

Abstract
BACKGROUND AND PURPOSE
Sigma-1 receptors are atypical receptors with potentially two transmembrane domains. Antagonists require doses significantly higher than their published affinities to have biological effects. We have reassessed the binding characteristics of these ligands and found antagonists bind to high- and low-affinity states not distinguished by agonists.
EXPERIMENTAL APPROACH
The affinities of sigma-1 receptor ligands was assessed using radioligand saturation and competition binding of [3H]-(+)-pentazocine to permeabilized MDA-MB-468 cells. This was compared with the effect of ligands on metabolic activity using an MTS-based assay and calcium signalling using cells loaded with the calcium dye, Fura-2.
KEY RESULTS
Sigma-1 receptor antagonists, but not agonists, show GTP- and suramin-sensitive high-affinity binding. Functional responses (calcium signalling and metabolic activity), while associated with sigma-1 receptor binding, required binding to an unidentified, low-affinity target.
CONCLUSIONS AND IMPLICATIONS
Sigma-1 receptors are coupled to G proteins. This interaction is only observed when analysing antagonist binding. The identity of the G protein remains to be resolved. The concept of agonist and antagonist at the sigma-1 receptor needs to be revisited.
Keywords: Sigma-1 receptor, agonist, antagonist, calcium, MTS, cholera toxin, IPAG, rimcazole, G protein
Introduction
The search for understanding of the sigma-1 receptor has been a long, mysterious and as yet incomplete journey. The receptor has mostly been studied within the CNS. However, more recent work over the past decade has shown the receptor to be abundant in neuronal, and non-neuronal tissues (Vilner et al., 1995) leading to its study in cancer biology (Spruce et al., 2004).
The sigma receptor was proposed as a novel fourth opioid receptor in 1976 to account for the behavioural effects of N-allylnormetazocine (SKF-10047), which could not be accounted for by the µ (morphine) or κ (ketocyclazocine) receptors (Martin et al., 1976). SKF-10047 was defined as an agonist as it raised pulse and respiration rates, as well as body temperature and caused dilatation of pupils in beagles. Interestingly, µ-receptor agonists lowered pulse and respiration rates, as well as body temperature and produced classic pinpoint pupils. Sigma-1 receptor antagonists were defined as agents that were able to prevent these responses. Later studies (e.g. Spruce et al., 2004) suggested that biochemically, agonists do not cause activation of the receptor but can block the effects of antagonists which do. More recently, data obtained using mice in which the sigma-1 receptor has been knocked out further cloud the picture; expression of these receptors acts as a prosurvival signal and treatment with antagonists causes apoptosis (Wang and Duncan, 2006).
The sigma-1 receptor, originally cloned from guinea-pig liver (Hanner et al., 1996), consists of a 223 amino acid long protein, which shares no homology with any other known mammalian protein. The subcellular location of this receptor is still under investigation: while Aydar et al. (2002) show plasma membrane localization of green fluorescent protein-labelled sigma-1 receptors, Hayashi and Su (2003) show equally compelling data for endogenous sigma-1 receptor in the endoplasmic reticulum. The endogenous ligand for the sigma-1 receptor has yet to be identified. Neurosteroids, in particular progesterone, are the main candidates for the endogenous ligand (Su et al., 1988; Ramamoorthy et al., 1995; Waterhouse and Collier, 1997). However, their affinities for this receptor appear to be too low for them to be endogenous ligands. The physiological levels of progesterone during pregnancy can reach up to 400 nM; however, only 2% of this is free to pass the blood–brain barrier (Pardridge et al., 1988; Schwarz et al., 1989), resulting in only approximately 8 nM free progesterone that is able to interact with sigma-1 receptors in the CNS (Schwarz et al., 1989). More recently, N,N-dimethyltryptamine (DMT) has been proposed as the endogenous ligand (Fontanilla et al., 2009). As with progesterone, the moderate affinity of DMT would suggest this trace amine is not the bona fide endogenous ligand.
Investigations have found that sigma-1 receptor antagonists modulate cytoplasmic calcium levels (Brent et al., 1996a,b;), and induce apoptosis (Brent and Pang, 1995; Vilner et al., 1995; Spruce et al., 2004), whereas the sigma-1 receptor agonists block these effects. This brings into question the biochemical nature of whether ligands are agonists or antagonists in the true sense.
Experiments using Ca2+-free buffers indicate that the sigma-1 receptor favours calcium influx mediated at the plasma membrane rather than Ca2+ release from intracellular stores (Spruce et al., 2004). Sigma-1 receptor antagonists induce a rapid increase in cytoplasmic [Ca2+], which can be inhibited by their agonists such as (+) pentazocine and SKF-10047 (Spruce et al., 2004). The spike in intracellular [Ca2+] caused by the antagonists leads to caspase-dependent apoptosis, which suggests that the presence of the sigma-1 receptor acts as a survival signal to the cell (Spruce et al., 2004). Sigma-1 receptor agonists alone do not induce calcium influx into the cell; however, they prevent influx mediated by depolarization (Tchedre et al., 2008).
The debate as to whether sigma-1 receptors are G protein-coupled is yet to be satisfactorily concluded. Early binding studies showed that the binding of sigma-1 receptor ligands could be altered by the addition of GTP and its analogues. Using rat brain membranes, Itzhak (1989) showed that the sigma-1 receptor ligand (+)-3-PPP binds high- and a low-affinity sites in the absence of a GTP analogue. Single-site binding was observed following the addition of GppNHp. Other similar data showed that GTP and its analogues are able to modify the binding characteristics of other sigma-1 receptor ligands (Beart et al., 1989; Connick et al., 1992). Studies have also shown that treatment with GPCR inhibitors such as Pertussis toxin inhibit high-affinity (+)-3-PPP binding, and remove the effect of GTP analogues on ligand binding (Itzhak, 1989). These data suggest that the sigma-1 receptor is a GPCR. However, this protein in no way resembles the classical 7 transmembrane GPCR. Also, other studies showed that GTPγS was unable to affect ligand binding at the sigma-1 receptor (Hong and Werling, 2000), and that doses of sigma-1 receptor agonists required to activate GTPase are much higher than those required to saturate the sigma-1 receptor (Tokuyama et al., 1997). In order to explain these contradictory results, it has been proposed that the sigma-1 receptor could fall into further subtypes with one subtype acting through G proteins and another G protein-independent (Bermack and Debonnel, 2005).
Sigma-1 receptor agonists have been shown to inhibit numerous types of K+ channel including voltage-gated K+ channels and Ca2+-activated K+ channels (Soriani et al., 1999; Aydar et al., 2002); these inhibitions are not sensitive to the G-protein inhibitor GDPβS or the G-protein activator GTPγS and require the sigma-1 receptor to be present (Lupardus et al., 2000; Aydar et al., 2002). Sigma-1 receptor agonists also cause the inhibition of Na+ channels and knocking down the sigma-1 receptor reduced the effect of these agonists (Johannessen et al., 2009).
All of the studies on the sigma-1 receptor's link to G proteins have used sigma-1 receptor agonists, which with regard to the sigma-1 receptor do not behave biochemically in the same way as true receptor agonists. They appear to behave more like typical antagonists, not having any effect at the sigma-1 receptor themselves. The sigma-1 receptor antagonists on the other hand appear to have activity at the sigma-1 receptor, with 9-{3-[(3R,5S)-3,5-dimethylpiperazin-1-yl]propyl}-9H-carbazole (rimcazole) and 1-(4-iodophenyl)-3-(2-adamantyl)guanidine (IPAG) causing an increase in cytoplasmic [Ca2+], which subsequently activates calcium-dependent PLC and inhibits PKB, leading to caspase-dependent apoptosis which could be inhibited by a 30 min preincubation with a sigma-1 receptor agonist, (+) SKF-10047 or (+) pentazocine (Spruce et al., 2004).
In normal healthy cells at any one time, there are specific signals that prevent programmed cell death (apoptosis); these are likely to have evolved to maintain correct spatiotemporal patterning during development (Kerr et al., 1972; Raff, 1992). There are also signals that lead to apoptosis, for example, when a cell is damaged or has reached the end of its lifespan. Cells with limited access to nutrients and survival factors may have evolved by expressing sigma-1 receptors to prevent them from undergoing premature apoptosis. Avoiding apoptosis is also important to tumour cell survival, and it has been suggested that tumours have hijacked the sigma-1 receptor as a potential driving force in cell survival (Wang et al., 2005; Wang and Duncan, 2006). As sigma-1 receptors are highly expressed in a number of cancers and tumours, they have been shown to be useful in tumour imaging, using labelled sigma-1 receptor ligands in positron emission tomography scans (Kawamura et al., 2005). Using a range of cancer cell lines, Spruce et al. (2004) showed that sigma-1 receptor antagonists, such as rimcazole and IPAG, were able to reduce tumour cell viability. Moreover, rimcazole was able to inhibit or reduce xenograft tumour growth. This indicates that the sigma-1 receptor plays a role in cell survival and that it is a potential target for cancer treatments. Sigma-1 receptor agonists such as cocaine and PRE-048 have also been shown to increase tumour growth (Zhu et al., 2003; Gardner et al., 2004).
In this paper, we show, for the first time, that sigma-1 receptors are coupled to G proteins. Unlike other receptor systems, antagonist binding is affected by the addition of GTP and suramin, whereas agonist binding is unaffected. Antagonists are able to raise intracellular calcium levels but only at doses at which the orthosteric binding site is saturated, suggesting there is a secondary site either on the receptor or on an accessory protein.
Methods
General materials
Tissue culture media (DMEM without HEPES, catalogue number 41965), antibiotics, trypsin and serum were purchased from Invitrogen (Paisley, UK). Nunc tissue culture ware was purchased from Fisher Scientific (Loughborough, UK). IPAG, purchased from Tocris Bioscience, Bristol, UK, was dissolved in DMSO. [3H]-(1S,9S,13S)-1,13-dimethyl-10-(3-methylbut-2-en-1-yl)-10-azatricyclotrideca-2,4,6-trien-4-ol [(+)-pentazocine; specific activity 32.2 Ci·mmol−1] was purchased from Perkin Elmer (Beaconsfield, UK).Other reagents were purchased from Sigma-Aldrich (Poole, UK). Drug and molecular target nomenclature conforms to the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2009).
Tissue culture
MDA-MB-468 cells (ATCC LGC Promotech, Teddington, UK) were maintained as previously described (Spruce et al., 2004). Note that no HEPES-based buffers were used as HEPES has sigma-1 receptor affinity (pKi± SEM 1.9 ± 0.5, Ki 12 mM, data not shown). The cells were permeabilized (five pulses at 3.75 KV·cm−1) and washed in Tris-buffered saline (10 mM Tris, 0.9% NaCl, pH 7.4) to remove any endogenous GTP. We have used permeabilized cells as many of the signalling pathways remain intact while allowing access to the cell interior (Safrany and Nahorski, 1994).
Saturation binding
Assays were performed in Tris-buffered saline using 0–300 nM [3H]-(+) pentazocine at room temperature for 2 h. Non-specific binding was determined using 0.1 mM rimcazole. The pKd± SEM of [3H]-(+)-pentazocine was determined as 7.8 ± 0.6, Kd 17 nM and the Bmax as 2300 ± 200 fmol·mg−1 (data not shown). Binding assays were also performed using intact cells at both 4°C and room temperature. No difference was observed in Bmax or Kd (data not shown). All experiments using intact cells were performed in a buffer comprising: 115 mM NaCl, 5 mM KCl, 1 mM NaH2PO4, 0.5 mM MgSO4, 11 mM glucose, 1.36 mM CaCl2, 0.1% bovine serum albumin, 50 mM Tris, pH 7.4.
Competition assays
Competition assays were performed using a final assay concentration of 30 nM [3H] (+) pentazocine. The assay was then allowed to equilibrate for 6 h at both 4°C and room temperature. After equilibration the cells were harvested using a Brandel M-24R cell harvester through GF/B fired glass fibre filters (Semat International, St Albans, UK), washing three times with 10 mL of buffer at room temperature. Non-specific [3H]-(+)-pentazocine binding was determined using 0.1 mM rimcazole. Under these conditions less than 10% of the [3H]-(+)-pentazocine was bound.
MTS cellular metabolic activity assay
Cellular metabolic activity was measured using the CellTiter 96® AQueous non-radioactive cell proliferation assay (MTS assay; Promega, Southampton, UK) as previously described (Spruce et al., 2004).
Fura-2 calcium measurements
Approximately 2 x 107 cells were resuspended in 2 mL of buffer. The cells were loaded with 5 µM Fura-2-acetoxymethyl ester (Fura-2-AM; Invitrogen; Grynkiewicz et al., 1985) and incubated at room temperature for 45 min.
The cells were then spun down and washed in the warmed calcium buffer three times before being made up to 106 cells·mL−1 and incubated at room temperature for a further 30 min to allow the acetoxymethyl groups to be removed by cellular esterases resulting in free Fura-2 in the cells. The cells were then kept in suspension in the dark at room temperature before use in calcium measurements. Two millilitre of Fura-2-loaded cells were placed in a cuvette with a magnetic stirring bar, and maintained at 37°C. The cells were excited at 340 nm and 380 nm and emissions were recorded at 510 nm using a Perkin Elmer LS 50B fluorescence spectrometer.
Drugs were pH balanced to 7.4. At the end of each individual experiment 20 µL of 1 mg·mL−1 digitonin was added to the cuvette to permeabilize the cells allowing calcium to flood the dye giving a maximum fluorescence, followed by 60 µL of 0.5 M EDTA pH 8.5 to chelate the calcium giving a minimum fluorescence. These values were then used to calculate the cytoplasmic calcium concentration of the cells (Grynkiewicz et al., 1985). Unloaded cells were used to check autofluorescence and drug fluorescence.
Sigma-1 receptor knockdown
The sigma-1 receptor RNAi pSilencer vector and the non-targeting RNAi control described previously (Aydar et al., 2006) were obtained from Dr Christopher Palmer (London Metropolitan University). MDA-MB-468 cells were transfected using Jet-PEI DNA transfection reagent using the protocol in the Jet-PEI handbook (Autogen Bioclear, Calne, UK). Sigma-1 receptor activity was assessed 24–48 h after transfection.
Data analysis
Saturation and competition data were analysed using non-linear regression analysis (GraphPad Prism, version 4.0 for Macintosh) to fit saturation curves and calculate maximal radioligand binding and dissociation constants. In order to give a more true representation of the drugs affinity for the receptor, the IC50 was converted to a Ki[where the Hill slope (nH) was near unity] or K50 (where the nH was not close to unity) value using the Cheng Prusoff equation (Cheng and Prusoff, 1973). Data are presented as mean ± SEM.
When comparing two binding models to see which fits the data better, the F-test was employed to compare the models. Fura-2 fluorescence data were collected using FL WinLab software (Perkin Elmer, Waltham, MA, USA). MTS and protein assays were read using a Versamax plate reader, and the data collected using Softmax Pro software.
Protein measurements
Protein amounts were determined using Bio-Rad Protein Dye Reagent, based on Coomassie Brilliant Blue G250 (Bio-Rad, Hemel Hempstead, UK). Bovine serum albumin was used to prepare standards.
Results
Cellular effects of antagonists at the sigma-1 receptor
IPAG is a ‘potent’ sigma-1 receptor antagonist with a published affinity of 2.8 nM (Wilson et al., 1991), yet doses of 10 µM or above are frequently used to see effects at the sigma-1 receptor. We have revisited binding, calcium and cell proliferation assays to investigate this anomaly. IPAG induced a dose-dependent increase in intracellular Ca2+ when added to stirred Fura-2-AM-loaded MDA-MB-468 cells (Figure 1). The pEC50 for IPAG in the calcium response assay was 3.9 ± 0.1, n = 6 (EC50 123 µM). IPAG also dose-dependently reduced cellular proliferation, determined using the MTS assay. We have previously shown this is due to apoptosis (Spruce et al., 2004). The pIC50 for IPAG in this assay was 4.6 ± 0.1, n = 5 (IC50 24 µM; Figure 2). The EC50 for IPAG in the calcium assay and the IC50 in the MTS assay are over 10 000 times higher than the published affinity for IPAG (Wilson et al., 1991).
Figure 1.
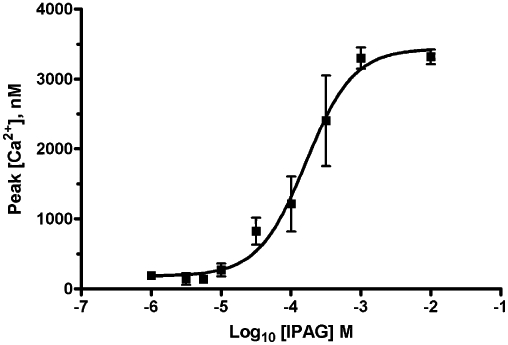
Cytoplasmic [Ca2+] increase in response to IPAG. Calcium response to IPAG in Fura-2-AM-loaded MDA-MB-468 cells. Error bars show SEM, n = 6.
Figure 2.
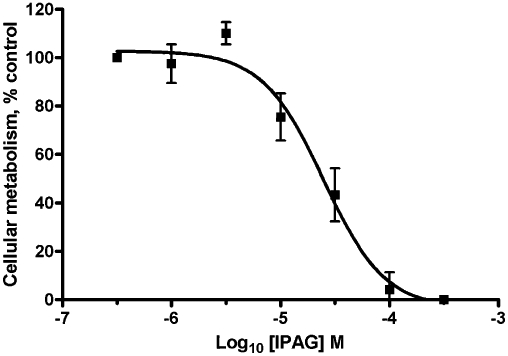
IPAG MTS assay dose–response. Cellular metabolic activity of MDA-MB-468 cells in response to IPAG, presented as a % of control. MTS was added 18 h after IPAG. Error bars show SEM, n = 5.
Knocking down the sigma-1 receptor by approximately 50% decreased the maximal Ca2+ response by 50%, but did not affect the EC50 (pEC50 4.08 ± 0.04, n = 3, EC50 80 µM; Figure 3). This suggests that the sigma-1 receptor is involved in the effects of IPAG and therefore the affinity of IPAG for this receptor was reassessed in the MDA-MB-468 cells. It also suggests that there is no receptor reserve for calcium signalling as reducing the receptor number by approximately 50% reduced the maximal response by 50%.
Figure 3.
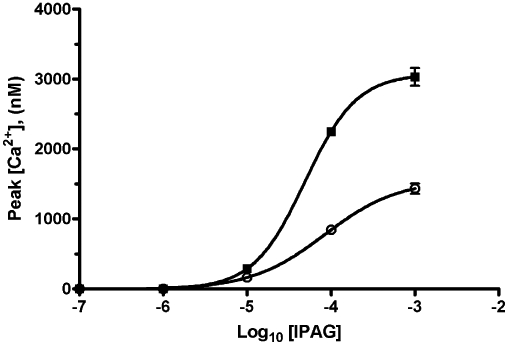
Effects of knocking down the sigma-1 receptor on cytoplasmic [Ca2+] response to IPAG. siRNA lowered maximal calcium response from 3100 ± 100 nM (non-targeting control) to 1600 ± 100 nM (mean ± SEM, n = 3). Error bars show SEM, n = 3. Parallel studies show receptor number was reduced from 1700 ± 100 fmol·mg−1 protein (non-targeting control) to 800 ± 200 fmol·mg−1 protein (mean ± SEM, n = 8).
Radioligand binding
To investigate the discrepancy between the published affinity of 2.8 nM for IPAG and the EC50 value observed in the calcium assay of 123 µM and the IC50 of 24 µM in the cell proliferation assay, the affinity of IPAG for the sigma-1 receptor was re-determined using [3H]-(+)-pentazocine competition binding (Figure 4). The radioligand binding assay did indeed give an affinity of IPAG for the sigma-1 receptor in the low nanomolar range pK50 8.6 ± 0.7, n = 5 (K50 2.5 nM; Figure 4A) compared to the published affinity of 2.8 nM. However, the single-site binding model had a very shallow nH of −0.3 ± 0.1, and the data did not appear to fit this model well. The two-site binding model fits the data better (Figure 4B), giving pKi high 12.8 ± 0.6, n = 5 (Kihigh 175 fM; 37% of total binding) and pKi low 6.8 ± 0.2, n = 5 (Ki low 168 nM; 63% total binding). Using the extra sum of squares F-test, the two-site model was preferred with an F-value of 31.1 and P-value <0.0001. Due to the high affinity of IPAG for this high-affinity site, it is anticipated that depletion of the competing ligand (IPAG) had occurred. The affinity for this high-affinity site must therefore be considered an estimate. During these studies, we found that HEPES was a ligand at the sigma-1 receptor, with pKi 1.9 ± 0.5, n = 5 (Ki 12 mM), so therefore chose to buffer solutions using Tris which was without effect on this receptor.
Figure 4.
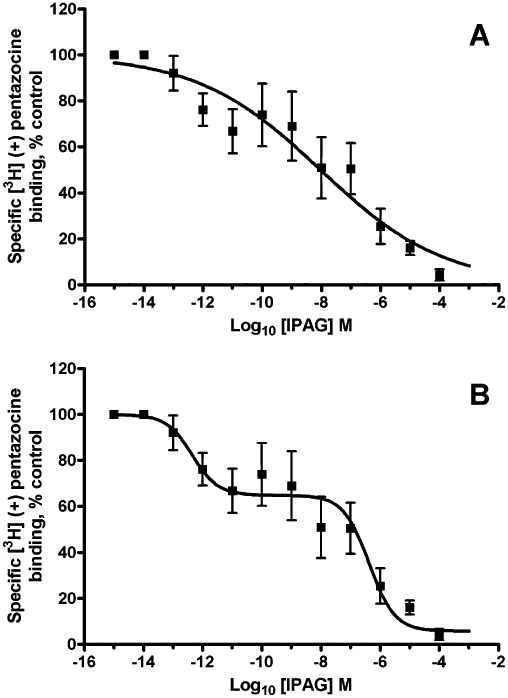
IPAG binding to the sigma-1 receptor. IPAG competition binding curve with 30 nM [3H]-(+)-pentazocine to permeabilized MDA-MB-468 cells. (A) Using a curve which has been created using a single-site analysis. (B) Using a curve which has been created using a multiple-site analysis. Error bars show SEM, n = 5.
In light of the observation that this competition curve resembles agonist competition curves binding to GPCRs (Itzhak, 1989; Connick et al., 1992), we examined the effects of G protein-uncoupling agents on agonist and antagonist binding. Assays were performed in the presence and absence of GTP [1 mM, 10 times the normal physiological concentration of GTP (Reinhardt et al., 2002)]. GTP was without effect on the binding of [3H]-(+)-pentazocine to the sigma-1 receptor in the permeabilized MDA-MB-468 cells; therefore, the competition assay with IPAG was repeated in the presence of GTP (Figure 5). The addition of GTP changed the shape of the binding curve, with a dramatic increase in the nH to −0.81 ± 0.05. The affinity of IPAG for the sigma-1 receptor also shifts to the low-affinity site (pKi 6.3 ± 0.2, Ki 500 nM) in the presence of GTP, which has no significant difference from the low-affinity site seen in the two-site fit in the absence of GTP (pKi 6.8; t-test P-value 0.183). Competition assays performed using intact cells also showed low-affinity binding to a single site [pKi 5.1 ± 0.2, n = 7 (Ki 8 µM)].
Figure 5.
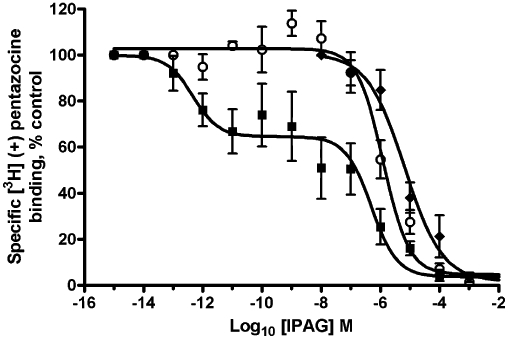
Effect of GTP (1 mM) on IPAG binding to the sigma-1 receptor. IPAG competition binding curve with 30 nM [3H]-(+)-pentazocine to permeabilized MDA-MB-468 cells washed with TBS to remove any endogenous GTP. Assays were performed in the absence (solid squares) or presence (open circles) of GTP. Competition binding to intact cells is also shown (solid diamonds). Error bars show SEM, n = 5.
We also tested a second sigma-1 receptor antagonist, rimcazole, which has a published affinity for this receptor of 0.9 µM (Gilmore et al., 2004) and has been previously shown to cause apoptosis in cancer cells through a sigma-1 receptor-dependent pathway (Spruce et al., 2004). We were unable to use rimcazole in the calcium assay as it interfered with the wavelengths at which Fura-2 is read, fluorescing intensely at 510 nm. Indeed, we were able to produce a dose–response curve in the absence of Fura-2-loaded cells with an apparent EC50 of approximately 10 µM (data not shown). However, rimcazole did not interfere with the reading of the MTS assay, and was found to inhibit metabolic activity of MDA-MB-468 cells with a pIC50 4.4 ± 0.2 (n = 5; IC50 45 µM) which is over 30 times higher than the published affinity for the sigma-1 receptor (0.9 µM; Gilmore et al., 2004). The affinity of rimcazole was therefore assessed, and the effect of GTP on rimcazole binding measured in Figure 6. The binding of rimcazole in the single-site binding model shows a shallow nH (−0.4 ± 0.1, n = 4) with a pK50 6.9 ± 0.5, n = 5 (K50 125 nM). The addition of GTP significantly increases in the nH (−1.0 ± 0.4, n = 5; P-value of 0.025) with a pKi 6.3 ± 0.3, n = 5 (K50 500 nM). Furthermore, the preferred model for binding of rimcazole in the absence of GTP is the two-site binding model; using the extra sum of squares F-test gave an F-value of 3.54 and a P-value of 0.028. The affinities for the sigma-1 receptor were pKi high 8.3 ± 0.4, n = 5 (Kihigh 5 nM), pKi low 5.9 ± 0.3, n = 5 (Ki low 1 µM), with the pKi high occupying 47% of the curve.
Figure 6.
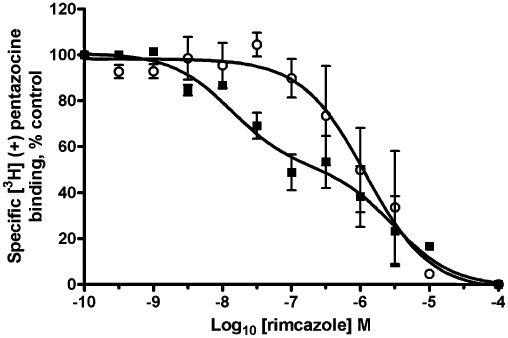
Effect of GTP (1 mM) on rimcazole binding to the sigma-1 receptor. Rimcazole competition binding curve with 30 nM [3H]-(+)-pentazocine to permeabilized MDA-MB-468 cells washed with TBS to remove any endogenous GTP. Assays were performed in the absence (solid squares) or presence (open circles) of GTP. Error bars show SEM, n = 5.
Suramin also uncouples G proteins (Beindl et al., 1996; Freissmuth et al., 1996; Hohenegger et al., 1998), suppressing the release of GDP from the α subunit, which is the rate-limiting step in G-protein activation. Suramin had no effect on pentazocine binding (data not shown); therefore, it was added to the radioligand competition assay with IPAG. In the presence of suramin (100 µM), a one-site fit was observed and nH increased to −0.8 ± 0.2, n = 5 (P-value 0.0342; Figure 7).
Figure 7.
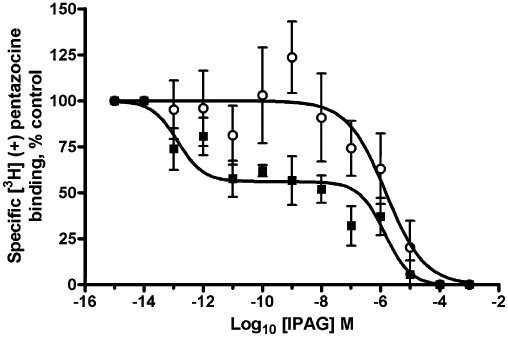
Effect of suramin (100 µM) on IPAG binding to the sigma-1 receptor. IPAG competition binding curve with 30 nM [3H]-(+)-pentazocine to permeabilized MDA-MB-468 cells washed with TBS to remove any endogenous GTP. Assays were performed in the absence (solid squares) or presence (open circles) of suramin. Error bars show SEM, n = 5.
In order to assess which G protein may be coupled to the sigma-1 receptor we treated MDA-MB-468 cells overnight with Pertussis toxin or cholera toxin. Such treatment would uncouple heterotrimeric G proteins of the Gi and Gs groups (Taylor, 1990). Neither treatment affected the binding of agonists or antagonists (data not shown).
Effects of cholera toxin
Cholera toxin treatment did, however, alter the calcium profile in response to IPAG. Peak [Ca2+]i to 100 µM IPAG was 600 ± 100 nM (n = 3 in control cells), whereas in cells treated with 100 µg·mL−1 cholera toxin overnight the peak response increased to 1600 ± 200 nM (n = 3; Figure 8). In contrast, such treatment blocked the ability of isoprenaline, acting on β2-adrenoceptors (Plummer et al., 2004), and dopamine, acting on D2-receptors (Lang et al., 2004), to raise cellular cAMP (data not shown).
Figure 8.
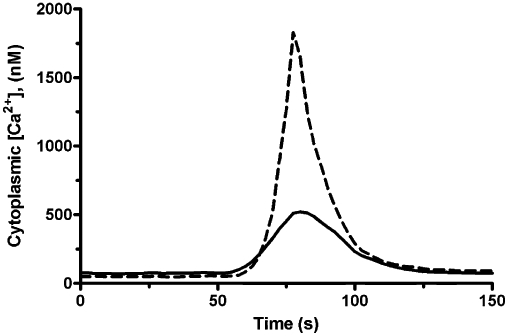
Effect of cholera toxin on IPAG-induced increase in cytoplasmic [Ca2+]. Representative (of n = 3) cytoplasmic Ca2+ response in Fura-2-loaded MDA-MB-468 cells following treatment with 100 µM IPAG. Solid line represents control cells; dashed line represents cells treated with 100 µg·mL−1 cholera toxin overnight. IPAG was added at 50 s.
Discussion and conclusions
Sigma-1 receptor ligands such as IPAG and rimcazole are thought of as antagonists, yet they are responsible for PKB inhibition and calcium influx into the cell leading to PLC activation, whereas sigma-1 receptor ligands such a (+)-SKF-10047 and (+)-pentazocine are thought of as agonists (due to the original effects seen in animals), yet they block the effects of the antagonists (Spruce et al., 2004). Considering the effects the sigma-1 receptor antagonists have on the cell, if there is G protein coupling with this receptor, the antagonists are the more likely tools to highlight it. Most previous studies that have looked at sigma-1 receptor coupling through G proteins have focused on their agonists. Sigma-1 receptor agonists have been shown to both stimulate (Maruo et al., 2000) and inhibit (Hong and Werling, 2000) GTPγS binding, whereas antagonists have no direct effect (Kim et al., 2010), but modulate GTPγS binding following classical GPCR activation. GTP-sensitive binding of antagonists to sigma-1 receptors has not previously been performed.
There is a large discrepancy between the IC50/EC50 (24 µM/123 µM) values observed for the response to IPAG in the MTS and calcium assay compared to its published affinity (2.8 nM; Wilson et al., 1991). This 10 000 times difference between the published affinity and the effective dose prompted a suspicion that either the published affinity was wrong or IPAG was having its effects through an allosteric site on the sigma-1 receptor or another receptor. Knocking down the sigma-1 receptor with the sigma-1 receptor-specific siRNA resulted in a reduction in the maximal response to IPAG in the calcium assay. This, taken with data from Spruce et al. (2004) showing sigma-1 receptor agonists blocking the effects of IPAG (although a 30 min pre-incubation is required), indicates that IPAG does indeed act through the sigma-1 receptor. Our attention turned therefore to the affinity of IPAG for the sigma-1 receptor. Wilson et al. (1991) identified IPAG as a high-affinity sigma-1 receptor ligand, although they do not present the competitive binding isotherm. The affinity of IPAG for the sigma-1 receptor was therefore reassessed using [3H]-(+)-pentazocine and permeabilized MDA-MB-468 cells. The competitive binding data revealed that when fitted with a single-site model the affinity was 2.5 nM, comparable to that previously described (Wilson et al., 1991); however, the nH for the single-site model was very low (−0.21).
Addition of GTP resulted in the nH approaching −1 and the affinity of IPAG and rimcazole for the sigma-1 receptor shifting to the low-affinity state (which still did not coincide with EC50 and IC50 doses for the calcium and MTS assays), suggesting G protein coupling. Furthermore, when suramin, which inhibits Gs (Hohenegger et al., 1998), Gi and Gq (Beindl et al., 1996), is added to the IPAG binding assay, the binding curve is again shifted to the low-affinity state with a nH approaching −1, providing yet more evidence that the sigma-1 receptor is coupled in some way to a G protein.
The model for agonists stimulating GPCRs is that they promote a conformational change in the receptor. Using circular dichroism, Kim et al. (2010) found that sigma-1 receptor antagonists promote a change in the C-terminal region of the protein, whereas agonists do not.
The affinity of IPAG determined for the sigma-1 receptor (8 µM, intact cells) still does not correspond to the functional EC50 for calcium signalling (123 µM) or IC50 for cellular proliferation (24 µM). We propose either that there is a second binding site, not recognized by [3H]-(+)-pentazocine, or that the affinity of IPAG for the sigma-1 receptor is altered as the receptor desensitizes. This second scenario is seen with nicotine acting at the nicotinic receptor: equilibrium binding studies show it has an affinity of 8 nM, whereas 86Rb2+ efflux studies show an EC50 of 10 µM (Xiao et al., 2006). A further explanation may involve comparing equilibrium binding where drug-receptor interactions are assessed after several hours (as observed for the binding and proliferation assays) with pre-equilibrium assays (a situation possible during calcium signalling assays), which could yield different values.
That cholera toxin did not alter the IPAG binding isotherm, while altering the biochemical response suggests that the involvement of G proteins is atypical. Furthermore, we have been unable to determine any changes in cAMP production in response to sigma-1 receptor agonists or antagonists (data not shown), which is the normal consequence of activating Gs.
In summary, we have shown that sigma-1 receptors show association with G proteins and that antagonists, not agonists, show GTP- and suramin-sensitive high-affinity binding. Gs may be involved as cholera toxin enhanced the calcium response to IPAG although it did not significantly affect binding of the antagonist. The nature of the G protein remains to be identified. Furthermore, a second target for sigma-1 receptor antagonists, be it on the receptor protein or elsewhere, may need to be identified.
Acknowledgments
We thank the Royal Society, the University of Bath and the University of Wolverhampton for financial support. We thank Dr Chris Palmer (London Metropolitan University) for siRNA reagents.
Glossary
Abbreviations
- (+)-3-PPP
(+)-3-(3-hydroxyphenyl)-N-1-(propyl)piperidine
- [Ca2+]
free concentration of Ca2+ ions
- DMT
N,N-dimethyltryptamine
- IPAG
1-(4-iodophenyl)-3-(2-adamantyl)guanidine
- SKF-10047
N-allylnormetazocine
Conflicts of interests
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158:S1–S239. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydar E, Palmer CP, Klyachko VA, Jackson MB. The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron. 2002;34:399–410. doi: 10.1016/s0896-6273(02)00677-3. [DOI] [PubMed] [Google Scholar]
- Aydar E, Onganer P, Perrett R, Djamgoz MB, Palmer CP. The expression and functional characterization of sigma (σ) 1 receptors in breast cancer cell lines. Cancer Letters. 2006;242:245–257. doi: 10.1016/j.canlet.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Beart PM, O'Shea RD, Manallack DT. Regulation of sigma-receptors: high- and low-affinity agonist states, GTP shifts, and up-regulation by rimcazole and 1,3-Di(2-tolyl)guanidine. J Neurochem. 1989;53:779–788. doi: 10.1111/j.1471-4159.1989.tb11773.x. [DOI] [PubMed] [Google Scholar]
- Beindl W, Mitterauer T, Hohenegger M, Ijzerman AP, Nanoff C, Freissmuth M. Inhibition of receptor/G protein coupling by suramin analogues. Mol Pharmacol. 1996;50:415–423. [PubMed] [Google Scholar]
- Bermack JE, Debonnel G. The role of sigma receptors in depression. J Pharmacol Sci. 2005;97:317–336. doi: 10.1254/jphs.crj04005x. [DOI] [PubMed] [Google Scholar]
- Brent PJ, Pang GT. Sigma binding site ligands inhibit cell proliferation in mammary and colon carcinoma cell lines and melanoma cells in culture. Eur J Pharmacol. 1995;278:151–160. doi: 10.1016/0014-2999(95)00115-2. [DOI] [PubMed] [Google Scholar]
- Brent PJ, Pang G, Little G, Dosen PJ, Van Helden DF. The sigma receptor ligand, reduced haloperidol, induces apoptosis and increases intracellular-free calcium levels [Ca2+]i in colon and mammary adenocarcinoma cells. Biochem Biophys Res Commun. 1996a;219:219–226. doi: 10.1006/bbrc.1996.0208. [DOI] [PubMed] [Google Scholar]
- Brent PJ, Saunders H, Dunkley PR. Intrasynaptosomal free calcium levels in rat forebrain synaptosomes: modulation by sigma (sigma) receptor ligands. Neurosci Lett. 1996b;211:138–142. doi: 10.1016/0304-3940(96)12711-7. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Connick JH, Hanlon G, Roberts J, France L, Fox PK, Nicholson CD. Multiple sigma binding sites in guinea-pig and rat brain membranes: G-protein interactions. Br J Pharmacol. 1992;107:726–731. doi: 10.1111/j.1476-5381.1992.tb14514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freissmuth M, Boehm S, Beindl W, Nickel P, Ijzerman AP, Hohenegger M, et al. Suramin analogues as subtype-selective G protein inhibitors. Mol Pharmacol. 1996;49:602–611. [PubMed] [Google Scholar]
- Gardner B, Zhu LX, Roth MD, Tashkin DP, Dubinett SM, Sharma S. Cocaine modulates cytokine and enhances tumor growth through sigma receptors. J Neuroimmunol. 2004;147:95–98. doi: 10.1016/j.jneuroim.2003.10.020. [DOI] [PubMed] [Google Scholar]
- Gilmore DL, Liu Y, Matsumoto RR. Review of the pharmacological and clinical profile of rimcazole. CNS Drug Rev. 2004;10:1–22. doi: 10.1111/j.1527-3458.2004.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, et al. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Intracellular dynamics of σ-1 receptors (σ1 binding sites) in NG108-15 cells. J Pharmacol Exp Ther. 2003;306:726–733. doi: 10.1124/jpet.103.051292. [DOI] [PubMed] [Google Scholar]
- Hohenegger M, Waldhoer M, Beindl W, Boing B, Kreimeyer A, Nickel P, et al. Gsα-selective G protein antagonists. Proc Natl Acad Sci U S A. 1998;95:346–351. doi: 10.1073/pnas.95.1.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong W, Werling LL. Evidence that the σ1 receptor is not directly coupled to G proteins. Eur J Pharmacol. 2000;408:117–125. doi: 10.1016/s0014-2999(00)00774-3. [DOI] [PubMed] [Google Scholar]
- Itzhak Y. Multiple affinity binding states of the sigma receptor: effect of GTP-binding protein-modifying agents. Mol Pharmacol. 1989;36:512–517. [PubMed] [Google Scholar]
- Johannessen M, Ramachandran S, Riemer L, Ramos-Serrano A, Ruoho AE, Jackson MB. Voltage-gated sodium channel modulation by sigma-receptors in cardiac myocytes and heterologous systems. Am J Physiol Cell Physiol. 2009;296:C1049–C1057. doi: 10.1152/ajpcell.00431.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura K, Kubota K, Kobayashi T, Elsinga PH, Ono M, Maeda M, et al. Evaluation of [11C]SA5845 and [11C]SA4503 for imaging of sigma receptors in tumors by animal PET. Ann Nucl Med. 2005;19:701–709. doi: 10.1007/BF02985120. [DOI] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim FJ, Kovalyshyn I, Burgman M, Neilan C, Chien CC, Pasternak GW. σ1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol. 2010;77:695–703. doi: 10.1124/mol.109.057083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang K, Drell TL, Lindecke A, Niggemann B, Kaltschmidt C, Zaenker KS, et al. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int J Cancer. 2004;112:231–238. doi: 10.1002/ijc.20410. [DOI] [PubMed] [Google Scholar]
- Lupardus PJ, Wilke RA, Aydar E, Palmer CP, Chen Y, Ruoho AE, et al. Membrane-delimited coupling between sigma receptors and K+ channels in rat neurohypophysial terminals requires neither G-protein nor ATP. J Physiol. 2000;526:527–539. doi: 10.1111/j.1469-7793.2000.00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WR, Eades CG, Thompson JA, Huppler RE, Gilbert PE. Effects of morphine-like and nalorphine-like drugs in nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- Maruo J, Yoshida A, Shimohira I, Matsuno K, Mita S, Ueda H. Binding of [35S]GTPγS stimulated by (+)-pentazocine sigma receptor agonist, is abundant in the guinea pig spleen. Life Sci. 2000;67:599–603. doi: 10.1016/s0024-3205(00)00651-2. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Eisenberg J, Fierer G, Musto NA. Developmental changes in brain and serum binding of testosterone and in brain capillary uptake of testosterone-binding serum proteins in the rabbit. Brain Res. 1988;466:245–253. doi: 10.1016/0165-3806(88)90050-8. [DOI] [PubMed] [Google Scholar]
- Plummer HK, 3rd, Yu Q, Cakir Y, Schuller HM. Expression of inwardly rectifying potassium channels (GIRKs) and beta-adrenergic regulation of breast cancer cell lines. BMC Cancer. 2004;4:93. doi: 10.1186/1471-2407-4-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy JD, Ramamoorthy S, Mahesh VB, Leibach FH, Ganapathy V. Cocaine-sensitive sigma-receptor and its interaction with steroid-hormones in the human placental syncytiotrophoblast and in choriocarcinoma cells. Endocrinology. 1995;136:924–932. doi: 10.1210/endo.136.3.7867601. [DOI] [PubMed] [Google Scholar]
- Reinhardt R, Manaenko A, Pissarek M, Wagner A, Illes P. Alterations of purine and pyrimidine nucleotide contents in rat corticoencephalic cell cultures following metabolic damage and treatment with openers and blockers of ATP-sensitive potassium channels. Neurochem Int. 2002;40:427–433. doi: 10.1016/s0197-0186(01)00102-4. [DOI] [PubMed] [Google Scholar]
- Safrany ST, Nahorski SR. A comparison between muscarinic receptor occupancy, inositol 1,4,5- trisphosphate accumulation and Ca2+ mobilization in permeabilized SH- SY5Y neuroblastoma cells. Neuropharmacology. 1994;33:837–846. doi: 10.1016/0028-3908(94)90179-1. [DOI] [PubMed] [Google Scholar]
- Schwarz S, Pohl P, Zhou GZ. Steroid binding at sigma-“opioid” receptors. Science. 1989;246:1635–1638. doi: 10.1126/science.2556797. [DOI] [PubMed] [Google Scholar]
- Soriani O, Foll FL, Roman F, Monnet FP, Vaudry H, Cazin L. A-Current down-modulated by sigma receptor in frog pituitary melanotrope cells through a G protein-dependent pathway. J Pharmacol Exp Ther. 1999;289:321–328. [PubMed] [Google Scholar]
- Spruce BA, Campbell LA, McTavish N, Cooper MA, Appleyard MV, O'Neill M, et al. Small molecule antagonists of the sigma-1 receptor cause selective release of the death program in tumor and self-reliant cells and inhibit tumor growth in vitro and in vivo. Cancer Res. 2004;64:4875–4886. doi: 10.1158/0008-5472.CAN-03-3180. [DOI] [PubMed] [Google Scholar]
- Su TP, London ED, Jaffe JH. Steroid binding at sigma receptors suggests a link between endocrine, nervous, and immune systems. Science. 1988;240:219–221. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]
- Taylor CW. The role of G proteins in transmembrane signalling. Biochem J. 1990;272:1–13. doi: 10.1042/bj2720001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchedre KT, Huang RQ, Dibas A, Krishnamoorthy RR, Dillon GH, Yorio T. Sigma-1 receptor regulation of voltage-gated calcium channels involves a direct interaction. Invest Ophthalmol Vis Sci. 2008;49:4993–5002. doi: 10.1167/iovs.08-1867. [DOI] [PubMed] [Google Scholar]
- Tokuyama S, Hirata K, Ide A, Ueda H. Sigma ligands stimulate GTPase activity in mouse prefrontal membranes: evidence for the existence of metabotropic sigma receptor. Neurosci Lett. 1997;233:141–144. doi: 10.1016/s0304-3940(97)00657-5. [DOI] [PubMed] [Google Scholar]
- Vilner BJ, John CS, Bowen WD. Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 1995;55:408–413. [PubMed] [Google Scholar]
- Wang L, Duncan G. Silencing of sigma-1 receptor induces cell death in human lens cells. Exp Cell Res. 2006;312:1439–1446. doi: 10.1016/j.yexcr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Wang L, Prescott AR, Spruce BA, Sanderson J, Duncan G. Sigma receptor antagonists inhibit human lens cell growth and induce pigmentation. Invest Ophthalmol Vis Sci. 2005;46:1403–1408. doi: 10.1167/iovs.04-1209. [DOI] [PubMed] [Google Scholar]
- Waterhouse RN, Collier TL. In vivo evaluation of [18F]1-(3-fluoropropyl)-4-(4-cyanophenoxymethyl)piperidine: a selective sigma-1 receptor radioligand for PET. Nucl Med Biol. 1997;24:127–134. doi: 10.1016/s0969-8051(96)00184-9. [DOI] [PubMed] [Google Scholar]
- Wilson AA, Dannals RF, Ravert HT, Sonders MS, Weber E, Wagner HN., Jr Radiosynthesis of sigma receptor ligands for positron emission tomography: 11C- and 18F-labeled guanidines. J Med Chem. 1991;34:1867–1870. doi: 10.1021/jm00110a017. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Fan H, Musachio JL, Wei ZL, Chellappan SK, Kozikowski AP, et al. Sazetidine-A, a novel ligand that desensitizes α4β2 nicotinic acetylcholine receptors without activating them. Mol Pharmacol. 2006;70:1454–1460. doi: 10.1124/mol.106.027318. [DOI] [PubMed] [Google Scholar]
- Zhu LX, Sharma S, Gardner B, Escuadro B, Atianzar K, Tashkin DP, et al. IL-10 mediates sigma 1 receptor-dependent suppression of antitumor immunity. J Immunol. 2003;170:3585–3591. doi: 10.4049/jimmunol.170.7.3585. [DOI] [PubMed] [Google Scholar]