

Abstract
BACKGROUND AND PURPOSE
Altered glutamatergic neurotransmission is linked to several neurological and psychiatric disorders. Metabotropic glutamate receptor 2 (mGlu2) plays an important role on the presynaptic control of glutamate release and negative allosteric modulators (NAMs) acting on mGlu2/3 receptors are under assessment for their potential as antidepressants, neurogenics and cognitive enhancers. Two new potent mGlu2/3 NAMs, RO4988546 and RO5488608, are described in this study and the allosteric binding site in the transmembrane (TM) domain of mGlu2 is characterized.
EXPERIMENTAL APPROACH
Site directed mutagenesis, functional measurements and β2-adrenoceptor-based modelling of mGlu2 were employed to identify important molecular determinants of two new potent mGlu2/3 NAMs.
KEY RESULTS
RO4988546 and RO5488608 affected both [3H]-LY354740 agonist binding at the orthosteric site and the binding of a tritiated positive allosteric modulator (3H-PAM), indicating that NAMs and PAMs could have overlapping binding sites in the mGlu2 TM domain. We identified eight residues in the allosteric binding pocket that are crucial for non-competitive antagonism of agonist-dependent activation of mGlu2 and directly interact with the NAMs: Arg3.28, Arg3.29, Phe3.36, HisE2.52, Leu5.43, Trp6.48, Phe6.55 and Val7.43. The mGlu2 specific residue HisE2.52 is likely to be involved in selectivity and residues located in the outer part of the binding pocket are more important for [3H]-LY354740 agonist binding inhibition, which is independent of the highly conserved Trp6.48 residue.
CONCLUSIONS AND IMPLICATIONS
This is the first complete molecular investigation of the allosteric binding pocket of mGlu2 and Group II mGluRs and provides new information on what determines mGlu2 NAMs selective interactions and effects.
Keywords: metabotropic glutamate receptor, mGlu2, mGluR, negative allosteric modulators, positive allosteric modulators, LY354740
Introduction
Glutamate, the main excitatory neurotransmitter in the brain, acts on two distinct classes of receptors, ionotropic (NMDA, AMPA, kainate) and metabotropic glutamate receptors (mGluR). The mGluRs, mGlu1–8, are divided into three groups based on sequence homology, second messenger coupling and pharmacology: group I (mGlu1 and mGlu5), group II (mGlu2 and mGlu3) and group III (mGlu4, mGlu6, mGlu7, mGlu8) (Lundström et al., 2010). mGluRs belong to the class C family of GPCRs and are characterized by a large extracellular N-terminal domain, containing a venus flytrap (VFT) module for agonist binding (orthosteric binding site) and a cystein-rich domain responsible for linking the two subunits of the homodimeric complex (Kunishima et al., 2000). The subsequent 7-transmembrane (TM) and C-terminal domains are involved in receptor activation and intracellular signalling through G-protein coupling. A binding pocket located in the 7-TM domain of class C GPCRs allows the modulation of receptor activity by allosteric ligands (Brauner-Osborne et al., 2007). Interestingly, homology models based on the high resolution crystal structure of bovine rhodopsin (Palczewski et al., 2000) have proven successful when studying allosteric interactions at class C GPCRs (Pagano et al., 2000; Petrel et al., 2003; Malherbe et al., 2003a; Bu et al., 2008). Hence, although the sequence identity of the class C family is distant to the rhodopsin-like class A GPCRs, the allosteric binding site of class C receptors share structural features with the orthosteric binding site of the class A family.
Allosteric modulation of membrane receptors is the most important indirect mechanism for the control of receptor function and the allosteric sites on GPCRs are of high interest as molecular targets for the development of new drugs (Conn et al., 2009). mGluR ligands acting at the allosteric site in the TM-domain offer greater possibilities for subtype selectivity and their modulating, rather than competing, effect on glutamate is likely to result in better tolerance (Lundström et al., 2010; Niswender and Conn, 2010). Group II mGluRs are predominantly located presynaptically, acting as inhibitory autoreceptors (Schoepp, 2001). Activation of mGlu2/3 receptors generally decreases glutamate signalling and lowers intracellular cAMP levels. For this reason, mGlu2 selective positive allosteric modulators (PAMs) are being developed as potential antipsychotics (Fraley, 2009). Selective competitive antagonists acting on group II mGluRs show antidepressant-like and cognitive enhancing properties (Chaki et al., 2004; Higgins et al., 2004; Yoshimizu and Chaki, 2004; Yoshimizu et al., 2006) and the therapeutic potential of mGlu2/3 allosteric antagonists [also referred to as negative allosteric modulators (NAMs) or non-competitive antagonists] is also under assessment (Campo et al., 2009; Woltering et al., 2010).Our group has extensively characterized, in vitro and in vivo, the pharmacological properties of mGlu2/3 NAM ligands (e.g. RO4491533 a 1,3-dihydro-benzo[b][1,4]diazepin-2-one derivative) showing that they dose-dependently improve working memory in rats challenged with either scopolamine or LY354740 (Gatti et al., 2001; Ballard et al., 2005; Woltering et al., 2010) and reverse the cognitive impairment induced by aging and APP/PS2 over-expression (Knoflach et al., 2005).
Previous studies that initiated the characterization of the allosteric binding site of group II mGluRs, have indicated the importance of three residues located in TM4 and TM5, Ser4.44 (688), Gly4.45 (689) and Asn5.46 (735) for mGlu2 receptor potentiation (Schaffhauser et al., 2003; Hemstapat et al., 2007; Rowe et al., 2008). In contrast, a NAM from the 1,3-dihydrobenzo[b][1,4]diazepin-2-one series was shown not to depend on Asn5.46 for antagonizing mGlu2 receptor function, suggesting distinct binding sites for PAMs and NAMs acting on mGlu2/3 receptors (Hemstapat et al., 2007). In the present study, we have performed a complete mutagenesis study of the mGlu2 receptor, targeting TM2-7 and the extracellular loop 2 (ECL2), to better characterize the allosteric binding pocket and the binding mode of mGlu2 NAMs. By comparing two novel mGlu2 NAMs RO4988546 (Gatti et al., 2006) and RO5488608 (Gatti et al., 2008) in two different functional assays: inhibition of agonist induced Ca2+ release and inhibition of [3H]-LY354740 agonist binding, we have identified several important molecular determinants in TM3, TM5, TM6, TM7 and ECL2 as well as specific interaction sites for the two different NAMs. A homology model was constructed from the crystal structure of the β2-adrenoceptor (Cherezov et al., 2007) in which the binding mode of the two NAMs was visualized. By using a 3H-PAM binding assay we furthermore showed that mGlu2 NAMs and PAMs could share an overlapping binding site in the TM domain of the receptor.
Methods
Materials
5-[7-Trifluoromethyl-5-(4-trifluoromethyl-phenyl)-pyrazolo[1,5-a]pyrimidin-3-ylethynyl]-pyridine-3-sulphonic acid (RO4988546) and 3′-(8-Methyl-4-oxo-7-trifluoromethyl-4,5-dihydro-3H-benzo[b][1,4]diazepin-2-yl)-biphenyl-3-sulphonic acid (RO5488608) (Figure 1), were synthesized at F. Hoffmann-La Roche by Dr T. Woltering (Gatti et al., 2006). L-Glutamate and LY487379 were purchased from Tocris Cookson (Bristol, UK). (1S,2R,5R,6S)-2-amino-bicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740), (2S)-2-amino-2-((1S,2S)-2-carboxycycloprop-1-yl)-3-(9-xanthyl)propanoic acid disodium salt (LY351495) (rac)-methyl-4-carboxyphenylglycine (MCPG) (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV), 2,2,2-trifluoroethyl [3-(1-methyl-butoxy)-phenyl]-pyridine-3-ylmethyl-sulphonamide (2,2,2-TEMPS) (Coleman et al., 2001; Lorrain et al., 2003; Barda et al., 2004) and 2,2,2-trifluoro-ethanesulphonic acid (4-phenoxy-phenyl)-pyridin-3-ylmethyl-amide (RO4388034) (Barda et al., 2004) were synthesized at F. Hoffmann-La Roche. All the compounds, except LY354740 and L-glutamate, were dissolved in dimethyl sulphoxide (DMSO) before dilution in assay buffer. The final concentration of DMSO (% v/v) used in each assay was: 2.7% in 3H-PAM binding and [35S]-GTPγS binding, 2% in [3H]-LY354740 binding and 0.1% in Ca2+ release measured by a fluorometric imaging plate reader (FLIPR). The radioligands [3H]-2,2,2-TEMPS ([3H]-PAM, 2,2,2-trifluoro-ethanesulphonic acid [3-(3,4-ditritio-1-methyl-butoxy)-phenyl]-pyridin-3-ylmethyl-amide) (specific activity 92 Ci·mmol−1), [3H]-LY354740 (specific activity 43 Ci·mmol−1) (Schaffhauser et al., 1998) and [3H]-HYDIA (Lundström et al., 2009) were synthesized at F. Hoffmann-La Roche by Drs D. Muri, P. Huguenin, J. Wichmann, T. Woltering and H. Stadler with a radiochemical purity >95%.
Figure 1.
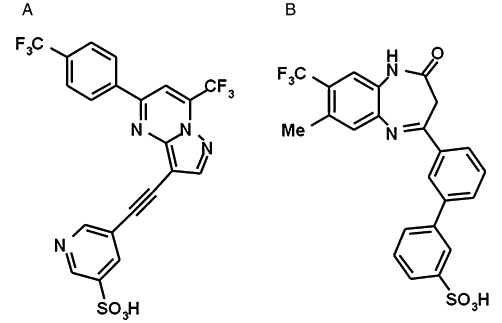
Chemical structures of two novel mGlu2/3 receptor NAMs used for mutagenesis studies. (A) RO4988546, 5-[7-trifluoromethyl-5-(4-trifluoromethyl-phenyl)-pyrazolo[1,5-a]pyrimidin-3-ylethynyl]-pyridine-3-sulphonic acid; molecular weight: 512.4 g·mol−1, calculated pKa: −1.82, clogP: 2.76, solubility at pH 6.5 >683 mg·L−1, intrinsic solubility >0.15 mg·L−1. (B) RO5488608, 3′-(8-methyl-4-oxo-7-trifluoromethyl-4,5-dihydro-3H-benzo[b][1,4]diazepin-2-yl)-biphenyl-3-sulphonic acid; molecular weight: 474.5 g·mol−1, calculated pKa: −0.8, clogP: 3.72.
Plasmids, cell culture and membrane preparations
cDNAs encoding for rat mGlu2 was initially provided by Professor S. Nakanishi (Kyoto, Japan). cDNA for human mGlu3 was purchased from the Missouri S&T cDNA Resource center (Rolla, MO, USA). All point mutations were constructed using the QuickChange Lightening Site-Directed Mutagenesis Kit (catalogue no. 210519, Stratagene, La Jolla, CA, USA). Mutations were confirmed by complete sequence of the receptor coding region from both strands (Microsynth, Balgach, Switzerland). CHO cells expressing the Gα16 protein, CHO-Gα16, and HEK-293 cells were transiently transfected with rat mGlu2 receptor mutants (20 µg) using Lipofectamine transfection reagent and Plus reagent according to manufacturer's protocol (Invitrogen, Carlsbad, CA, USA). CHO-Gα16 cells were maintained in HAM F-12 medium supplemented with 10% fetal calf serum (FCS), 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, 100 µg·mL−1 hygromycin B. HEK-293 cells were maintained in Dulbecco's modified Eagle medium (DMEM) medium supplemented with 10% FCS and 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin. After mGlu2 transfection, cells were cultured in medium containing dialyzed FCS and in the presence of 500 µM MCPG. CHO cells permanently expressing rat mGlu2 were maintained in DMEM medium supplemented with 10% dialyzed FCS, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, 36 mg·L−1 L-proline, 1 mM L-Glutamine, 300 µg·mL−1 G418 and 500 µM MCPG. Flp-in T-rex Gα16 HEK-293 human mGlu3 cells were maintained in DMEM high glucose medium supplemented with 10% dialyzed FCS, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, 15 µg·mL−1 blasticidine, 1 mM L-glutamine, 500 µg·mL−1 G418, 75 µg·mL−1 hygromycin B and 500 µM MCPG. For cell membrane preparation, mGlu2 or mGlu3 expressing cells were harvested and washed three times in ice-cold PBS. The pellet was resuspended in ice-cold 20 mM HEPES buffer containing 10 mM EDTA pH 7.4 and homogenized with a polytron (PT3100 Kinematica AG, Littau, Switzerland) for 10 s at 10 000 r.p.m.. After centrifugation at 48 000×g for 30 min at 4°C, the pellet was resuspended in 20 mM HEPES buffer containing 0.1 mM EDTA pH 7.4 and homogenized and centrifuged as above. The pellet obtained was resuspended in ice-cold 20 mM HEPES buffer containing 0.1 mM EDTA pH 7. Protein concentrations were determined using the Pierce BCA protein kit (Thermo scientific, Waltham, MA, USA) and the membrane homogenate were frozen at −80°C before use.
[3H]-LY354740 agonist affinity studies
Membrane homogenates were centrifuged at 48 000×g for 10 min at 4°C and resuspended in binding buffer (50 mM Tris and 2 mM MgCl2 at pH 7.4) to a final content of 25 µg protein per well. Saturation isotherms were performed with various concentrations of [3H]-LY354740 in a total reaction volume of 1 mL for 3 h incubation at room temperature (RT), 23°C. Binding displacement studies were carried out using 10 nM or 40 nM [3H]-LY354740 on mGlu2 and mGlu3, respectively, and with increasing concentrations of ligands in a total reaction volume of 1 mL for 1 h incubation at RT. Nonspecific binding was measured in the presence of 10 µM DCG-IV. The reaction was terminated by rapid filtration over GF/B glass fibre filters and washed with ice-cold assay buffer using a Brandel harvester (Biomedical Research and Development Laboratories Inc., Gaithersburg, MD, USA) (Schaffhauser et al., 1998). The radioactivity on the filters was measured by liquid scintillation on a beta counter in the presence of Ultima-gold scintillation fluid (Canberra Packard SA, Zürich, Switzerland) after 24 h incubation.
[3H]-PAM binding ([3H]-2,2,2-TEMPS)
Membrane homogenates were centrifuged at 48 000×g for 10 min at 4°C and resuspended in binding buffer (20 mM HEPES and 2 mM MgCl2 at pH 7.4) to a final content of 10 µg protein per well. Membranes were preincubated with Poly-lysine Coated Yttrium Silicate SPA beads (0.5 mg per well) (catalogue no. RPNQ0010, PerkinElmer, Waltham, MA, USA) for 1 h, shaking at 800 r.p.m. at RT. Saturation isotherms were performed with increasing concentrations of radioligand [3H]-PAM in a total reaction volume of 180 µL, shaking at 350 r.p.m. for 3 h incubation at RT. Displacement studies were carried out in the presence of 3 nM [3H]-PAM, with increasing concentrations of ligands in a total reaction volume of 180 µL shaking at 350 r.p.m. for 1 h incubation at RT. The compound with best nonspecific binding properties, RO5488608, was selected among several available NAM and PAM ligands and used at 10 µM. SPA beads were allowed to settle for 1 h before measurement on a Top-count (Packard, Zürich, Switzerland) with quench correction. The assay was performed in 96-well OptiPlates (PerkinElmer, Waltham, MA, USA). Saturation and inhibition binding data were analysed by Prism 5.0 (GraphPad Software, San Diego, CA, USA), using equations Y = Bmax× X/(Kd+ X) and Y = Bottom + (Top-Bottom)/(1 + 10^((LogEC50-X)*HillSlope)) respectively. Ki values were calculated from the IC50 values using the Cheng Prusoff equation.
[35S]-GTPγS binding
LY354740-stimulated [35S]-GTPγS binding was performed on CHO mGlu2 cell membranes according to Cartmell et al. (1998). Briefly, the effect of increasing concentrations of agonist was measured using 10 µg membrane protein in the presence of 2 µM GDP, 1 mg of Weatgerm Agglutinin SPA beads (GE Healthcare, Buckinghamshire, UK) and and 0.3 nM [35S]-GTPγS (Perkin Elmer, Waltham, MA, USA) in assay buffer containing 20 mM HEPES, 100 mM NaCl and 10 mM MgCl2. The reaction was carried out for 1 h under shaking and beads were allowed to settle for 1 h before being counted on a Top-count (Packard, Zürich, Switzerland) with quench correction.
Intracellular Ca2+ release measurements
CHO-Gα16 cells, grown to 80% confluence, were transfected with the wild-type (WT) or mutant mGlu2 receptor. Twenty-four hours post transfection, the cells were harvested and seeded at 5 × 104 cells per well in a black poly-d-lysine treated clear bottomed 96-well plates (BD Biosciences, Palo Alto, CA, USA) in culture medium containing 10% dialyzed FCS and 500 µM MCPG. On the day of assay, 48 h post transfection, the cells were loaded with 2 µM Fluo-4 AM (Cataloge No. F-14202, Molecular Probes, Eugene, OR, USA) in assay buffer; HBSS supplemented with 20 mM HEPES and 2.5 mM probenecid (Sigma-Aldrich, St. Louis, MO, USA) and incubated at RT for 90 min. Cells were washed five times with assay buffer before intracellular Ca2+ release was measured using a FLIPR (Molecular Devices, Sunnyvale, CA, USA) as previously described (Porter et al., 1999). The antagonistic potency of the NAMs was determined in the presence of an EC80 concentration of the LY354740 agonist, which was individually determined for each receptor mutant (see Table S1) and ranged from 20–70 nM. RO4988546 and RO5488608 were dissolved in DMSO and diluted in assay buffer to a 5 × working solution, which was added to the cells, resulting in a final concentration of 0.1% v v-1 DMSO. Compounds with antagonistic properties where always applied 5 min prior to agonist application. The Ca2+ release responses were measured as peak increase in fluorescence minus basal, normalized to the maximal stimulating effect induced by 10 µM LY354740 or to the effect induced by the EC80 concentration of LY354740. Inhibition curves were fitted using Prism 5.0 (GraphPad Software, San Diego, CA, USA), equation Y = Bottom + (Top-Bottom)/(1 + 10^((LogIC50-X)*HillSlope)). The mean ± SEM values of six to nine dose–response curves from three to five different transfections were calculated. The relative efficacy (Emax) values of LY354740 were calculated as fitted maximum of the dose–response of each mutated receptor expressed as a percentage of fitted maximum of the WT dose–response curve from cells transfected and assayed on the same day.
Residue numbering scheme
The position of each amino acid residue in the 7-TM domain was identified both by its sequence number and by its generic numbering system proposed by Ballesteros–Weinstein (Ballesteros and Weinstein, 1995) which is shown as superscript. In this numbering system, amino acid residues in the 7-TM domain are given two numbers: the first refers to the TM domain number; the second indicates its position relative to a highly conserved residue of family 1 GPCRs in that TM which is arbitrarily to 50. The amino acids in the extracellular loop EC2 are labelled E2. The highly conserved cysteine thought to be disulphide bonded, was given the index number E2.50 and the residues within the EC2 loop are then indexed relative to the ‘50’ position.
Alignment and building of mGlu2 homology model
The amino acid sequences of the rat mGlu2 (accession number: P31421), rat mGlu3 (accession number: P31422) and rat mGlu5 (accession number: P31424) were retrieved from the Swiss-Prot database. These amino acid sequences were aligned using the ClustalW multiple alignment program. A slow pairwise alignment using the BLOSUM matrix series and a gap opening penalty of 15.0 were chosen for aligning the amino acid sequences. Other parameters were those given as default. The aligned mGluR sequences were then manually aligned against the sequence of the human β2-adrenoceptor (accession number: P07550) such that the alignment of class A and class C receptors was reproduced as previously published (Bissantz et al., 2004). Using this alignment and the X-ray structure of the human β2-adrenoceptor [pdb code 2rh1 (Cherezov et al., 2007)] as template, the software package MOE (MOE v.2009.10, Chemical Computing Group, Montreal, QC, Canada) was used to generate a three-dimensional model of the rat mGlu2. The two NAMs were then manually docked into the TM cavity in order to achieve the best complementarily to the binding site in terms of shape and electrostatic properties. It should be noted that the ligand alignment between the two NAMs is straightforward because the sulphonic acid functions and trifluormethyl substituents have to be superimposed. This pregenerated ligand alignment was only slightly adjusted to best fit the pocket when transferring the NAMs into the binding cavity. The resulting protein-ligand complexes were then minimized using MOE in order to prevent any clashes between ligand atoms and protein side chains. Backbone atoms were hereby kept fixed. A total of 30 residues surrounding the docked NAMs were then chosen for site-direct mutagenesis studies to get information about the different residues involved in NAM binding and their non-selectivity towards rat mGlu3. As no prior knowledge about the allosteric mGlu2 NAM binding site was available, we distributed the mutations over the complete TM cavity.
Results
The mGlu2/3 NAMs; RO4988546 and RO5488608
The binding mode of NAMs to the mGlu2 receptor was investigated in order to understand which residues are important for this interaction and for the effects on receptor function. Two chemically different mGlu2/3 receptor selective NAMs were chosen: RO4988546 (Figure 1A) and RO5488608 (Figure 1B). Both compounds are very soluble and potent, therefore suitable for in vitro pharmacology and mutagenesis studies. Compounds belonging to the same chemical series as RO5488608 (Figure 1B) were examined in previous studies (Woltering et al., 2007; 2008a,b;), whereas this is the first time a mGlu2/3 receptor NAM containing a pyrazolo[1,5-a]pyrimidine heteronucleus is described (Figure 1A). Both compounds were initially characterized in vitro in affinity and functional studies using CHO cells permanently expressing rat mGlu2 (coupling to the native Gi protein) as described by Woltering et al. (2008a). No interaction or effect on other mGluRs was detected when RO4988546 and RO5488608 were tested at concentrations up to 10 µM (data not shown). See legend to Figure 1 for further details.
Functional and affinity studies characterizing the pharmacological properties of mGlu2 NAMs
In our experimental conditions [3H]-LY354740 exhibited a Kd of 9.1 nM for rat mGlu2 and a Kd of 65 nM for human mGlu3 (data not shown), which is in agreement with previous reports (Schweitzer et al., 2000). The EC50 values for LY354740 in mGlu2 functional studies ranged between 15 nM ([35S]-GTPγS binding, Figure 2B and C, Table 1), 9.8 nM (Ca2+ release assay, Figure 6, Table S1) and 7 nM (cAMP assay in presence of 10 µM forskolin, data not shown). In all assays LY354740 behaved as full agonist when compared with L-glutamate and 1S,3R-ACPD and the agonistic properties were not influenced by the receptor coupling (native Gi/o vs Gα16). In vitro characterization of NAMs' properties and site directed mutagenesis studies were all carried out using LY354740 (radioligand and agonist) to take advantage of the properties of this mGlu2 agonist already described by Cartmell et al. (1998). In fact, in our experimental conditions the relationship between receptor occupancy and cellular effect was linear with about 50% occupancy required to obtain 50% agonistic effect in the different assays, limiting in this way the possible impact of receptor reserve in our experimental conditions. Moreover, no clear indication of mGlu2 constitutive receptor activity was obtained in any of the relevant assays.
Figure 2.
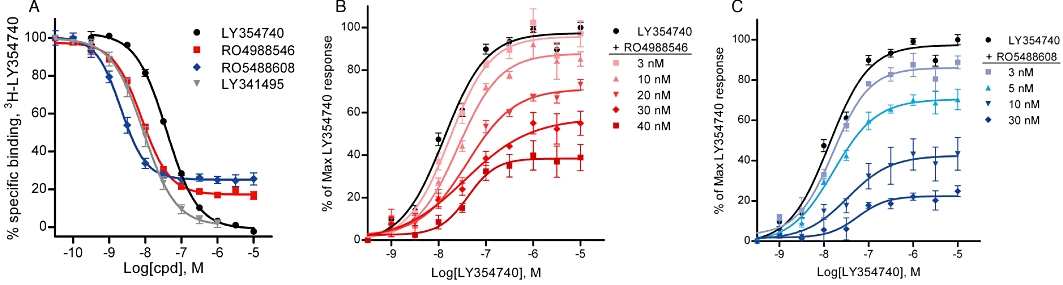
Effect of mGlu2 NAMs on [3H]-LY354740 agonist binding and LY354740-induced [35S]-GTPγS binding. (A) By binding to an allosteric binding site RO4988546 and RO5488608 concentration-dependently inhibited [3H]-LY354740 binding at the orthosteric site with a residual 20% specific binding at the mGlu2 receptor. Calculated IC50 and nH values for the dose–response curves are 8.7 ± 0.6 nM and −1.2 ± 0.1 for RO4988546, and 2.5 ± 0.3 nM and −1.5 ± 0.1 for RO5488608, respectively. The competitive agonist (LY354740) and antagonist (LY341495) completely displaced [3H]-LY354740 binding with Ki and nH values of 19.6 ± 0.3 nM, −1.0 ± 0.0 for LY354740 and 5.1 ± 0.9 nM, −1.1 ± 0.1 for LY341495. Each curve represents the mean ± SEM of eight to nine dose–response measurements from three to five separate experiments performed in duplicate on membranes from CHO rat mGlu2 cells. (B and C) Schild plot analysis of LY354740 effects on [35S]-GTPγS binding with increasing concentrations of RO4988546 and RO5488608 reducing the maximum response to LY354740 at mGlu2 and inducing a shift to the right of the dose–response curve (See Table 1 for values). Each curve represents the mean ± SEM of three dose–response measurements from separate experiments performed in duplicate on membranes from CHO rat mGlu2 cells.
Table 1.
Effect of mGlu2 NAMs on agonist induced [35S]-GTPγS binding at mGlu2
| EC50 (nM) | Maximum responses (%) | |
|---|---|---|
| LY354740 | 15.5 ± 3.2 | 100 |
| RO4988546 | ||
| +3 nM | 18.4 ± 4.1 | 95.7 ± 5.8 |
| +10 nM | 27.3 ± 6.1 | 88.1 ± 6.0 |
| +20 nM | 42.6 ± 5.4 | 71.9 ± 2.4 |
| +30 nM | 36.6 ± 2.5 | 59.3 ± 9.0 |
| +40 nM | 34.2 ± 5.8 | 39.2 ± 6.4 |
| RO5488608 | ||
| +3 nM | 16.0 ± 3.0 | 85.4 ± 5.3 |
| +5 nM | 17.5 ± 2.9 | 71.0 ± 4.5 |
| +10 nM | 36.6 ± 3.8 | 43.0 ± 7.8 |
| +30 nM | 113.1 ± 63.6 | 19.7 ± 3.1 |
Calculated EC50 and maximum responses for the LY354740 agonist dose–response curves (Schild plot) in [35S]-GTPγS binding in the absence or presence of mGlu2 NAMs shown in Figure 2B–C. The dose–response curve of LY354740 has an EC50 value of 15.5 nM and a Hill slope of 1.0 and the maximum response is 97% above basal stimulation (in agreement with Cartmell et al., 1998). Dose–response curves in the presence of NAM are normalized to the maximum response of the LY354740 agonist. Values are mean ± SEM of three dose–response measurements, performed in duplicate, from individual experiments.
Figure 6.
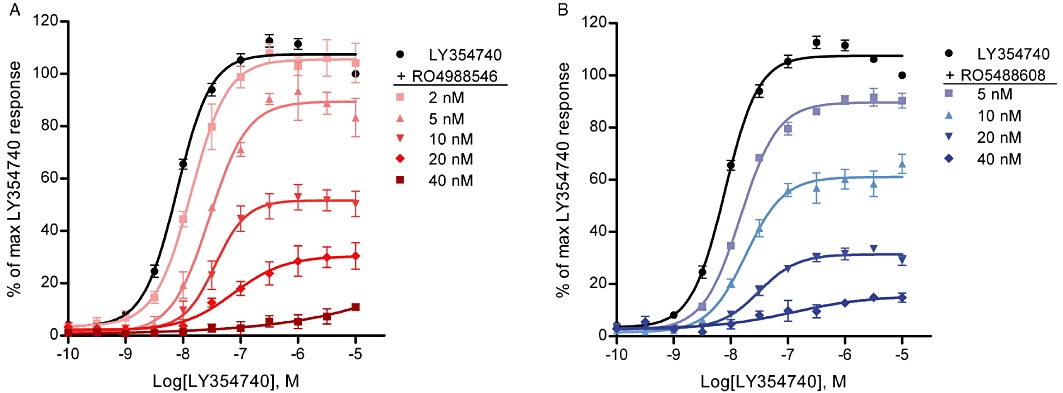
Effect of mGlu2 NAMs on agonist-induced Ca2+ release response at the mGlu2 WT receptor. Increasing concentrations of mGlu2 NAMs caused a rightward shift as well as a reduction in maximal response of the LY354740 dose–response curve. EC50 values and maximal responses were as follows; (A) LY354740; 9.8 ± 0.6 nM 100%, 2 nM RO4988546; 13.9 ± 0.7 nM 105 ± 5%, 5 nM RO4988546; 28.7 ± 3.6 nM 90 ± 1%, 10 nM RO4988546; 38.3 ± 6.8 nM 51 ± 4%, 20 nM RO49888546; 84.7 ± 16.5 nM 31 ± 5%. (B) LY354740; 9.8 ± 0.6 nM 100%, 5 nM RO5488608; 14.9 ± 0.8 nM 90 ± 2%, 10 nM RO5488608; 18.8 ± 1.9 nM 61 ± 4%, 20 nM RO5488608; 33.5 ± 5.8 nM 32 ± 1%. Each curve represents the mean ± SEM of three to six dose–response measurement, performed in duplicate, from three to five independent transfections and the data are normalized to the maximum response of LY354740.
RO4988546 and RO5488608 partially inhibited [3H]-LY354740 agonist binding at the orthosteric binding site of mGlu2, leaving approximately 20% residual specific binding, while competitive ligands like the LY354740 agonist and the LY341495 antagonist completely displaced [3H]-LY354740 (Figure 2A). Both NAMs showed high potency in functional binding inhibition studies with IC50 values of 8.7 nM for RO4988546 and 2.5 nM for RO5488608, respectively (Figure 2A). The residual specific binding of the NAMs, indicative of their allosteric binding at the receptor, was 27% for RO5488608, and 19% for RO4988546. Interestingly, for both mGlu2 NAMs the Hill slope was >1 further suggesting an effect mediated by an interaction with an allosteric site. In functional binding inhibition studies of [3H]-LY354740 at mGlu3 both RO4988546 and RO5488608 have higher IC50 values, 77 nM and 155 nM, respectively, with 45–51% residual specific binding (data not shown). In agreement with the interaction of the two NAMs with an allosteric site no effect was observed on the specific binding at mGlu2 of the competitive [3H]-HYDIA antagonist (50 nM), at NAM concentrations up to 10 µM (data not shown). The non-competitive mechanism of RO4988546 and RO5488608 antagonism at mGlu2 is shown in the [35S]-GTPγS binding studies, where the maximal response of LY354740 was reduced by both NAMs in a concentration-dependent manner (Figure 2B and C, Table 1). Further functional studies with intracellular measurements of cAMP were also carried out on CHO rat mGlu2 cells in the presence of 10 µM forskolin and 10 µM 1S,3R-ACPD to assure consistency in IC50 values between different cellular assays. IC50 values for the inhibition of forskolin-induced cAMP release was 7 nM and 14 nM for RO4988546 and RO5488608, respectively (data not shown, see also Woltering et al., 2007).
In order to better understand how negative allosterism at mGlu2 influences agonist binding at the orthosteric binding site, the [3H]-LY354740 saturation isotherm was determined in the presence of varying concentrations of RO4988546 and RO5488608. The presence of increasing concentrations of NAMs caused a concentration-dependent inhibition of [3H]-LY354740 agonist binding, reducing the affinity (Kd) as well as the number of binding sites occupied by the agonist (Bmax) (Figure 3A and B, Table 2). Specifically, the presence of RO4988546 clearly reduced the affinity of the LY354740 agonist to the mGlu2 receptor, while only a minor reduction was observed on the level of occupied binding sites (Figure 3A). In contrast, the presence of RO5488608 caused a marked reduction in the number of receptors that was occupied by the [3H]-LY354740 agonist accompanied by increasing Kd values (Figure 3B).
Figure 3.
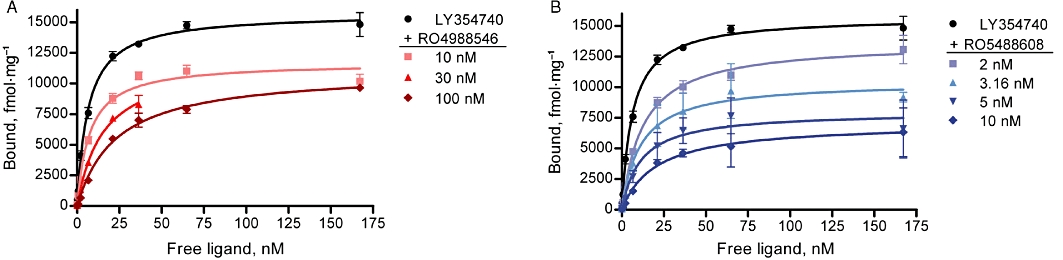
Effect of mGlu2 NAMs on [3H]-LY354740 saturation binding at mGlu2. Isothermal curves of [3H]-LY354740 (Kd = 9.1 nM) in the presence of RO4988546 and RO5488608 showing a dose-dependent reduction of the affinity of LY354740 to the orthosteric binding site. Increasing concentrations of RO4988546 induced a minor reduction in the Bmax value (A) while in the presence of RO5488608, the Bmax was reduced to 50% at 10 nM (B) (See Table 2 for values). Saturation binding was performed on CHO mGlu2 membranes and non-linear regression is the mean ± SEM from individual experiments performed in duplicate (LY354740 n = 8, RO4988546 n = 2, RO5488608 3.16 nM and 10 nM n = 2, RO5488608 2 nM and 5 nM n = 3).
Table 2.
Effect of mGlu2 NAMs on [3H]-LY354740 saturation binding at mGlu2
| Kd (nM) | Bmax (fmol·mg−1) | |
|---|---|---|
| LY354740 | 9.1 ± 0.9 | 14632 ± 151 |
| +10 nM RO4988546 | 12.3 ± 2.0 | 13540 ± 420 |
| +30 nM RO4988546 | 26.0 ± 1.5 | 13870 ± 170 |
| +100 nM RO4988546 | 39.7 ± 3.2 | 13175 ± 515 |
| +2 nM RO5488608 | 14.6 ± 1.1 | 14830 ± 410 |
| +3.16 nM RO5488608 | 11.1 ± 0.3 | 11515 ± 185 |
| +5 nM RO5488608 | 21.5 ± 1.0 | 9034 ± 150 |
| +10 nM RO5488608 | 24.1 ± 1.4 | 7465 ± 171 |
Calculated Kd and Bmax values for the saturation isotherms of [3H]-LY354740 in the absence and presence of mGlu2 NAMs shown in Figure 3. Values are mean ± SEM from individual experiments performed in duplicate (LY354740 n = 8, RO4988546 n = 2, RO5488608 3.16 nM and 10 nM n = 2, RO5488608 2 nM and 5 nM n = 3).
The physicochemical properties of the mGlu2 NAMs, discovered and characterized above, limit the possibility of obtaining precise direct affinity measures for these compounds at the mGlu2 receptor. However, by utilizing a 3H-PAM selective for mGlu2 it was possible to determine whether mGlu2 NAMs could displace other allosteric ligands from their mGlu2 receptor binding site. The mGlu2 PAM (2,2,2-TEMPS) used here as a radioligand has a chemical structure similar to the mGlu2 PAM LY487379, which is more widely used as reference compound for in vitro studies. The main advantage with 2,2,2-TEMPS is its higher potency that makes it more suitable as a radioligand (Barda et al., 2004). In equilibrium binding studies, 3H-PAM displayed high affinity for mGlu2 with a Kd value of 4.2 nM while no specific binding could be detected at mGlu3. Furthermore, the number of binding sites detected by the 3H-PAM corresponds to about 40% of the number of binding sites recognized by the [3H]-LY354740 ligand on the same membranes (L. Lundström et al., unpubl obs). Binding of 3H-PAM at the allosteric site of mGlu2 was completely displaced by the two NAMs RO4988546 and RO5488608, with Ki values of 2.3 and 1.9 nM, respectively (Figure 4). Under the same experimental conditions, two PAM ligands; the non-tritiated 2,2,2-TEMPS and RO4388034 had lower affinity, Ki values of 5.5 and 318.5 nM, respectively (Figure 4). The ability of mGlu2 NAMs to completely displace the binding of 3H-PAM indicates that the allosteric modulators have an overlapping binding site(s) at the mGlu2 receptor. In addition, a 25–45% increase in 3H-PAM specific binding was seen in the presence of increasing concentrations of LY345740 and L-glutamate while not in the presence of increasing concentrations of the orthosteric antagonists, LY341495 and HYDIA, further confirming the presence of a modulatory interaction between orthosteric and allosteric binding sites when occupied with agonist and potentiating modulator respectively (L. Lundström et al., unpubl obs). The results described here, while characterizing the different pharmacological properties of two mGlu2 NAMs are indicative of an interesting interplay between agonist and modulator sites at the mGlu2 receptor (Kenakin, 2009) that could be further addressed by site mutagenesis studies at the allosteric binding site.
Figure 4.
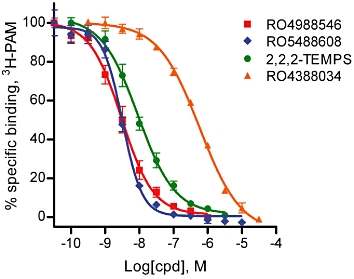
Effects of mGlu2 NAMs on 3H-PAM binding at the allosteric binding site of mGlu2. RO4988546 and RO5488608 completely inhibited 3H-PAM binding to the allosteric binding site with high affinity, Ki of 2.3 ± 0.7 nM (nH−1.1 ± 0.1) and 1.9 ± 0.3 nM (nH−1.5 ± 0.1), respectively. mGlu2 PAMs, 2,2,2-TEMPS and RO4388034 displayed lower affinity at the allosteric site, Ki values of 5.5 ± 0.9 nM (nH−0.8 ± 0.0) and 318.5 ± 14.7 nM (nH−0.8 ± 0.0) respectively. Data represent the mean ± SEM of five to seven dose–response curves, performed in duplicate, from four different experiments.
Sequence alignment and mutagenesis strategy of mGlu2
The results obtained indicate that allosteric modulators at mGluRs share overlapping binding sites with the receptor. When selecting residues for the mGlu2 NAM mutagenesis study, the well-characterized allosteric binding site of mGlu5 (Malherbe et al., 2003a; Muhlemann et al., 2006) was serving as guidance. An alignment of the mGlu2 TM domain was made against the closely related mGlu3 receptor as well as the mGlu5 receptor for further comparison (Figure 5). In the cavity formed in the TM bundle only five residues differ between mGlu2 and mGlu3 (Figure 5), which were mutated to the corresponding residue in mGlu3. Overall, residues targeted by site-directed mutagenesis were selected to constitute the large binding cavity characterized in class A GPCRs (Cherezov et al., 2007) and thus the 30 selected mutations were distributed over six TM domains and in ECL2 (Figure 5).
Figure 5.

Sequence alignment of the amino acid residues in the TM and ECL2 of rat mGlu2 relative to mGlu3 and mGlu5. Residues highlighted in grey are located in the binding pocket of the mGlu2 homology model constructed from the crystal structure of the β2-adrenoceptor [pdb code 2rh1 (Cherezov et al., 2007)]. Amino acids mutated in mGlu2 are underlined and residues identified to be important for NAM binding are shown in bold and tagged with its amino acid number. •, Residues conserved within Group II mGluRs; *, residues identical between mGlu2 and Glu5. Critical residues for 2-methyl-6-(phenylethynyl)-pyridine binding to the allosteric site of mGlu5 are shown in bold (Malherbe et al., 2003a). Ballesteros–Weinstein numbering scheme of the mGlu2 residues are shown at the bottom.
Expression of mGlu2 receptor mutants in CHO-Gα16 cells determined by Western blot analysis
The polyclonal anti mGlu2 antibody used in this study recognizes an epitope in the C-terminus of the protein corresponding to the amino acids 829-845 of rat mGlu2 and correctly stains in the cell lysate a faint band of approximately 100 kDa, corresponding to the homomeric form of the receptor, and a main band of approximately 200 kDa, corresponding to the homodimeric form of the receptor. Using gradient conditions and in CHO cell lysates it is also possible to detect the prominent glycosylation profile of the homodimeric form of the receptor. For all mGlu2 receptor mutants it was possible to achieve a reproducible and comparable expression level of the mGlu2 homodimer. The level of expression for a representative group of mutations is shown in Figure S1 where levels of expression are compared with the mGlu2 WT receptor in a semi-quantitative manner.
Key residues for NAM inhibition of agonist-induced intracellular Ca2+ release
After transient transfection of the mGlu2 WT receptor into CHO-Gα16 cells the agonists L-glutamate and LY354740 elicited a concentration-dependent increase in intracellular Ca2+ release with EC50 values of 1.3 µM and 9.8 nM, respectively (see Figure 6 for the dose–response curve of LY354740). For L-glutamate, the potency is in agreement with previously reported data (Johnson et al., 2003; Galici et al., 2005) while for LY354740 it correlates well with the reported pharmacology (Schoepp et al., 1997; Schaffhauser et al., 1998) but is higher than previously published data using promiscuous G-proteins, that obatined an EC50 value of 34 nM in intracellular Ca2+ release (Kowal et al., 2003). The relative efficacy (Emax) of L-glutamate was 93.2 ± 6.7% of Emax for LY354740; hence, LY354740, the more potent agonist, was selected and used in the study. To confirm the non-competitive mode of antagonism by RO4988546 and RO5488608 also via the Gα16 protein, the effect of increasing concentrations of NAM on the LY354740 dose–response curve was assessed in the intracellular Ca2+ release assay. Similar to the [35S]-GTPγS binding assay, RO4988546 and RO5488608 shifted the LY354740 dose–response curve to the right with a main characteristic decrease in the relative efficacy (Figure 6A and B). When determined on a sub-maximal concentration (EC80) of LY354740 agonist, RO4988546 and RO5488608 elicited a concentration-dependent antagonism of the induced Ca2+ release with IC50 values of 5.5 nM and 11.4 nM, respectively (Figure 7). Notable here is the difference in relative potency between the two NAMs compared with the functional [3H]-LY354740 binding inhibition assay, where RO5488608 was the more potent of the two compounds. Neither RO4988546 nor RO5488608 exhibited agonist properties when tested in the absence of a mGlu2 agonist. On all the mGlu2 mutants generated, LY354740 elicited a concentration-dependent increase in intracellular Ca2+. The potency (EC50), Hill coefficient (nH) and relative efficacy (Emax), calculated from the concentration-response curves for LY354740 on the mGlu2 receptor mutants are presented in Table S1. On mutation T620A2.61, Y647V3.40, L732A5.43, T769V6.44, W773A6.48 and V798A7.43, the EC50 and Emax values of LY354740 were slightly different from those of the WT receptor (Table S1). However, considering the intrinsic variability in a functional readout assay after transient transfections, the mGlu2 mutants elicited concentration–response curves for the LY354740 agonist with potencies and relative efficacies in close agreement with those obtained with the WT mGlu2 receptor (Table S1). Thus the inserted mutations did not seem to have a significant effect on the orthosteric agonist binding site and all constructed receptors were considered to be functionally active.
Figure 7.
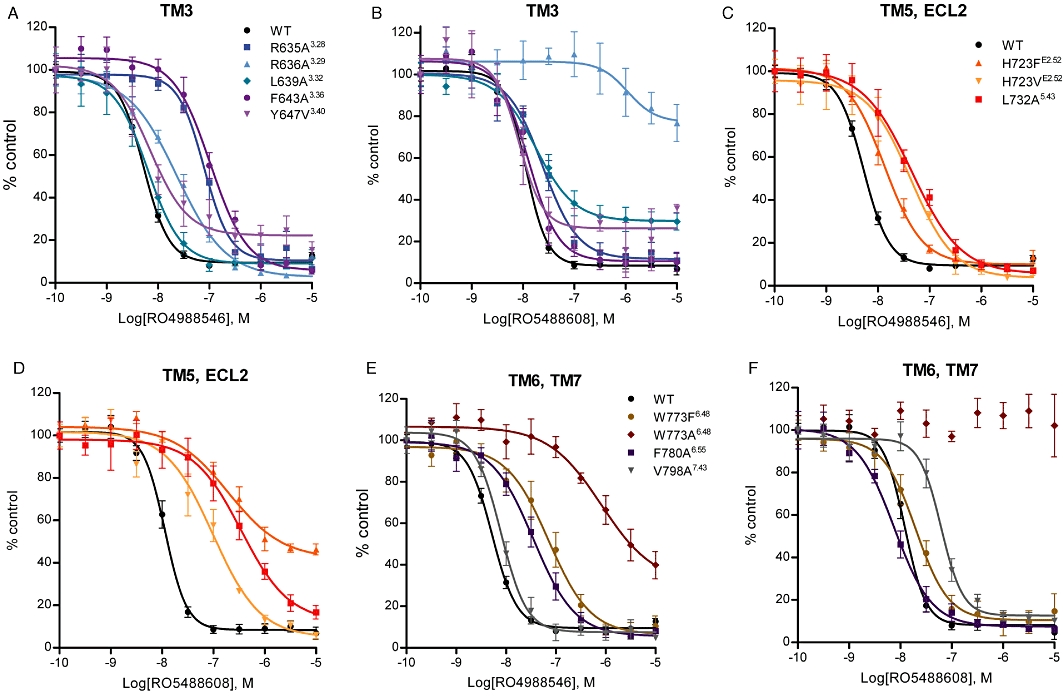
Effect of mGlu2 mutations on NAM inhibition of LY354740-induced intracellular Ca2+ release. Concentration-dependent inhibition by RO4988546 (A, C, E) and RO5488608 (B, D, F) of effects of a sub-maximal (EC80) concentration of LY354740 at WT and mutant mGlu2 receptors. mGlu2 receptor mutants on which the inhibitory response was reduced compared with WT are shown graphically here and the calculated IC50 values for all studied mutants are presented in Table 3. Inhibition of LY354740-induced intracellular Ca2+ responses were determined after transient transfection into CHO-Gα16 cells. Responses are normalized to the control response (EC80) and each curve represents the mean ± SEM of three to eight dose–response curves, performed in duplicate, from a minimum of three independent transfections.
The calculated potency (IC50) and Hill coefficients (nH) for the concentration-dependent antagonism of agonist-induced Ca2+ release by RO4988546 and RO5488608 on mGlu2 WT and mutants are shown in Table 3, and the mutants on which the inhibition by the NAMs were clearly different from WT are shown in Figure 7A–F. Two Arginine residues in TM3 were shown to be of significant importance for inhibition of Ca2+ release and an increase in IC50 value was seen for both NAMs. At mutation R636A3.29 the inhibitory potency of RO5488608 was severely reduced, having an IC50 value >1000 nM and with only 20% inhibition of agonist response at 10 µM. In contrast, the functional potency of RO4988546 was only reduced by 4.1-fold (Figure 7A and B). The inhibitory potency of RO4988546 also showed moderate reduction on mutation F643A3.36 and to a smaller extent on Y647V3.40 (Table 3). Notable is also the incomplete inhibition of the agonist response by RO4988546 and RO5488608 on Y647V3.40 and by RO5488608 on L639A3.32 (Figure 7A and B). Residue HisE2.52 located in ECL2 was sensitive for mutation, particularly for inhibition by RO5488608, which had a 28.3-fold and an 8.2-fold increase in IC50 on the H723FE2.52 and H723VE2.52 mutations respectively (Figure 7D). Additionally, RO5488608 (up to 10 µM) only inhibited 60% of the agonist-induced Ca2+release at the H723F mutant (Figure 7D). For RO4988546 an 8.2-fold increase in IC50 was seen on H723VE2.52 while H723FE2.52 only had a 2.9-fold increase compared with WT (Figure 7C). On mutant L732A5.43, located in TM5, inhibition of Ca2+ release by RO5488608 was markedly reduced whereas only a moderate reduction was observed for RO4988546 (Figure 7C and D). Inhibition of the agonist-induced Ca2+ release by mGlu2 NAMs was largely affected by mutagenesis of residue Trp6.48. RO4988546 showed a 178.9-fold increase in potency and a complete attenuation of inhibition was seen by RO5488608 at the W773A6.48 mutation (Figure 7E–F). With the maintenance of an aromatic residue at this position (W773F), RO5488608 became functional, showing a 1.9-fold reduction compared with WT and RO4988546 only showed a moderate reduction in potency (Figure 7E–F). On mutation F780A6.55 RO4988546 showed a moderate reduction in potency while no change was observed for RO5488608 (Figure 7E–F). RO5488608 showed a reduction in inhibition of Ca2+ release of 5.5-fold compared with WT at mutation V798A7.43 in TM7 (Figure 7E–F), while no change was observed for RO4988546. In contrast to the reduced potency seen in most mutants, inhibition of the agonist-induced Ca2+ release by RO4988546 and RO5488608 was enhanced at two receptor mutants in TM5, M728A5.39 and V736A5.47 (Table 3).
Table 3.
Effect of mGlu2 mutations on NAM inhibition of LY354740-induced intracellular Ca2+ release
| RO4988546 | RO5488608 | ||||||
|---|---|---|---|---|---|---|---|
| Mutant | Position | IC50 (nM) | Ratio vs. WT | nH | IC50 (nM) | Ratio vs. WT | nH |
| WT | 5.5 ± 0.5 | −2.0 ± 0.2 | 11.4 ± 1.1 | −2.4 ± 0.8 | |||
| C616S | 2.57 | 5.3 ± 0.7 | 1.0 | −2.4 ± 0.6 | 8.9 ± 1.4 | 0.8 | −1.8 ± 0.5 |
| Y617F | 2.58 | 7.2 ± 0.8 | 1.3 | −1.5 ± 0.2 | 12.3 ± 4.1 | 1.1 | −1.6 ± 0.2 |
| T620A | 2.61 | 4.4 ± 0.7 | 0.8 | −1.7 ± 0.3 | 11.8 ± 1.8 | 1.0 | −1.7 ± 0.3 |
| R635A | 3.28 | 83.5 ± 20.6 | 15.2 | −1.7 ± 0.2 | 26.7 ± 6.8 | 2.3 | −1.8 ± 0.4 |
| R636A | 3.29 | 22.8 ± 1.5 | 4.1 | −1.0 ± 0.2 | >1000 | >100 | |
| L639A | 3.32 | 6.6 ± 1.5 | 1.2 | −1.5 ± 0.2 | 19.9 ± 4.6 | 1.7 | −1.3 ± 0.3 |
| A642S | 3.35 | 5.8 ± 1.4 | 1.1 | −1.8 ± 0.1 | 18.0 ± 2.6 | 1.6 | −2.3 ± 0.2 |
| F643A | 3.36 | 106.4 ± 21.0 | 19.3 | −1.3 ± 0.2 | 13.6 ± 3.1 | 1.2 | −2.0 ± 0.5 |
| S644A | 3.37 | 4.9 ± 1.2 | 0.9 | −1.9 ± 0.2 | 12.9 ± 2.9 | 1.1 | −2.0 ± 0.1 |
| Y647V | 3.40 | 15.8 ± 8.2 | 2.9 | −1.2 ± 0.3 | 9.5 ± 2.8 | 0.8 | −2.2 ± 0.4 |
| S688L-G689V | 4.44–45 | 11.5 ± 1.9 | 2.1 | −1.8 ± 0.2 | 26.7 ± 4.2 | 2.3 | −4.5 ± 0.4 |
| H723F | E2.52 | 15.9 ± 4.1 | 2.9 | −1.4 ± 0.1 | 322.2 ± 175.5 | 28.3 | −0.9 ± 0.1 |
| H723V | E2.52 | 45.2 ± 8.0 | 8.2 | −1.0 ± 0.2 | 93.3 ± 23.3 | 8.2 | −1.0 ± 0.1 |
| M728A | 5.39 | 1.8 ± 0.4 | 0.3 | −2.3 ± 0.5 | 3.5 ± 0.1 | 0.3 | −2.5 ± 0.3 |
| S731A | 5.42 | 3.9 ± 0.3 | 0.7 | −2.1 ± 0.2 | 10.9 ± 2.1 | 0.9 | −2.1 ± 0.3 |
| L732A | 5.43 | 50.0 ± 11.8 | 9.1 | −1.0 ± 0.1 | 348.9 ± 39.1 | 30.6 | −1.0 ± 0.2 |
| N735D | 5.46 | 7.6 ± 1.1 | 1.4 | −1.9 ± 0.3 | 14.5 ± 3.0 | 1.3 | −1.9 ± 0.2 |
| V736A | 5.47 | 2.7 ± 0.4 | 0.5 | −1.8 ± 0.3 | 5.3 ± 0.7 | 0.5 | −1.8 ± 0.2 |
| T769S | 6.44 | 2.3 ± 0.5 | 0.4 | −1.8 ± 0.1 | 10.1 ± 3.0 | 0.9 | −2.6 ± 0.6 |
| T769V | 6.44 | 2.7 ± 0.6 | 0.5 | −1.9 ± 0.03 | 9.5 ± 1.7 | 0.8 | −2.0 ± 0.2 |
| W773F | 6.48 | 62.8 ± 11.9 | 11.4 | −1.1 ± 0.1 | 21.2 ± 4.2 | 1.9 | −1.5 ± 0.4 |
| W773A | 6.48 | 984.1 ± 232.9 | 178.9 | −1.3 ± 0.6 | x | ||
| L777A | 6.52 | 4.9 ± 0.4 | 0.9 | −1.9 ± 0.6 | 10.9 ± 1.8 | 1.0 | −1.3 ± 0.1 |
| F780A | 6.55 | 43.7 ± 11.2 | 7.9 | −1.2 ± 0.1 | 9.0 ± 2.6 | 0.8 | −1.2 ± 0.2 |
| T793A | 7.38 | 5.2 ± 1.0 | 0.9 | −1.6 ± 0.1 | 10.0 ± 1.4 | 0.9 | −1.8 ± 0.2 |
| M794A | 7.39 | 3.9 ± 1.2 | 0.7 | −1.6 ± 0.1 | 12.4 ± 2.2 | 1.1 | −2.1 ± 0.4 |
| C795A | 7.40 | 3.4 ± 0.4 | 0.6 | −1.8 ± 0.1 | 13.2 ± 3.4 | 1.2 | −2.4 ± 0.7 |
| S797A | 7.42 | 7.0 ± 1.7 | 1.3 | −1.5 ± 0.1 | 17.4 ± 3.2 | 1.5 | −2.7 ± 0.2 |
| V798A | 7.43 | 8.1 ± 1.2 | 1.5 | −1.8 ± 0.1 | 62.7 ± 11.4 | 5.5 | −2.4 ± 0.3 |
The IC50 value, ratio versus WT and Hill coefficient of RO4988546 and RO5488608 inhibition of LY354740-induced Ca2+ release on mGlu2 WT and mutant receptors after transient transfection into CHO-Gα16 cells. Affected mutants are shown in bold. Data are presented as mean ± SEM and for RO4988546 the calculated values represent three to eight dose–response measurements from three to eight independent transfections and for RO5488608 the values represent three to five dose–response measurements from three to four independent transfections.
Modelling of RO4988546 and RO5488608 binding in the allosteric binding pocket of the homology model of rat mGlu2
The mutagenesis study of mGlu2 was conducted to obtain a better understanding of the interaction of mGlu2 NAMs in the 7-TM domain and to identify their molecular determinants. We know from the [3H]-PAM binding studies that mGlu2 NAMs and PAMs share an overlapping binding site in the receptor and residue Asn5.46, Ser4.44 and Gly4.45 had previously been shown to be important for PAM function at mGlu2 (Schaffhauser et al., 2003). However, when building the homology model of mGlu2 it was obvious that the adjacent residues Ser4.44 and Gly4.45 were located outside the characterized binding cavity of class A GPCRs and consequently, our studies did not reveal them to be involved in NAM binding (Table 3). The results obtained with mGlu2 NAMs in the agonist-induced intracellular Ca2+ release assay on the mGlu2 mutants were used to guide modelling of the two NAMs in the binding cavity. In the homology model of mGlu2, the sulphonic acid of RO4988546 and RO5488608 are located close to Arg3.28 and Arg3.29 in the binding cavity. Specifically, for RO4988546 the sulphonic group is positioned in between the two Arginines whereas for RO5488608 it is situated closer to the Arg3.29 residue (Figure 8). The trifluoromethylgroup of RO4988546 is located around the aromatic CH groups of Phe3.36, locating the Fluor atoms in a distance of ≈3 Å to the aromatic carbon, a typical geometry of aromatic CH---F interactions (Figure 8). The trifluoromethylgroup of RO4988546 is furthermore positioned in the proximity of the Trp6.48 residue, which is known to be important for GPCR function, although in the mGlu2 model the trypthophane residue is too distant to form a direct interaction with the NAM (Figure 8A). In the proposed binding mode Leu5.43 has a direct interaction with the 7-membered ring of RO5488608 as the side-chain is located 3.5 Å above the π-system of the 7-membered ring which is typical for a favourable conjugated π–CH interaction (Figure 8B). In contrast, for RO4988546, the Leu5.43 residue comes in close proximity to but the interaction is much weaker as the side-chain is located at a distance of 3.6 Å in the aromatic plane of RO4988546 and thus only a weaker Van der Waals interaction is formed (Figure 8A). Around the Phe6.55 residue, RO4988546 can be positioned to gain from the favourable edge-2-face interaction with the aromatic residue (Figure 8A). The orientation of RO4988546 and RO5488608 in the extracellular part of the binding pocket is diverse, which is clearly seen in their different functional response on the H723VE2.52 and H723FE2.52 mutants (Table 3). A valine substitution, corresponding to mGlu3, results in a moderate reduction in Ca2+ release for both NAMs whereas RO5488608 is mainly affected by the phenylalanine substitution. This difference for the H723F mutation shows that the two NAMs interact differently with this residue. This fits with our docking pose, which shows that the orientation of the aromatic moieties of the ligands are turned differently in the environment close to HisE2.52. In the case of RO4988546, the terminal phenyl ring is optimally situated to interact with HisE2.52 by an edge-to-face π–π interaction (Figure 8A). This interaction is preserved when His is substituted by Phe, which explains the lack of effect of this mutation on the activity of RO4988546. In the case of RO5488608, the His residue cannot form a strong aromatic interaction with the ligand because the terminal aromatic moiety is turned such that the aromatic CHs of the ligand point not onto the aromatic interface of HisE2.52 but onto its aromatic edge only (Figure 8B). However, according to our docking pose, HisE2.52 can form a hydrogen bond with its ring NH to the sp2 nitrogen in the bicyclic structure of the ligand (Figure 8B). As this hydrogen bond is lost in the phenylalanine mutant, this can explain the activity loss observed for RO5488608 on the H723F mutation.
Figure 8.
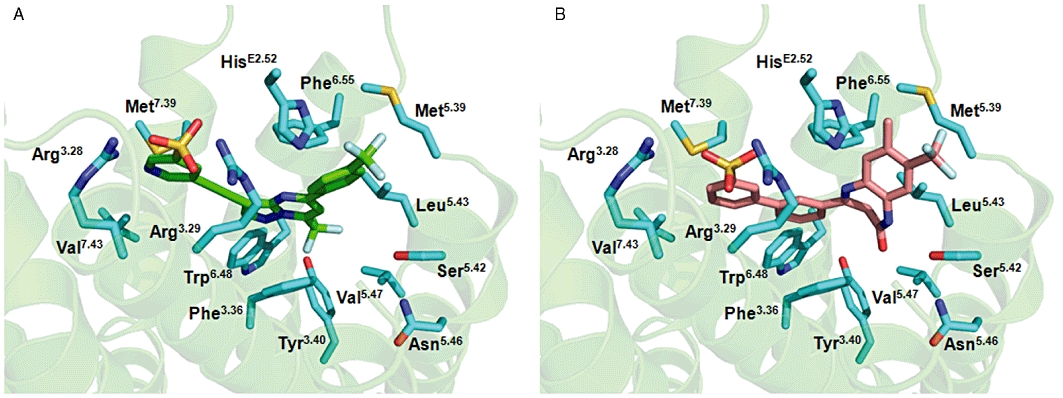
Molecular modelling of RO4988546 and RO5488608 in the allosteric binding pocket of mGlu2. Proposed binding mode of (A) RO4988546 (green) and (B) RO5488608 (salmon) in the allosteric binding pocket of the mGlu2 receptor. The three-dimensional model of rat mGlu2 was generated using the X-ray structure of the human β2-adrenoceptor [pdb code 2rh1 (Cherezov et al., 2007)] and the two NAMs were manually docked into the TM cavity in order to achieve the best interaction with the binding site in terms of shape and electrostatic properties. Residues from the binding pocket, visualized in the picture, are labelled with Ballesteros–Weinstein numbering. See text for details on specific interactions.
Key residues for NAM inhibition of [3H]-LY354740 agonist binding at the orthosteric site
Based on their location in the allosteric binding site and their effect on mGlu2 NAM inhibition of LY354740-induced Ca2+ release, a set of receptor mutants were selected for further analysis in the functional [3H]-LY354740 agonist binding inhibition assay. The calculated potencies (IC50 values) and Hill coefficient (nH) for inhibition of agonist binding for all the mutants studied are shown in Table 4, while results with selected mutants are shown graphically in Figure S2. Similar to the intracellular Ca2+ release data, the two adjacent arginines in TM3, Arg3.28 and Arg3.29, play an important role in inhibition of [3H]-LY354740 agonist binding. The relevance of the two residues are in the same range, 18-fold and 22-fold reduction in potency compared with WT for RO4988546 and 36-fold and 38-fold for RO5488608 (Table 4). Consequently, Arg3.29 had an increased importance for RO4988546 while Arg3.28 was shown to be more important to RO5488608 in functional binding inhibition compared to Ca2+ release. The aromatic residue Phe3.36, which was found to be specific for RO4988546 inhibition of Ca2+ release was less vital for binding inhibition, displaying only a 6-fold reduction compared with WT (Table 4). Mutation of HisE2.52 and Leu5.43 had a larger effect on RO5488608 inhibition of [3H]-LY354740 agonist binding than inhibition of LY354740-induced Ca2+ release; 52-fold compared with 8.2-fold on H723VE2.52 and 40.3-fold compared with 30.6-fold on L732A5.43 (Tables 3 and 4). This is indicative of an increased importance of HisE2.52 and Leu5.43 in RO5488608 inhibition of agonist binding. Interestingly and specific for agonist binding, mutagenesis of Asn5.46 reduced the potency of NAM inhibition. RO4988546 showed a subtle 4-fold reduction whereas a moderate reduction of 10-fold was seen for RO5488608 (Table 4). The most dramatic effect on receptor function and difference between the two functional assays was seen on Trp6.48, which is known to be crucial for GPCR activation. Mutagenesis of Trp6.48 had major effect on Ca2+ release while no reduction in functional potency was seen with [3H]-LY354740 agonist binding (Tables 3 and 4). RO4988546 even showed enhanced binding inhibition on W773A6.48, which is similar to the effect observed on M728A5.39 and S731A5.42 (Table 4). The enhanced binding potency of W773A6.48 was accompanied by a reduction in residual binding (Figure S2). The Phe6.55 residue, which was shown to be specific for RO4988546-induced inhibition of Ca2+ release, was important for both RO4988546 and RO5488608 in [3H]-LY354740 agonist binding inhibition (Tables 3 and 4). Mutation of Val7.43 resulted in a weak reduction of potency in binding inhibition for both RO4988546 and RO5488608, while only the latter was affected when inhibition of LY354740-induced intracellular Ca2+ release was investigated (Table 4).
Table 4.
Effect of mGlu2 mutations on NAM inhibition of [3H]-LY354740 agonist binding at the orthosteric binding site
| RO4988546 | RO5488608 | ||||||
|---|---|---|---|---|---|---|---|
| Mutant | Position | IC50 (nM) | Ratio vs. WT | nH | IC50 (nM) | Ratio vs. WT | nH |
| WT | 7.0 ± 1.2 | −1.1 ± 0.2 | 1.0 ± 0.4 | −1.4 ± 0.4 | |||
| R635A | 3.28 | 125.7 ± 11.8 | 18.0 | −1.4 ± 0.3 | 38.3 ± 3.1 | 38.3 | −0.8 ± 0.1 |
| R636A | 3.29 | 152.8 ± 65.0 | 21.8 | −1.2 ± 0.1 | 36.0 ± 2.6 | 36.0 | −1.1 ± 0.2 |
| F643A | 3.36 | 41.2 ± 13.7 | 5.9 | −1.1 ± 0.2 | 3.4 ± 1.1 | 3.4 | −0.9 ± 0.3 |
| Y647V | 3.40 | 23.8 ± 6.4 | 3.4 | −0.9 ± 0.2 | 1.8 ± 0.5 | 1.8 | −1.0 ± 0.4 |
| H723F | E2.52 | 24.6 ± 12.2 | 3.5 | −1.0 ± 0.1 | 45.9 ± 8.0 | 45.9 | −0.9 ± 0.1 |
| H723V | E2.52 | 48.5 ± 11.0 | 6.9 | −1.1 ± 0.1 | 52.0 ± 8.1 | 52.0 | −0.9 ± 0.1 |
| M728A | 5.39 | 1.1 ± 0.3 | 0.2 | −1.4 ± 0.1 | 0.9 ± 0.3 | 0.9 | −1.4 ± 0.2 |
| S731A | 5.42 | 0.85 ± 0.4 | 0.1 | −1.0 ± 0.1 | 1.5 ± 0.1 | 1.5 | −1.4 ± 0.3 |
| L732A | 5.43 | 19.3 ± 5.4 | 2.8 | −1.1 ± 0.1 | 40.3 ± 7.8 | 40.3 | −1.0 ± 0.0 |
| N735D | 5.46 | 30.50 ± 7.9 | 4.3 | −1.0 ± 0.1 | 10.4 ± 1.6 | 10.4 | −1.5 ± 0.2 |
| V736A | 5.47 | 4.2 ± 1.1 | 0.6 | −0.6 ± 0.1 | 1.9 ± 0.05 | 1.9 | −1.5 ± 0.2 |
| W773F | 6.48 | 47.7 ± 2.8 | 6.8 | −1.1 ± 0.1 | 3.8 ± 0.5 | 3.8 | −1.1 ± 0.1 |
| W773A | 6.48 | 1.7 ± 0.1 | 0.2 | −1.0 ± 0.03 | 2.1 ± 0.1 | 2.1 | −1.2 ± 0.1 |
| F780A | 6.55 | 145.7 ± 34.3 | 20.8 | −0.9 ± 0.04 | 11.0 ± 3.7 | 11.0 | −0.8 ± 0.1 |
| M794A | 7.39 | 20.1 ± 4.0 | 2.9 | −1.5 ± 0.2 | 3.2 ± 1.5 | 3.2 | −1.2 ± 0.3 |
| V798A | 7.43 | 39.8 ± 12.1 | 5.7 | −1.1 ± 0.1 | 4.1 ± 0.7 | 4.1 | −2.3 ± 0.2 |
IC50 value, ratio versus WT and Hill coefficient of RO4988546 and RO5488608 inhibition of [3H]-LY354740 binding at the orthosteric binding site on mGlu2 WT and mutant receptors after transient transfection into HEK293 cells. Affected mutants are shown in bold. Data are presented as mean ± SEM of three to seven dose–response measurements from two to four experiments performed in duplicate on membranes of HEK293 cells transiently transfected with mGlu2 WT or mutant receptors.
Discussion and conclusions
In the present study we characterized two novel NAMs that act as potent non-competitive antagonists on group II mGluRs and determined their molecular interactions within the allosteric binding pocket required for functional inhibition of mGlu2 receptor agonist-dependent activation. The results confirm those from two previous reports in which compounds similar to RO5488608, were shown to have negative allosteric properties on the mGlu2 receptor (Hemstapat et al., 2007; Woltering et al., 2007). Despite the allosteric site of action, shown here in both [35S]-GTPγS binding and intracellular Ca2+ release, RO4988546 and RO5488608 are able to inhibit [3H]-LY354740 agonist binding at the orthosteric site of the receptor. Moreover, none of the two NAMs affected antagonist binding at this site when tested with the [3H]-HYDIA competitive antagonist, confirming data presented by Hemstapat and colleagues (Hemstapat et al., 2007). Taken together, these findings suggest that RO4988546 and RO5488608 binding at the allosteric site result in conformation changes specifically affecting agonist interaction at the orthosteric site. Interestingly, a more detailed analysis of the [3H]-LY354740 binding inhibition mechanism(s) shows the mGlu2 NAMs to reduce both the affinity (Kd) of the agonist and the number of available binding sites to which the agonist can bind (Bmax). Furthermore, the influence on [3H]-LY354740 agonist binding was different between the two NAMs as the effect of RO5488608 resulted in a larger reduction in the number of agonist binding sites. In the presence of 10 nM RO5488608, [3H]-LY354740 was only seen to bind to half the number of binding sites. This observation could possibly be a result of different interactions by the NAMs in the allosteric binding site, having diverse effects on the orthosteric site and agonist binding. The ability of allosteric modulators of mGluRs to influence orthosteric agonist affinity has previously been described for PAMs (Knoflach et al., 2001; Schaffhauser et al., 2003), whereas it has been reported that non-competitive antagonists do not modify agonist binding (Litschig et al., 1999; Pagano et al., 2000). Furthermore, our findings indicate that orthosteric agonists have a positive allosteric effect on [3H]-PAM binding, which indicate the allosterism between the binding sites at mGlu2 to function in both directions. To obtain full activity of a mGluR both VFTs must bind an agonist and adopt the closed conformation whereas binding of only one agonist results in partial activation of the receptor (Kniazeff et al., 2004; Muto et al., 2007). On the other hand, full mGluR activation via PAM interaction in the TM domain requires the binding of only one PAM per receptor dimer and with only one TM domain adopting the active conformation state (Goudet et al., 2005). By using a 3H-PAM we further developed this theory; we observed that the 3H-PAM only recognised half the number of binding sites as the agonist [3H]-LY354740. This suggests that not only one PAM is required for full receptor activation but also only one TM domain of mGlu2 can bind a PAM and adopt its active conformation at the time. In contrast, efficient inhibition of an agonist-induced response by the interaction of NAM at the TM domain has been suggested to require the binding of two NAMs to the receptor dimer (Hlavackova et al., 2005) and consequently, we assumed that one NAM binds to each subunit of the mGlu2 receptor dimer in the system we studied.
When combining functional measurements of NAM inhibition of intracellular Ca2+ release on mGlu2 mutants with mGlu2 homology binding we identified eight residues in the allosteric binding pocket that are crucial for non-competitive antagonism: Arg3.28, Arg3.29, Phe3.36, HisE2.52, Leu5.43, Trp6.48, Phe6.55 and Val7.43. The two arginines at position 3.28 and 3.29 were shown to play a critical role for mGlu2 NAM inhibition of intracellular Ca2+ release which was supported by the close interaction of the sulphonic group of RO4988546 and RO5488608 with the residues. Interestingly, Arg3.29 is conserved within the mGluR family and in mGlu5 this residue is important for the binding affinity and potency of fenobam while for 2-methyl-6-(phenylethynyl)-pyridine the alanine mutation causes a small increase in binding affinity and potency (Malherbe et al., 2003a, 2006). Mutation of Phe3.36 had a detrimental and specific effect on intracellular Ca2+ release inhibition by RO4988546 and a direct interaction could be seen between its trifluoromethylgroup and the phenylalanine residue (Figure 8). Tyr3.40 is completely conserved among mGluRs and of great importance for NAM binding and function in mGlu1 and mGlu5 (Malherbe et al., 2003a,b;). In contrast, our data only indicate a moderate reduction in Ca2+ inhibition with RO4988546 (Table 3) and Tyr3.40 is not located in the nearby vicinity to directly interact with the mGlu2 NAMs in the pocket (distance of 5.5 Å to the closest ligand atom of RO4988546 and distance of 7.3 Å to the closest ligand atom of RO5488608) (Figure 8). With the presentation of a high resolution crystal structure of the human β2-adrenoceptor class A GPCR it became feasible to model the ECL2, at least the residues after the conserved cysteine in the sequence, which is defined as the entrance of the ligand binding pocket of GPCRs (Cherezov et al., 2007). From this model, the amino acid at position E2.52 is known to be pointing into the binding pocket and be involved in ligand binding. The residue at this position is not conserved among mGluRs, not even within group II, hence HisE2.52 pointing into the binding pocket is specific for the mGlu2 receptor. Because of its close location to the mGlu2 NAMs in the binding pocket and as the only mGlu2 specific residue within the binding cavity that showed an effect on NAM inhibition of Ca2+ release (see Figure 5 for alignment), HisE2.52 is likely to be important for mGlu2-selective interactions. Due to a shift of one turn in TM5 in the most recent mGluR sequence alignment the residue previously referred to as 5.47 is now located at position 5.43 (Malherbe et al., 2003b). This position has been proposed as strategic for gating the effect of allosteric modulators of group I mGluRs, being crucial for inhibition by NAMs while its alanine substitution enhances potentiation by PAMs (Knoflach et al., 2001; Malherbe et al., 2003a,b; Muhlemann et al., 2006). A similar mechanism, at least for the NAM effect, could be concluded for Leu5.43 in mGlu2 as its alanine mutant resulted in a clear reduction in Ca2+ release inhibition, which was supported by the identification of direct interactions with both RO4988546 and RO5488608. The difference in functional response and interaction seen for the two NAMs at the L732A5.43 mutant is explained by the slight turn of the RO5488608 structure around its 7-membered ring. Subsequently, the two NAMs also have a different orientation around the Phe6.55 residue where only RO4988546 can be positioned to make a strong interaction and its potency is affected by mutation at this position. Position 6.55 has also been shown crucial for NAMs acting at group I mGluRs (Malherbe et al., 2003a,b;). Correct interaction in the space around Trp6.48 is generally known to be crucial for GPCR activation (Shi et al., 2002; Schwartz et al., 2006). While a complete loss of function is observed with RO5488608 at the W773A6.48 mutant, the presence of the trifluoromethylgroup in RO4988546 is likely to contribute to the overall receptor stability in this mutant, comprising the space of the large tryptophane residue, and therefore partly restoring the functional response to RO4988546 (Table 3, Figure 8). In the case of a phenyalanine substitution, W773F6.48, covering a significantly larger volume in the cavity compared with alanine and possibly increasing receptor stability, the functional response of the mGlu2 NAMs was markedly enhanced (Table 3). A reduction in functional inhibition was seen for RO5488608 at the V798A7.43 mutant while no direct interaction could be identified due to its distant location from the NAM and its effect is therefore probably contributing to the overall stability of the allosteric binding site of the receptor. On three of the mGlu2 mutants that had the largest effect on NAM inhibition of intracellular Ca2+ release, R636A3.29, H723FE2.52 and W773A6.48, the inhibition was incomplete, leaving 40–80% residual receptor activity (Figure 7). This effect is most likely explained by the inability of the mGlu2 NAMs to have a favourable interaction in the binding site of the mutated receptor. Incomplete inhibition, although only 20–30%, was also seen on the L639A3.32 and Y647V3.40 mutants where both NAMs exhibited inhibition potencies close to the WT receptor (Table 4). For the Y647V3.40 mutant this could possibly be a consequence of the reduced agonist potency and efficacy that was observed on this receptor (Table S1), which could be an indication of Tyr3.40 being important for receptor stability and/or for transmission between the VFT and TM domains.
Studying allosteric binding by NAMs and PAMs at the mGlu2 receptor helps to improve the understanding of the interaction between the two binding sites present in the receptor. Previous studies have shown the 7-TM domain of mGluRs, when expressed without its extracellular domain, to function in a similar way as class A GPCRs as their PAMs become full agonists (Goudet et al., 2004). Although this is an important observation in itself, one of the main functional characteristics of mGluRs is the complex allosteric interaction between the VFT and TM domains. The cysteine-rich domain of the mGluR prevents a direct interaction between the VFT and the TM domain but has been suggested to play a role in transmitting the conformational changes induced by ligand binding at either site (Rondard et al., 2006; Muto et al., 2007). In order to determine whether specific interactions within the allosteric binding pocket are involved in transmitting the signal of mGlu2 NAM binding to the orthosteric binding site, we utilized a selection of the mGlu2 mutants for the [3H]-LY354740 agonist binding inhibition assay. We know from the saturation isotherm (Figure 3) that the interaction of RO4988546 and RO5488608 in the allosteric binding site results in diverse effects at the orthosteric site. From the [3H]-LY354740 agonist binding inhibition studies on the selected mGlu2 mutants it became obvious that residues located in the extracellular part of the allosteric binding pocket are of increased importance for this mechanism compared with inhibition of intracellular Ca2+ release. For RO4988546 not only Arg3.28 but also Arg3.29 was important while Phe6.55 was found more important than observed in the intracellular Ca2+ release assay. For RO5488608, which showed a more pronounced effect on the agonist Bmax value, Arg3.28 and Arg3.29 were both of significance and Arg3.28, HisE2.52, Phe6.55 and Leu5.43 all displayed increased importance compared with the observations made in the intracellular Ca2+ release assay. In contrast, Trp6.48, which is completely conserved in class A GPCRs and in mGluRs and known to be important for receptor activation did not specifically influence [3H]-LY354740 agonist binding inhibition by the NAMs. In contrast, a minor enhanced potency in binding inhibition was observed for RO4988546 on W773A6.48. Also located relatively deep in the binding site and close to Trp6.48, Phe3.36 did not affect the functionality of [3H]-LY354740 agonist binding for RO4988546 as was seen in the intracellular Ca2+ release assay. However, an extracellular location does not completely correlate with importance for binding inhibition because Asn5.46, located relatively deep in the pocket, was found to be important for binding inhibition for RO5488608. Due to its distant location from the mGlu2 NAM, Asn5.46 is most likely involved in receptor stabilization as no direct interaction could be observed in molecular modelling (Figure 8). Notable is that Asn5.46 was found to be crucial for PAM activation of the mGlu2 receptor (Schaffhauser et al., 2003).
Since the difference in agonist binding inhibition between the two mGlu2 NAMs was found to be most significant on the HisE2.52 mutants, it is tempting to speculate that the increased effect seen on the agonist Bmax value with RO5488608 involves its favourable interaction with this residue. Furthermore, HisE2.52 is the only residue within the binding cavity important for NAM function that is specific for mGlu2 over mGlu3 and interestingly, IC50 values for [3H]-LY354740 agonist binding inhibition on H723VE2.52 was shifted towards the IC50 values observed at the mGlu3 receptor (77 and 155 nM for RO4988546 and RO5488608, respectively) (Table 4). Consequently, HisE2.52 appears to have increased importance for the transmission from the allosteric to the orthosteric binding site as well as functioning as a selectivity filter for negative allosteric ligands acting on mGlu2 and mGlu3 receptors.
A number of mGlu2 mutants resulted in enhanced potency of RO4988546 and RO5488608 in the functional assays. Both NAMs showed enhanced potency as antagonists in the intracellular Ca2+ release assay on mutation M728A5.39 and V736A5.47 and RO4988546 showed enhanced potency on M728A5.39, S731A5.42 and W773A6.48 in agonist binding. A decreased residual binding of both RO4988546 and RO5488608 was seen on mutations M728A5.39, S731A5.42, L732A5.43 and W773A6.48. With the exception of Trp6.48, the identified residues (Met5.39, Ser5.42, Leu5.43 and Val5.47) are located in TM5. In adrenoceptors and dopamine receptors this region contains three highly conserved serines, at position 5.42, 5.43 and 5.46, that are involved in agonist binding and crucial for receptor activation (Liapakis et al., 2000). Also in some mGluRs, mGlu2, -3, -7 and -8, a serine residue is located at position 5.42. This region of mGlu2 is therefore likely to be involved in the agonist (PAM) interaction and function, and mutagenesis of these residues would therefore affect the receptor conformation which could be a possible explanation for the enhanced potency of mGlu2 NAMs on these mutants.
In conclusion, this study is the first pharmacological report on two novel and potent mGlu2 NAMs describing in detail their interaction in the allosteric binding pocket of the mGlu2 receptor. Although NAMs are defined as modulators of agonist affinity and signalling potency this is the first study clearly demonstrating that mGluR NAMs affect the binding features of an orthosteric agonist and do not only influence functional activation. This observation is valuable for the complete understanding of the cross-talk between the two sites of interaction in mGluRs, orthosteric and allosteric. A binding mode of RO4988546 and RO5488608 NAMs is proposed in an mGlu2 homology model, based on the β2-adrenoceptor crystal structure, identifying specific molecular determinants for the two compounds. By comparing two functional assays we are able to suggest interaction points in the allosteric binding site that are crucial for antagonism of agonist-induced intracellular Ca2+ release or for inhibition of [3H]-LY354740 agonist binding. As the allosteric binding pocket of the mGlu2 receptor is an attractive drug target, both for NAMs and PAMs, the data presented in this report should further help to improve the development of potent and selective ligands for the mGlu2 receptor.
Acknowledgments
The authors would like to thank Dr P. Huguenin for the synthesis of tritiated radioligands, Monique Dellenbach for excellent technical assistance and Dr Pari Malherbe and Dr Hervé Schaffhauser for critical comments on the paper.
Glossary
Abbreviations
- ECL
extracellular loop
- FLIPR
fluorometric imaging plate reader
- mGluR
metabotropic glutamate receptor
- NAM
negative allosteric modulator
- PAM
positive allosteric modulator
- TM
transmembrane
- VFT
venus flytrap
- WT
wild-type
Conflicts of interest
All authors work at F. Hoffmann-La Roche Ltd, Basel, Switzerland.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Western blot analysis on mGlu2 WT and mutant receptors. Representative Western blot analysis on a selection of mGlu2 mutants to illustrate the expression of the receptors in CHO-Gα16 cells after transient transfection. Cell lysates of CHO-Gα16 cells transfected with rat mGlu2 WT or mutant receptor (for 48 h) were prepared using a lysis buffer containing 1 mM Tris, 1.25% w/v Triton X-100, 1 mM EGTA, 120 mM NaCl and a cocktail of protease inhibitors (F. Hoffmann-La Roche Diagnostics GmbH, Mannheim, Germany). Samples were prepared with NuPage Loading Dye and NuPage Reducing Agent (Invitrogen, Carlsbad, CA, USA) and warmed at 70°C for 10 min. In total, 20 mg cell lysate was separated on a 3–8% w/v Tris-Acetate gel (Invitrogen, Carlsbad, CA, USA) and transferred to a PVDF membrane. Anti mGlu2 rabbit antibody (Millipore, Billerica, MA, USA, 1:1000 dilution in blocking buffer) was incubated over night at 4°C and goat Anti-Rabbit IgG POD antibody (GE Healthcare, Buckinghamshire, UK, 1:5000) was incubated for 1 h at RT before detection with ECL Plus detection (GE Healthcare, Buckinghamshire, UK). The prestained SDS-PAGE molecular marker High Range was used (Bio-Rad, Hercules, CA, USA) and the relevant sizes in kDa are indicated to the left. The specificity of the mGlu2 polyclonal antibody in our experimental conditions was previously described (Lundström et al., 2009) and was confirmed in mock-transfected CHO-Gα16 cells (data not shown), see also manufacturer's instructions.
Figure S2 Effect of mGlu2 mutations on NAM inhibition of 3[H]-LY354740 agonist binding at the orthosteric binding site. Concentration-dependent inhibition of 3[H]-LY354740 agonist binding at the orthosteric binding site by RO4988546 (A, C, E) and RO5488608 (B, D, F) at mGlu2 WT and mutant receptors. mGlu2 receptor mutants on which the function inhibition response was reduced compared with WT are shown graphically here while the calculated IC50 values for all studied mutants are presented in Table 4. Inhibition of functional agonist binding was determined after transient transfection into HEK293 cells. Responses are normalized to the control response and each curve represents the mean ± SEM of three to seven dose–response measurements, performed in duplicate, from two to four experiments.
Table S1 Effect of mGlu2 mutations on LY354740 agonist induced intracellular Ca2+ release
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ballard TM, Gatti S, Goetschi E, Wichmann J, Woltering TJ. Combination of mGluR2 antagonist and ache inhibitor for treatment of acute and/or chronic neurological disorders. 2005. WO2005014002A1, F. Hoffmann-La Roche AG.
- Ballesteros JA, Weinstein H. Integrated Methods for construction three-dimentional models and computational probing of structure-function relations in G protein-coupled recepotrs. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- Barda DA, Wang ZQ, Britton TC, Henry SS, Jagdmann GE, Coleman DS, et al. SAR study of a subtype selective allosteric potentiator of metabotropic glutamate 2 receptor, N-(4-phenoxyphenyl)-N-(3-pyridinylmethyl)ethanesulfonamide. Bioorg Med Chem Lett. 2004;14:3099–3102. doi: 10.1016/j.bmcl.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Bissantz C, Logean A, Rognan D. High-throughput modeling of human G-protein coupled receptors: amino acid sequence alignment, three-dimensional model building, and receptor library screening. J Chem Inf Comput Sci. 2004;44:1162–1176. doi: 10.1021/ci034181a. [DOI] [PubMed] [Google Scholar]
- Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets. 2007;8:169–184. doi: 10.2174/138945007779315614. [DOI] [PubMed] [Google Scholar]
- Bu L, Michino M, Wolf RM, Brooks CL., 3rd Improved model building and assessment of the Calcium-sensing receptor transmembrane domain. Proteins. 2008;71:215–226. doi: 10.1002/prot.21685. [DOI] [PubMed] [Google Scholar]
- Campo B, Girard F, Celanire S, Legrand C, Rizzo O, Bessif A, et al. In-vivo characterization of mGlu2/3 negative allosteric modulators in forced swim test, a rodent model depression, and comparison with reference mGlu2/3 orthosteric antagonists (343.8/Q11) 2009. Annual Meeting of the Society for Neuroscience: Washington DC.
- Cartmell J, Adam G, Chaboz S, Henningsen R, Kemp JA, Klingelschmidt A, et al. Characterization of [3H]-(2S,2′R,3′R)-2-(2′,3′-dicarboxy-cyclopropyl)glycine ([3H]-DCG IV) binding to metabotropic mGlu2 receptor-transfected cell membranes. Br J Pharmacol. 1998;123:497–504. doi: 10.1038/sj.bjp.0701647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, et al. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46:457–467. doi: 10.1016/j.neuropharm.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman DS, Jagdmann GEJ, Johnson KW, Johnson MP, Large TH, Monn JA, et al. Preparation of N-phenyl-N-alkylsulfonyl(pyridylmethyl)amines as potentiators of glutamate receptors. 2001. WO 2001056990, Eli Lilly.
- Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley ME. Positive allosteric modulators of the metabotropic glutamate receptor 2 for the treatment of schizophrenia. Expert Opin Ther Pat. 2009;19:1259–1275. doi: 10.1517/13543770903045009. [DOI] [PubMed] [Google Scholar]
- Galici R, Echemendia NG, Rodriguez AL, Conn PJ. A selective allosteric potentiator of metabotropic glutamate (mGlu) 2 receptors has effects similar to an orthosteric mGlu2/3 receptor agonist in mouse models predictive of antipsychotic activity. J Pharmacol Exp Ther. 2005;315:1181–1187. doi: 10.1124/jpet.105.091074. [DOI] [PubMed] [Google Scholar]
- Gatti S, Goetschi E, Palmer WS, Wichmann J, Woltering T. Acetylenyl-pyrazolo-pyrimidine derivates as mGlu2 antagonists. 2006. WO/2006/099972, F. Hoffmann-La Roche AG.
- Gatti S, Knoflach F, Kew JNC, Adam G, Goetschi E, Wichmann J, et al. 8-Arylethynyl-1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives are potent non-competitive metabotropic glutamate receptor 2/3 antagonists. 2001. Annual Meeting of the Society for Neuroscience; San Diego, CA. 2001.
- Gatti S, Wichmann J, Woltering TJ. Preparation of dihydrobenzo[b][1,4]diazepin-2-one biphenylsulfonamides as group II mGlu receptor antagonists. 2008. US2008261957, F. Hoffmann-La Roche AG.
- Goudet C, Gaven F, Kniazeff J, Vol C, Liu J, Cohen-Gonsaud M, et al. Heptahelical domain of metabotropic glutamate receptor 5 behaves like rhodopsin-like receptors. Proc Natl Acad Sci U S A. 2004;101:378–383. doi: 10.1073/pnas.0304699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet C, Kniazeff J, Hlavackova V, Malhaire F, Maurel D, Acher F, et al. Asymmetric functioning of dimeric metabotropic glutamate receptors disclosed by positive allosteric modulators. J Biol Chem. 2005;280:24380–24385. doi: 10.1074/jbc.M502642200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemstapat K, Da Costa H, Nong Y, Brady AE, Luo Q, Niswender CM, et al. A novel family of potent negative allosteric modulators of group II metabotropic glutamate receptors. J Pharmacol Exp Ther. 2007;322:254–264. doi: 10.1124/jpet.106.117093. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Ballard TM, Kew JN, Richards JG, Kemp JA, Adam G, et al. Pharmacological manipulation of mGlu2 receptors influences cognitive performance in the rodent. Neuropharmacology. 2004;46:907–917. doi: 10.1016/j.neuropharm.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Hlavackova V, Goudet C, Kniazeff J, Zikova A, Maurel D, Vol C, et al. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. Embo J. 2005;24:499–509. doi: 10.1038/sj.emboj.7600557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MP, Baez M, Jagdmann GE, Jr, Britton TC, Large TH, Callagaro DO, et al. Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine. J Med Chem. 2003;46:3189–3192. doi: 10.1021/jm034015u. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. 7TM receptor allostery: putting numbers to shapeshifting proteins. Trends Pharmacol Sci. 2009;30:460–469. doi: 10.1016/j.tips.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prezeau L, Pin JP. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat Struct Mol Biol. 2004;11:706–713. doi: 10.1038/nsmb794. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Ballard T, Goetschi E, Huwyler J, Wichmann J, Woltering T, et al. R1315, A potent orally actve non-competitive group II metabotropic glutamate receptor antagonist. 2005. 5th international meeting on metabotropic receptors Taormina, Italy. 2005.
- Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, et al. Positive allosteric modulators of metabotropic glutamate 1 receptor: characterization, mechanism of action, and binding site. Proc Natl Acad Sci U S A. 2001;98:13402–13407. doi: 10.1073/pnas.231358298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal D, Nawoschik S, Ochalski R, Dunlop J. Functional calcium coupling with the human metabotropic glutamate receptor subtypes 2 and 4 by stable co-expression with a calcium pathway facilitating G-protein chimera in Chinese hamster ovary cells. Biochem Pharmacol. 2003;66:785–790. doi: 10.1016/s0006-2952(03)00399-x. [DOI] [PubMed] [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, et al. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- Liapakis G, Ballesteros JA, Papachristou S, Chan WC, Chen X, Javitch JA. The forgotten serine. A critical role for Ser-2035.42 in ligand binding to and activation of the beta 2-adrenergic receptor. J Biol Chem. 2000;275:37779–37788. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- Litschig S, Gasparini F, Rueegg D, Stoehr N, Flor PJ, Vranesic I, et al. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- Lorrain DS, Schaffhauser H, Campbell UC, Baccei CS, Correa LD, Rowe B, et al. Group II mGlu receptor activation suppresses norepinephrine release in the ventral hippocampus and locomotor responses to acute ketamine challenge. Neuropsychopharmacology. 2003;28:1622–1632. doi: 10.1038/sj.npp.1300238. [DOI] [PubMed] [Google Scholar]
- Lundström L, Kuhn B, Beck J, Borroni E, Wettstein JG, Woltering TJ, et al. Mutagenesis and molecular modeling of the orthosteric binding site of the mGlu2 receptor determining interactions of the group II receptor antagonist (3)H-HYDIA. ChemMedChem. 2009;4:1086–1094. doi: 10.1002/cmdc.200900028. [DOI] [PubMed] [Google Scholar]
- Lundström L, Spooren W, Gatti S. Excitatory amino acids and their antagonists. In: Stolerman IPE, editor. Encyclopedia of Psychopharmacology. Vol. 1. Berlin, Heidelberg: Springer; 2010. pp. 508–516. [Google Scholar]
- Malherbe P, Kratochwil N, Knoflach F, Zenner MT, Kew JN, Kratzeisen C, et al. Mutational analysis and molecular modeling of the allosteric binding site of a novel, selective, noncompetitive antagonist of the metabotropic glutamate 1 receptor. J Biol Chem. 2003b;278:8340–8347. doi: 10.1074/jbc.M211759200. [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Muhlemann A, Zenner MT, Fischer C, Stahl M, et al. Comparison of the binding pockets of two chemically unrelated allosteric antagonists of the mGlu5 receptor and identification of crucial residues involved in the inverse agonism of MPEP. J Neurochem. 2006;98:601–615. doi: 10.1111/j.1471-4159.2006.03886.x. [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, et al. Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol. 2003a;64:823–832. doi: 10.1124/mol.64.4.823. [DOI] [PubMed] [Google Scholar]
- Muhlemann A, Ward NA, Kratochwil N, Diener C, Fischer C, Stucki A, et al. Determination of key amino acids implicated in the actions of allosteric modulation by 3,3′-difluorobenzaldazine on rat mGlu5 receptors. Eur J Pharmacol. 2006;529:95–104. doi: 10.1016/j.ejphar.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Muto T, Tsuchiya D, Morikawa K, Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proc Natl Acad Sci U S A. 2007;104:3759–3764. doi: 10.1073/pnas.0611577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, et al. The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem. 2000;275:33750–33758. doi: 10.1074/jbc.M006230200. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Petrel C, Kessler A, Maslah F, Dauban P, Dodd RH, Rognan D, et al. Modeling and mutagenesis of the binding site of Calhex 231, a novel negative allosteric modulator of the extracellular Ca(2+)-sensing receptor. J Biol Chem. 2003;278:49487–49494. doi: 10.1074/jbc.M308010200. [DOI] [PubMed] [Google Scholar]
- Porter RH, Benwell KR, Lamb H, Malcolm CS, Allen NH, Revell DF, et al. Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. Br J Pharmacol. 1999;128:13–20. doi: 10.1038/sj.bjp.0702751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondard P, Liu J, Huang S, Malhaire F, Vol C, Pinault A, et al. Coupling of agonist binding to effector domain activation in metabotropic glutamate-like receptors. J Biol Chem. 2006;281:24653–24661. doi: 10.1074/jbc.M602277200. [DOI] [PubMed] [Google Scholar]
- Rowe BA, Schaffhauser H, Morales S, Lubbers LS, Bonnefous C, Kamenecka TM, et al. Transposition of three amino acids transforms the human metabotropic glutamate receptor (mGluR)-3-positive allosteric modulation site to mGluR2, and additional characterization of the mGluR2-positive allosteric modulation site. J Pharmacol Exp Ther. 2008;326:240–251. doi: 10.1124/jpet.108.138271. [DOI] [PubMed] [Google Scholar]
- Schaffhauser H, Richards JG, Cartmell J, Chaboz S, Kemp JA, Klingelschmidt A, et al. In vitro binding characteristics of a new selective group II metabotropic glutamate receptor radioligand, [3H]LY354740, in rat brain. Mol Pharmacol. 1998;53:228–233. [PubMed] [Google Scholar]
- Schaffhauser H, Rowe BA, Morales S, Chavez-Noriega LE, Yin R, Jachec C, et al. Pharmacological characterization and identification of amino acids involved in the positive modulation of metabotropic glutamate receptor subtype 2. Mol Pharmacol. 2003;64:798–810. doi: 10.1124/mol.64.4.798. [DOI] [PubMed] [Google Scholar]
- Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- Schoepp DD, Johnson BG, Wright RA, Salhoff CR, Mayne NG, Wu S, et al. LY354740 is a potent and highly selective group II metabotropic glutamate receptor agonist in cells expressing human glutamate receptors. Neuropharmacology. 1997;36:1–11. doi: 10.1016/s0028-3908(96)00160-8. [DOI] [PubMed] [Google Scholar]
- Schwartz TW, Frimurer TM, Holst B, Rosenkilde MM, Elling CE. Molecular mechanism of 7TM receptor activation – a global toggle switch model. Annu Rev Pharmacol Toxicol. 2006;46:481–519. doi: 10.1146/annurev.pharmtox.46.120604.141218. [DOI] [PubMed] [Google Scholar]
- Schweitzer C, Kratzeisen C, Adam G, Lundstrom K, Malherbe P, Ohresser S, et al. Characterization of [(3)H]-LY354740 binding to rat mGlu2 and mGlu3 receptors expressed in CHO cells using semliki forest virus vectors. Neuropharmacology. 2000;39:1700–1706. doi: 10.1016/s0028-3908(99)00265-8. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Woltering TJ, Adam G, Alanine A, Wichmann J, Knoflach F, Mutel V, et al. Synthesis and characterization of 8-ethynyl-1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: new potent non-competitive metabotropic glutamate receptor 2/3 antagonists. Part 1. Bioorg Med Chem Lett. 2007;17:6811–6815. doi: 10.1016/j.bmcl.2007.10.026. [DOI] [PubMed] [Google Scholar]
- Woltering TJ, Adam G, Wichmann J, Goetschi E, Kew JN, Knoflach F, et al. Synthesis and characterization of 8-ethynyl-1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: Part 2. New potent non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg Med Chem Lett. 2008a;18:1091–1095. doi: 10.1016/j.bmcl.2007.12.005. [DOI] [PubMed] [Google Scholar]
- Woltering TJ, Wichmann J, Goetschi E, Adam G, Kew JN, Knoflach F, et al. Synthesis and characterization of 1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: Part 3. New potent non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg Med Chem Lett. 2008b;18:2725–2729. doi: 10.1016/j.bmcl.2008.02.076. [DOI] [PubMed] [Google Scholar]
- Woltering TJ, Wichmann J, Goetschi E, Knoflach F, Ballard TM, Huwyler J, et al. Synthesis and characterization of 1,3-dihydro-benzo[b][1,4]diazepin-2-one derivatives: Part 4. In vivo active potent and selective non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg Med Chem Lett. 2010;20:6969–6974. doi: 10.1016/j.bmcl.2010.09.125. [DOI] [PubMed] [Google Scholar]
- Yoshimizu T, Chaki S. Increased cell proliferation in the adult mouse hippocampus following chronic administration of group II metabotropic glutamate receptor antagonist, MGS0039. Biochem Biophys Res Commun. 2004;315:493–496. doi: 10.1016/j.bbrc.2004.01.073. [DOI] [PubMed] [Google Scholar]
- Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl) 2006;186:587–593. doi: 10.1007/s00213-006-0390-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.