-S. Sohn et al., unpubl. obs.). More striking is IS cells' inability to tolerate iron loads, as they undergo growth arrest by exposure to FAC or V under conditions that do not affect the WT cells. Similarly, endocytosis of macromolecular iron or phagocytosis of aged RBC produces marked oxidative-toxicity in IS, but not in WT cells. These deleterious effects may be explained in part by an enhanced rate of internalization of particulate iron complexes and erythrocytes (Figure 2), which may also explain the markedly elevated total iron content of IS cells after iron loading (Figure 3).
In addition to excessive endocytosis, the propensity of IS cells to generate elevated LCI (Figure 3) and cytosolic ROI (Figure 4 and Table 2) following exposure to iron sources, indicates that these cells also have an impaired capacity to process the internalized iron, despite robust induction of ferritin and ferroportin (Figure 3), the two principal proteins involved in protection from iron toxicity. Importantly, the fact that ferroportin down-regulation by hepcidin exacerbates IS cell iron loading (Figure 3) and ensuing toxicity (Figure 1) indicates that ferroportin also plays a role in iron overload management in these cells, as it does in macrophages generally. It also indicates that the IS cells used in this study provide an extreme but useful experimental model for situations in which macrophage ferroportin is chronically down-regulated even after erythrophagocytosis, due to elevated circulating hepcidin and cytokine levels (Weiss and Goodnough, 2005; Beaumont and Delaby, 2009). Thus, the IS cells experimentally reproduce one of the characterisitic features of ACD-affected macrophages – a restricted ability to eliminate excessively accumulated iron. That restricted ability is accentuated following erythrophagocytosis or exposure to iron polymers used for parenteral iron supplementation. Together, these features make the IS cells a highly sensitive model for quantitatively assessing the ability of iron scavenging agents to prevent cellular iron load in the presence of a ferroportin blockade and to restore cell activities.
Rescue of cells from the detrimental effects of iron loading by a variety of chelators has been demonstrated previously (Hider et al., 1994; Kalinowski and Richardson, 2005; Glickstein et al., 2006) and was also observed here for DFP, DFR and DFO applied to IS cells challenged with FAC, V (±hepcidin) or RBC (Figure 5). However, for chelators to be useful in systemic iron deficiency they need to perform as iron redistribution agents. Such agents must be endowed with: (a) the ability to maintain any chelated iron in a biologically usable form and thereby (b) permit continued cell growth (Breuer and Cabantchik, 2009; Kakhlon et al., 2010) of both iron-loaded cells and be permissive for growth of iron-replete as well as iron-deprived cells. This requirement has been shown to be partially fulfilled by DFP in in vitro cell models of Friedreich's ataxia (Kakhlon et al., 2010) and cardiomyocyte iron overload (Glickstein et al., 2006). Here we extended the notion to an experimental cell model of iron retention by macrophages and tested the ability of chelating agents to mediate iron transfer to iron-deprived human erythroleukaemia cells (Figure 7A and B). Other chelators with a relatively high cell iron scavenging ability, but lesser ability to transfer complexed iron to apo-Tf (Breuer et al., 2001), such as DFR, were less efficacious than DFP in both rescuing iron overloaded macrophages and at the same time supporting cell growth of iron deprived K562 cells. We attribute the resumption of K562 cell growth in the presence of iron loaded IS cells and DFP to DFP-mediated redistribution of iron between the two cell populations. At present it is not clear whether DFP conveyed iron to the K562 cells directly by transmembrane permeation of DFP-iron complexes and donation to acceptor proteins, or via extracellular transfer to transferrin followed by receptor mediated uptake. Based on previous studies (Sohn et al., 2008), both mechanisms are plausible, although in vivo only the latter is likely to be relevant. The possibility that RAW cells might secrete growth stimulating factors following addition of DFP but not DFR or DFO, cannot be ignored, although the fact that the DFP stimulating effect was observed only when iron loaded RAW cells were used, renders it unlikely in the context of the described experiment.
One possible objection to the use of iron-redistributing reagents in ACD associated with infection by pathogens is the potential for donation of iron to the invading microorganisms. This question was addressed in the present work (Table 3) by assessing the effect of DFP on intracellular multiplication of Salmonella typhimurium in WT macrophages exposed to FAC and V. Intracellular bacterial growth was not enhanced but even slightly inhibited by DFP indicating that iron transfer from DFP-Fe chelates to endobacteria is unlikely. Experiments carried out with the IS cell line yielded a similar pattern of results (data not shown); however, bacterial survival in these cells was considerably lower than in WT, a finding currently under investigation.
In this work we reinforced the principle of iron redistribution (Breuer and Cabantchik, 2009; Kakhlon et al., 2010) observed with cell models of Friedreich's ataxia, whereby DFP managed to restore most of the cell functions affected by frataxin deficiency. The effect of DFP was not restricted to detoxification of mitochondrial labile iron as it also provided support for de novo iron-sulfur cluster synthesis (Kakhlon et al., 2008). Clinical studies with DFP in Friedreich's ataxia patients showed amelioration of complications associated with iron accumulation in the heart and in the brain (Boddaert et al., 2007; Velasco-Sánchez et al., 2011). Importantly, the iron status of patients could be largely maintained with moderate DFP treatments even in the absence of overt RES iron accumulation. In early studies on rheumatoid arthritis, an ACD resulting condition, DFP produced slight increases in haemoglobin levels (Vreugdenhil et al., 1990), consistent with the notion that DFP can mobilize sizeable amounts of iron sequestered in the RES and/or that it can induce erythropoietin formation, as does DFO (Zhu and Bunn, 1999). Moreover, the increasing use of long-term, high-dose i.v. iron supplementation to treat anaemia of ACD could become complicated by eventual systemic iron loading due to inefficient utilization of the infused iron (Besarab et al., 2009). In such situations, iron-redistributing agents could prove particularly useful for reducing iron overload while achieving higher iron availability for haemoglobinization.
Clearly, the ability of some chelators to conservatively redistribute iron within and/or between cells can have both beneficial as well as detrimental effects (Kawabata et al., 2007). In the case of DFP, its idiosyncratic neutropaenic effects have limited its therapeutic potential. However, the novel approach of conservative iron redistribution for treating ACD and of the experimental platform used in this work, could serve as a basis for screening novel agents with better performance as rescuers of iron-affected cells and possibly also potential reversers of cellular iron deficiency.
Acknowledgments
This work was supported by the Israel Science Foundation (141/06) and the Framework 6 (LSHM-CT-2006-037296 Euroiron1) and by the Canadian Friends of the Hebrew University.
Glossary
Abbreviations
- AB
Alamar Blue
- ACD
anaemia of chronic disease
- Apo-Tf
apo-transferrin
- CALB
calcein blue (methylumbelliferone-8-methyleneiminodiacetic acid)
- CALG
calcein green {3,3'-bis[N,N-bis(carboxymethyl) aminomethyl] fluorescein}
- CDCHF
6-carboxy-2',7'-dichlorofluorescein diacetate-acetoxymethyl ester
- CO
carbonyl
- DFO
deferrioxamine
- DFP
deferiprone
- DFR
deferasirox
- DHR
dihydrorhodamine 123
- FAC
ferric ammonium citrate
- IRIDA
iron-refractory iron deficient anaemia
- IS
iron sensitive
- LCI
labile cell iron
- PI
propidium iodide
- RBC
red blood cell
- ROI
reactive oxygen intermediates
- ROS
reactive oxygen species
- SIH
salicyl isonicotinoyl hydrazone
- V
venofer
Conflicts of interest
None.
References
- Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Ann Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- Agarwal N, Prchal JT. Anemia of chronic disease (anemia of inflammation) Acta Haematol. 2009;122:103–108. doi: 10.1159/000243794. [DOI] [PubMed] [Google Scholar]
- Babitt JL, Lin HY. Molecular mechanisms of hepcidin regulation: implications for the anemia of CKD. Am J Kidney Dis. 2010;55:726–741. doi: 10.1053/j.ajkd.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117:1933–1939. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont C, Delaby C. Recycling iron in normal and pathological states. Semin Hematol. 2009;46:328–338. doi: 10.1053/j.seminhematol.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Besarab A, Hörl WH, Silverberg D. Iron metabolism, iron deficiency, thrombocytosis, and the cardiorenal anemia syndrome. Oncologist. 2009;14(Suppl 1):22–33. doi: 10.1634/theoncologist.2009-S1-22. [DOI] [PubMed] [Google Scholar]
- Boddaert N, Le Quan Sang KH, Rotig A, Leroy-Willig A, Gallet S, Brunelle F, et al. Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood. 2007;110:401–408. doi: 10.1182/blood-2006-12-065433. [DOI] [PubMed] [Google Scholar]
- Breuer W, Cabantchik ZI. Disorders affecting iron distribution: causes, consequences and possible treatments. BloodMed. 2009 http://www.bloodmed.com/800000/mini-reviews1.asp?id = 253&p = 1&v = 1. [Google Scholar]
- Breuer W, Ermers MJ, Pootrakul P, Abramov A, Hershko C, Cabantchik ZI. Desferrioxamine-chelatable iron, a component of serum non-transferrin-bound iron, used for assessing chelation therapy. Blood. 2001;97:792–798. doi: 10.1182/blood.v97.3.792. [DOI] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, Kaplan J. Specific iron chelators determine the route of ferritin degradation. Blood. 2009;114:4546–4551. doi: 10.1182/blood-2009-05-224188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RW, Sharma M, Ogwang W, Patel KJ, Bartlett AN, Kontoghiorghes GJ. The effect of alphaketohydroxypyridine chelators on transferrin saturation in vitro and in vivo. Drugs Today (Barc) 1992;28:19–23. [Google Scholar]
- Finberg KE. Iron-refractory iron deficiency anemia. Semin Hematol. 2009;46:378–386. doi: 10.1053/j.seminhematol.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Germann UA. Detection of expressed recombinant protein based on multidrug resistance: P-glycoprotein. Methods Mol Biol. 1997;63:139–159. doi: 10.1385/0-89603-481-X:139. [DOI] [PubMed] [Google Scholar]
- Glickstein H, Ben El R, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators. A fluorescence study of chelator action in living cells. Blood. 1994;106:3242–3250. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- Glickstein H, Ben El R, Link G, Breuer W, Konijn AM, Hershko C, et al. Action of chelators in iron-loaded cardiac cells: accessibility to intracellular labile iron and functional consequences. Blood. 2006;108:3195–3203. doi: 10.1182/blood-2006-05-020867. [DOI] [PubMed] [Google Scholar]
- Hider RC, Porter JB, Singh S. The design of therapeutically useful iron chelators. In: Bergeron RJ, Brittenham GM, editors. The Development of Iron Chelators for Clinical Use. Boca Raton, FL: CRC Press; 1994. pp. 353–358. [Google Scholar]
- Kakhlon O, Manning H, Breuer W, Melamed-Book N, Cortopassi G, Munnich A, et al. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood. 2008;112:5219–5227. doi: 10.1182/blood-2008-06-161919. [DOI] [PubMed] [Google Scholar]
- Kakhlon O, Breuer W, Munnich A, Cabantchik ZI. Iron redistribution as a therapeutic strategy for treating diseases of localized iron accumulation. Can J Physiol Pharmacol. 2010;88:187–196. doi: 10.1139/Y09-128. invited review. [DOI] [PubMed] [Google Scholar]
- Kalinowski DS, Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- Kawabata H, Tomosugi N, Kanda J, Tanaka Y, Yoshizaki K, Uchiyama T. Anti-interleukin 6 receptor antibody tocilizumab reduces the level of serum hepcidin in patients with multicentric Castleman's disease. Haematologica. 2007;92:857–858. doi: 10.3324/haematol.10794. [DOI] [PubMed] [Google Scholar]
- Kemma EHJM, Tjalsma H, Willems HL, Swinkels DW. Hepcidin: from discovery to differential diagnosis. Hematologica. 2009;93:90–97. doi: 10.3324/haematol.11705. doi: 10.3324/haematol. 11705. [DOI] [PubMed] [Google Scholar]
- Konijn AM, Glickstein H, Vaisman B, Meyron-Holtz EG, Slotki IN, Cabantchik ZI. Thecellular labile iron pool and intracellular ferritin in K562 cells. Blood. 1999;94:2128–2134. [PubMed] [Google Scholar]
- Lescoat G, Chantrel-Groussard K, Pasdeloup N, Nick H, Brissot P, Gaboriau F. Antiproliferative and apoptotic effects in rat and human hepatoma cell cultures of the orally active iron chelator ICL670 compared to CP20: a possible relationship with polyamine metabolism. Cell Prolif. 2007;40:755–767. doi: 10.1111/j.1365-2184.2007.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwiczek S, Aigner E, Theurl I, Weiss G. Cytokine-mediated regulation of iron transport in human monocytic cells. Blood. 2003;101:4148–4154. doi: 10.1182/blood-2002-08-2459. [DOI] [PubMed] [Google Scholar]
- Macdougall IC. Iron supplementation in the non-dialysis chronic kidney disease (ND-CKD) patient: oral or intravenous? Curr Med Res Opin. 2010;26:473–482. doi: 10.1185/03007990903512461. [DOI] [PubMed] [Google Scholar]
- Mair SM, Weiss G. New pharmacological concepts for the treatment of iron overload disorders. Curr Med Chem. 2009;16:576–590. doi: 10.2174/092986709787458434. [DOI] [PubMed] [Google Scholar]
- Nairz M, Theurl I, Ludwiczek S, Theurl M, Mair SM, Fritsche G, et al. The co-ordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007;9:2126–2140. doi: 10.1111/j.1462-5822.2007.00942.x. [DOI] [PubMed] [Google Scholar]
- Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, et al. Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood. 2009;114:3642–5131. doi: 10.1182/blood-2009-05-223354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E. Targeting the hepcidin-ferroportin axis in the diagnosis and treatment of anemias. Adv Hematol. 2010;2010:750643. doi: 10.1155/2010/750643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Paradkar PN, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron depletion limits intracellular bacterial growth in macrophages. Blood. 2008;112:866–874. doi: 10.1182/blood-2007-12-126854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recalcati S, Minotti G, Cairo G. Iron regulatory proteins: from molecular mechanisms to drug development. Antioxid Redox Signal. 2010;13:1593–1616. doi: 10.1089/ars.2009.2983. [DOI] [PubMed] [Google Scholar]
- Reznick AZ, Packer L. Oxidative damage to proteins: spectrophotometric method for carbonyl assay. Methods Enzymol. 1994;223:357–363. doi: 10.1016/s0076-6879(94)33041-7. [DOI] [PubMed] [Google Scholar]
- Sasu BJ, Hainu M, Boone TC, Cooke KS, Arvedson TL, Plewa C, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood. 2010;115:3616–3624. doi: 10.1182/blood-2009-09-245977. [DOI] [PubMed] [Google Scholar]
- Schaible U, Kaufman SH. Iron and microbial infection. Nat Rev Microbiol. 2004;2:946–953. doi: 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- Sohn YS, Breuer W, Munnich A, Cabantchik ZI. Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood. 2008;113:1690–1699. doi: 10.1182/blood-2007-07-102335. [DOI] [PubMed] [Google Scholar]
- Theurl I, Aigner E, Theurl M, Nairz M, Seifert M, Schroll A, et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood. 2009;113:5277–5286. doi: 10.1182/blood-2008-12-195651. [DOI] [PubMed] [Google Scholar]
- Velasco-Sánchez D, Aracil A, Montero R, Mas A, Jiménez L, O'Callaghan M, et al. Combined therapy with idebenone and deferiprone in patients with Friedreich ataxia. Cerebellum. 2011;10:1–8. doi: 10.1007/s12311-010-0212-7. [DOI] [PubMed] [Google Scholar]
- Voigt W. Sulforhodamine B assay and chemosensitivity. Methods Mol Med. 2005;110:9–48. doi: 10.1385/1-59259-869-2:039. [DOI] [PubMed] [Google Scholar]
- Vreugdenhil G, Swaak AJ, de Jeu-Jaspers C, van Eijk HG. Correlation of iron exchange between the oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one(L1) and transferrin and possible antianaemic effects of L1 in rheumatoid arthritis. Ann Rheum Dis. 1990;49:956–957. doi: 10.1136/ard.49.11.956-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Goodnough T. Anemia of chronic disease. New Engl J Med. 2005;352:1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- Yang F, Liu XB, Quinones M, Melby PC, Ghio A, Haile DJ. Regulation of reticuloendothelial iron transporter MTP1 (Slc11a3) by inflammation. J Biol Chem. 2002;277:39786–39791. doi: 10.1074/jbc.M201485200. [DOI] [PubMed] [Google Scholar]
- Yu PB, Hong CC, Sachidanandan C. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Bunn HF. Oxygen sensing and signaling: impact on the regulation of physiologically important genes. Respir Physiol. 1999;115:239–247. doi: 10.1016/s0034-5687(99)00024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]