

Abstract
BACKGROUND AND PURPOSE
Most patients at elevated cardiovascular risk receive long-term aspirin (ASA) anti-platelet treatment. The present study specifically addresses the pharmacological interactions between selective COX-2 inhibitors and ASA and the possible consequences for the thrombotic risk during long-term treatment.
EXPERIMENTAL APPROACH
New Zealand white rabbits were fed a standard laboratory diet supplemented with 1% cholesterol (CON) for 12 weeks. Age-matched control rabbits were fed the same standard diet without addition of cholesterol (SD). Rabbits were randomly assigned to one of the following groups: rofecoxib (ROFE, 25 mg·kg−1, bid), acetylsalicylic acid (ASA, 5 mg·kg−1, bid) or a combination of both (ASA + ROFE). At the end of the feeding period, the severity of atherosclerotic plaque formation was assessed in the aorta. Thrombus formation was assessed in the left carotid artery using a modified Folts procedure.
KEY RESULTS
Treatment of cholesterol-fed rabbits with ASA significantly reduced plaque formation. This reduction in lesion size was not observed in animals treated with the combination of rofecoxib and ASA. In the modified Folts model, treatment with either rofecoxib or ASA increased the total blood flow above that of untreated animals. This increase was statistically significant in the case of ASA, while cotreatment with rofecoxib abolished this ASA effect completely and reduced the total flow rate to the levels seen in untreated hypercholesterolaemic controls.
CONCLUSIONS
COX-2 inhibition by rofecoxib attenuates the antithrombotic and anti-atherosclerotic effects of ASA during long-term treatment in cholesterol-fed rabbits.
Keywords: atherosclerosis, acetylsalicylic acid, rofecoxib, cyclo-oxygenase, thromboxane, prostacyclin
Introduction
Coxibs, selective inhibitors of COX-2, were originally designed to separate the anti-inflammatory/analgesic actions of COX-2 inhibition from the physiological effects of COX-1-derived prostaglandins on haemostasis and the gastrointestinal (GI) tract. Indeed, coxibs caused less GI intolerance, specifically reduced GI bleeding and ulcer formation. With increasing knowledge about the complex regulation and function of COX-2-derived prostaglandins, it became apparent that this concept was incomplete, and COX-2-derived prostaglandins might well have important physiological functions. This was convincingly demonstrated in COX-2 knockout animals, which exhibited severely disturbed tissue homeostasis and blood perfusion (Morham et al., 1995). Moreover, in human vascular endothelium (McAdam et al., 1999) and smooth muscle cells (Rimarachin et al., 1994), vasodilator and anti-platelet prostaglandins such as PGI2 and PGE2 are mainly derived from COX-2. These findings suggest that COX-2-dependent prostaglandin production in addition to its pathological role in inflammation may also have physiological functions by controlling vessel tone and haemostasis.
Several prospective randomized trials have now demonstrated that coxibs increase the risk for thromboembolic cardiovascular events, mainly myocardial infarction. They might also be associated with an increase in the severity of pre-existing atherosclerotic vessel injury. For example, there was a doubling of cardiovascular risk after 18 months of treatment with rofecoxib in the APPROVe (adenomatous polyp prevention on Vioxx) trial in patients without overt cardiovascular disorders (Fitzgerald, 2004; Juni et al., 2004), while in high-risk cardiovascular patients undergoing coronary artery bypass grafting (CABG), a doubling of the cardiovascular events was seen even after 1 week of treatment with coxibs (Nussmeier et al., 2005). This suggests that prevention of ‘reactive’ COX-2-derived prostaglandin formation is associated with an increased risk of thromboembolic cardiovascular events, and that the extent of this effect is dependent on the severity of pre-existing atherosclerosis.
Interestingly, the patients in the CABG trial (Nussmeier et al., 2005) did receive ASA as a prophylaxis; however, this was unable to protect the patients from the enhanced thromboembolic risk. Cheng et al. (2002) have shown that mice lacking the prostacyclin receptor exhibited increased thromboxane generation. This suggests that endogenous PGI2 might control the (prothrombotic) action of thromboxane via the prostacyclin receptor. Such an effect might become particularly relevant for coxibs that, by definition, do block COX-2-dependent PGI2 generation in the vascular endothelium but not COX-1-dependent thromboxane formation in platelets. It has been shown that conventional nonsteroidal anti-inflammatory drugs (NSAIDs) such as indomethacin (Livio et al., 1982) or ibuprofen (Rao et al., 1983) can completely block the anti-platelet/antithrombotic effects of ASA in vitro. Similar data were reported recently for dipyrone and other pyrazolinones (Hohlfeld et al., 2008).
To date, there are no data available on the possible interactions between selective COX-2 inhibitors and ASA with regard to their anti-platelet/antithrombotic activity. Hence, in the present study we investigated this issue in a standard animal model of atherosclerosis, the cholesterol-fed rabbit. In this model, treatment with ASA has been shown to reduce the severity of atherosclerotic vascular lesions (Prasad and Lee, 2003). However, there are no data available on the consequences of this effect for thrombotic risk and, specifically, on the possible pharmacological interactions between ASA and coxibs during long-term treatment.
Methods
Animals
New Zealand White rabbits of either sex (mean body weight 2–3 kg, male and female in equal parts) were fed a standard laboratory diet (Altromin®, Lage, Germany) supplemented with 1% cholesterol (CON) for 12 weeks. Age-matched control rabbits were fed the same diet without cholesterol (SD). In all rabbits, a blood lipid analysis was performed before and at the end of the feeding period. Rabbits were randomly assigned to one of the following groups: rofecoxib (ROFE, Vioxx®, MSD Sharp & Dohme GmbH, Haar, Germany, 25 mg·kg−1, n = 8), aspirin (ASA, Aspisol®, Bayer Vital GmbH, Leverkusen, Germany, 5 mg·kg−1, n = 8) or the combination of both (ASA + ROFE, n = 8). Cholesterol-fed animals without treatment (CON, n = 13) and animals fed a standard diet without cholesterol (SD, n = 11) were used as controls. The drugs were dissolved in water and given directly into the oropharyngeal cavity at a volume of 1 mL·kg−1, 7 days a week in the morning and evening, the last dose was administered 12 h prior to the acute experiment. The drugs were administered over the whole 12 weeks feeding period. The effective dose of rofecoxib was determined in preliminary dose-finding studies in untreated rabbits by measuring the PGE2 synthesis in monocytes after stimulation with LPS. All animal care and experimental procedures followed Guidelines of the German Animal Protection Act and were approved by the Animal Care Committee of the state of Thüringen (Germany).
Quantification of atherosclerotic lesions
The extent of atherosclerosis development was assessed using the ‘en face’ surface lesion analysis. At the end of the experiment, the rabbits were killed, and the aorta was perfusion-fixed in PBS supplemented with 4% paraformaldehyde and 5% sucrose. Afterwards, the aorta was carefully dissected from the iliac bifurcation up to the heart, opened longitudinally and pinned to expose the entire intimal surface. The aorta was placed in paraformaldehyde–sucrose solution for 120 min at 4°C, then it was rinsed three times with PBS and stained with Sudan IV (Paigen et al., 1987). Images of stained aortas were taken with a digital camera. Morphometric analysis was carried out using Leica Q500/W (Leica Microsystems, Wetzlar, Germany) image analysis software. The extent of atherosclerosis is expressed as percentage of the aortic surface area covered by lesions.
Measurement of thrombus formation
In the acute thrombosis experiments, rabbits were anaesthetized by i.v. administration of sodium pentobarbital (bolus injection of 35 mg·kg−1 followed by infusion of 6 mg·kg−1 h−1). Based on the model described by Folts et al. in stenosed canine coronary arteries (Folts et al., 1976; Folts, 1991), we established a model in the rabbit carotid artery where damage of the intimal vessel wall was combined with a defined reduction in blood flow. Vessel wall damage and stenosis stimulated platelet activation and coagulation that resulted in either recurrent vessel occlusions seen as cyclic flow variations (CFVs) or a complete thrombotic occlusion of the artery with permanent interruption of blood flow. In preliminary studies, we defined the optimal conditions to get a reproducible thrombotic effect in the rabbit carotid artery. Briefly, the right carotid artery of the rabbit was isolated, and a Doppler flow probe was placed on the vessel to continuously measure blood flow. Distal to the flow probe, the artery was gently squeezed with a rubber-covered surgical forceps to produce intimal and medial damage. Following injury, a cylindrical plastic constrictor was placed around the damaged area leading to a stenosis of the vessel equivalent to a 30–40% reduction of the carotid artery blood flow. To evaluate the effect of the drugs studied, we determined both the number of cyclic flow reductions and the amount of blood flowing through the carotid artery within 60 min after vessel injury. The last was calculated by measuring the area under the blood flow versus time curve using the Leica Q500/W image analysis software (Leica Microsystems). In addition, the number of total vessel occlusions was determined. In this setting, the number of cyclic flow variations indicates the frequency of thrombotic events, while total blood flow indicates the total extent of blood flow restriction.
Measurement of TXB2, PGE2 and 6-oxo-PGF1α
Before and at the end of the feeding period, blood (1 mL) was drawn from the ear artery, transferred to non-siliconized glass tubes and allowed to clot for 60 min at 37°C. Serum was separated by centrifugation (1000×g for 10 min), indomethacin (10 µg·mL−1) was added and aliquots were stored frozen at −20°C until radioimmunoassay for the stable degradation products thromboxane B2 and 6-oxo-PGF1α as previously described (Schrör and Seidel, 1988).
PGE2 generation was determined as a parameter for COX-2 activity. Heparin-treated blood (10 IU·mL−1) drawn 2 h after oral administration of the drugs was incubated with LPS (10 µg·mL−1) for 24 h at 37°C (Patrignani et al., 1994). Plasma was separated by centrifugation (1000×g for 10 min) and stored in aliquots at −80°C. PGE2 was measured by elisa (Cayman Chemicals Company, Ann Arbor, MI, USA).
RT-qPCR
Total RNA was extracted from the aorta after removal of the adventitial layer using TriReagent (Sigma-Aldrich, Deisenhofen, Germany) and reverse transcribed by the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA) according to the manufacturer's instructions. COX-2 expression was analysed by TaqMan Gene Expression Assay (Applied Biosystems, Oc03398291_m1) normalized to GAPDH (Oc03823402_g1).
Immunoblotting
Western blot analysis of COX-2 expression in the abdominal aorta was performed using primary anti-COX-2 polyclonal antibody (goat, Santa Cruz, Heidelberg, Germany; 1:1 000). Quantification was performed using fluorescent secondary antibodies and the Odyssey Infrared Imaging System (1:10.000, LI-COR Biosciences, Lincoln, NE).
Immunohistochemistry
For cryosectioning, tissue samples were completely embedded in TissueTek® (Sakura Finetek Germany GmbH, Staufen, Germany) and frozen at −40°C in isopentane. Fourteen-micrometre-thick unfixed cryosections were adsorbed to glass slides. After pretreatment with water containing 3% H2O2 in order to block endogenous peroxidases and 1% bovine albumin serum in PBS in order to block free binding sites, primary antibodies were diluted as indicated and tissue samples were incubated overnight at 4°C. After being rinsed, sections were incubated with horseradish peroxidase-linked secondary antibodies from mouse (tissue factor, TF; plasminogen activator inhibitor-1, PAI-1; 1:50) or goat (COX-2, 1:500; thrombomodulin, TM, 1:200) for 60 min (RT) and rinsed twice with PBS, before the final staining was developed with diaminobenzidine (Sigma-Aldrich). Bright-field images were taken using a ColorViewII and AnalySIS 3.2 software (Soft Imaging System; Münster, Germany). The expression of the following proteins was determined: COX-2, PAI-1 (both Santa Cruz Biotechnology, Heidelberg, Germany), TF and thrombomodulin (both American Diagnostica, Pfungstadt, Germany). Due to the limitations of immunohistochemistry, a semiquantitative scaling was used for quantification: no staining (−), mild (+), moderate (++), strong (+++) and intensive (++++). The degree of staining was evaluated by five independent observers in a blinded fashion for each tissue specimen.
Statistics
The data are presented as mean ± SEM of n different animals. Statistical analysis was performed using one-way anova followed by Bonferroni's multiple comparisons test. P-values for α of <0.05 were considered significant.
Results
Atherosclerotic lesion formation
Cholesterol feeding resulted in a marked increase in plasma cholesterol, from 1.7 ± 0.2 mmol·L−1 measured in all cholesterol-fed animals (n = 37) before to 25.6 ± 1.4 mmol·L−1 (n = 37) at 6 weeks and 46.2 ± 2.8 mmol·L−1 (n = 32) at 12 weeks. There were no significant differences between the various groups of untreated and drug-treated animals; that is, neither ASA nor rofecoxib affected the plasma cholesterol concentrations. Plasma concentrations at 12 weeks were as follows: control 45.4 ± 4.6 mmol·L−1 (n = 13); ASA 51.1 ± 5.6 mmol·L−1 (n = 8); rofecoxib 42.7 ± 5.1 mmol·L−1 (n = 6); ASA + ROFE 44.6 ± 8.0 mmol·L−1 (n = 5). It should be noted that from the 37 animals included in cholesterol feeding, four animals did not survive the treatment period of 12 weeks (one rabbit in the rofecoxib, three rabbits in the combination group). Additionally, one rabbit from the rofecoxib group completed the feeding period but died immediately before starting the experiment so that only organ but no blood parameters could be determined in this animal. Cholesterol feeding was associated with an extensive development of atherosclerotic lesions, covering 71 ± 5% (n = 13) of total aortic luminal surface in untreated animals. No plaque formation was seen in rabbits fed a standard diet without cholesterol supplementation (data not shown). Treatment of cholesterol-fed rabbits with ASA significantly reduced plaque formation by about two-thirds, to 29 ± 8% (n = 8, P < 0.001). Rofecoxib also reduced plaque formation to 52 ± 11% (n = 7), but, in contrast to ASA, this effect was not statistically significant. Interestingly, the combined administration of ASA and rofecoxib (n = 5) did not result in any further reduction of plaque formation. In contrast, the reduced plaque coverage by ASA was significantly attenuated by rofecoxib; in rabbits with combined treatment, 65 ± 10% (n = 5) of the total aortic surface were covered by atherosclerotic plaques. This lesion size was similar to that in untreated hypercholesterolaemic controls but significantly (P < 0.05) greater than that in rabbits treated with ASA alone (Figure 1).
Figure 1.
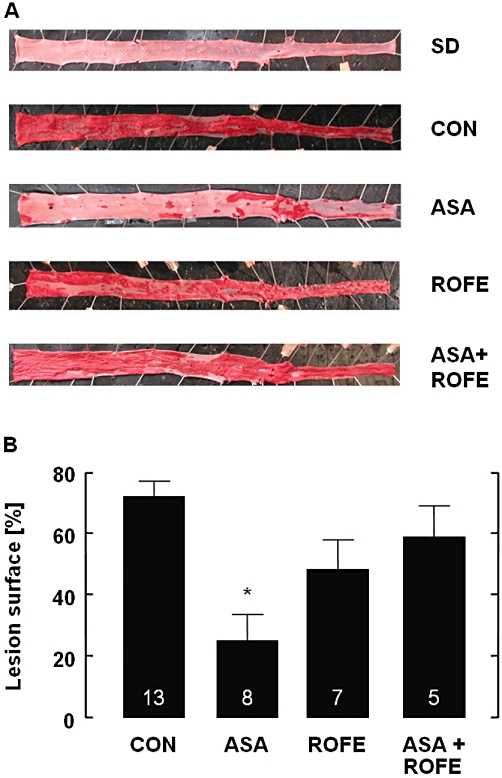
Severity of aortic plaque formation in cholesterol-fed animals as compared with animals on standard diet. (A) Original en face view of Sudan IV stained aortas of rabbits receiving standard laboratory diet (SD), cholesterol-enriched diet (CON) or cholesterol-enriched diet and the respective treatment for 12 weeks. (B) Reduction of aortic plaque formation, after 12 weeks treatment of cholesterol-fed rabbits, by ASA (5 mg·kg−1 per os, bid) and its prevention by rofecoxib cotreatment (25 mg·kg−1 per os, bid). The extent of atherosclerosis was calculated by morphometric analysis expressed as percentage of the aortic surface area covered by lesions. The data are mean ± SEM of the number of experiments indicated in the columns; *P < 0.05 (ASA vs. CON and vs. ASA + ROFE).
Thrombus formation
Mechanical injury of the carotid artery with subsequent partial stenosis reduced carotid blood flow in all cholesterol-fed animals from 37.3 ± 3.2 to 24.3 ± 2.1 mL·min−1 (n = 32), equivalent to an average reduction of 35% (P < 0.05). There were no significant differences between cholesterol- and standard-fed (from 34.4 ± 3.8 to 21.9 ± 3.1 mL·min−1; n = 11) animals. All animals on standard diet and cholesterol-enriched diet showed cyclic flow variations and/or occluding thrombi that, in some cases, redissolved spontaneously during the observation period of 60 min. Treatment with rofecoxib did not change the incidence of thrombotic events, whereas ASA reduced them by 62%; that is, in only three out of eight animals cyclic flow variations or complete thrombotic occlusions were found. Combined treatment with ASA and rofecoxib resulted in a marked attenuation of this effect – all but one rabbit exhibited CFVs or thrombotic occlusion (Table 1).
Table 1.
Concentrations of PGE2 in peripheral blood of rabbits before treatment and after 12 weeks of cholesterol and drug treatment
| PGE2 (ng·mL−1; mean ± SEM) | n | P-value | |
|---|---|---|---|
| Before treatment | 6.4 ± 0.4 | 37 | – |
| After treatment | |||
| Control | 7.0 ± 0.4 | 13 | n.s. |
| ASA | 5.0 ± 1.2 | 8 | n.s. |
| Rofecoxib | 3.7 ± 0.5 | 6 | <0.05 |
| ASA + rofecoxib | 3.2 ± 1.3 | 5 | <0.05 |
n.s., not significant.
This abolition of the antithrombotic effect of ASA by rofecoxib became even more apparent when the effect of treatment was evaluated in terms of the total blood flow rates in the stenosed carotid artery over a defined time after vessel injury (Figure 2, Table 2). Treatment with either rofecoxib or ASA increased the total blood flow above that in untreated animals on either a standard or cholesterol-enriched diet. Although this increase was statistically significant for both drugs (P < 0.05 for ASA and P < 0.05 for rofecoxib), cotreatment with ASA and rofecoxib abolished the antithrombotic effect completely and reduced the global flow rate to levels in untreated controls (Figure 2).
Figure 2.
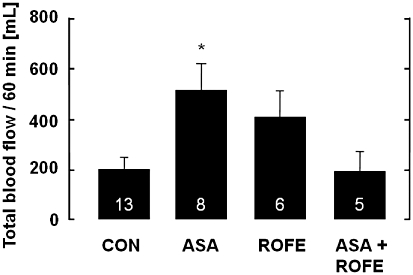
Improvement of carotid artery blood flow after mechanical injury of the vessel wall endothelium, after 12 weeks of treatment of cholesterol-fed rabbits, by ASA (5 mg·kg−1 per os, bid) and its prevention by cotreatment with rofecoxib (25 mg·kg−1 per os, bid). The amount of blood flowing through the carotid artery within 60 min after vessel injury was calculated by measuring the area under the blood flow versus time curve. Carotid blood flow was not different between cholesterol-fed rabbits and those fed a standard diet (not shown). The data are mean ± SEM of the number of experiments indicated in the columns; *P < 0.05 (ASA and ROFE vs. CON).
Table 2.
Effect of ASA (25 mg·kg−1 per os, bid) with and without rofecoxib cotreatment (25 mg·kg−1 per os, bid) on the incidence of thrombotic events (cyclic flow variations, CFVs; occluding thrombi, OT) after mechanical injury plus stenosis of the carotid artery in rabbits on standard (standard) or cholesterol-enriched (Chol) diet
| Thrombotic events [n] | ||||||
|---|---|---|---|---|---|---|
| Diet | Treatment | n | CFVs | OT | None | [%] |
| Standard | None | 11 | 3 | 8 | 0 | 100 |
| Chol | None | 13 | 5 | 8 | 0 | 100 |
| Chol | Rofecoxib | 6 | 4 | 3 | 0 | 100 |
| Chol | ASA | 8 | 2 | 1 | 5 | 38* |
| Chol | ASA + rofecoxib | 5 | 1 | 3 | 1 | 80 |
P < 0.05 (treatment vs. no treatment).
PGI2, PGE2 and thromboxane generation
Rofecoxib alone had no effect on COX-1 activity, as demonstrated by the unchanged thromboxane levels in serum, while ASA significantly reduced this parameter by about 50%. Interestingly, the combination of both rofecoxib and ASA did not show any inhibition but rather an increase in thromboxane formation that was statistically significant (Figure 3A). Somewhat different results were obtained for 6-oxo-PGF1α. The levels of 6-oxo-PGF1α were unchanged by ASA in hypercholesterolaemic rabbits but markedly reduced by rofecoxib treatment, confirming the involvement of COX-2 in PGI2 biosynthesis. Consequently, cotreatment with rofecoxib and ASA did not affect the reduced 6-oxo-PGF1α generation (Figure 3B).
Figure 3.
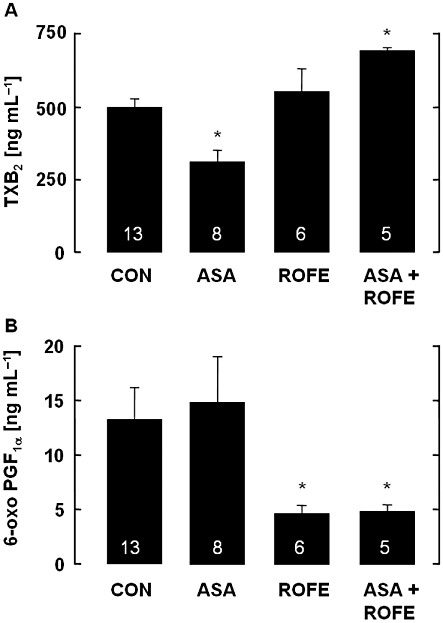
(A) Levels of TXB2 in serum of rabbits fed an atherogenic diet for 12 weeks and its modification by ASA (5 mg·kg−1 per os, bid), rofecoxib (25 mg·kg−1 per os, bid) or a combination of both. Aspirin significantly attenuated TXB2 formation, while rofecoxib had no effect. After cotreatment with ASA, rofecoxib significantly increased serum TXB2. The data are mean ± SEM of the number of experiments indicated in the columns; *P < 0.05 (ASA vs. CON and ASA + ROFE). (B) Levels of PGI2 (6-oxo PGF1α) in serum of cholesterol-fed rabbits and its modification by ASA (5 mg·kg−1 per os, bid), rofecoxib (25 mg·kg−1 per os, bid) or a combination of both. Rofecoxib significantly attenuated 6-oxo PGF1α accumulation, while ASA had no effect. Cotreatment of rofecoxib with ASA did not modify the action of rofecoxib. The data are mean ± SEM of the number of experiments indicated in the columns; *P < 0.05 (treatment vs. CON).
The LPS-induced PGE2 formation was used as a measure of COX-2-dependent prostaglandin formation. As shown in Table 1, treatment with rofecoxib significantly reduced PGE2 formation by about 50% as compared with control rabbits fed the atherogenic diet for 12 weeks. A slight and non-significant inhibition was observed in ASA-treated animals, while the combined treatment with ASA and rofecoxib gave the same result as with rofecoxib alone.
Vascular expression of COX-2
Cholesterol feeding resulted in a strong up-regulation of COX-2 mRNA (sevenfold) and protein (threefold) expression in the abdominal aorta as compared with animals on standard diet. Rofecoxib did not change COX-2 mRNA in animals receiving standard diet (data not shown) but reduced the elevated COX-2 expression in cholesterol-fed animals almost to control levels. The same effect was seen after its cotreatment with ASA, whereas ASA alone had no inhibitory effect on cholesterol-induced COX-2 expression (Figure 4A and B).
Figure 4.
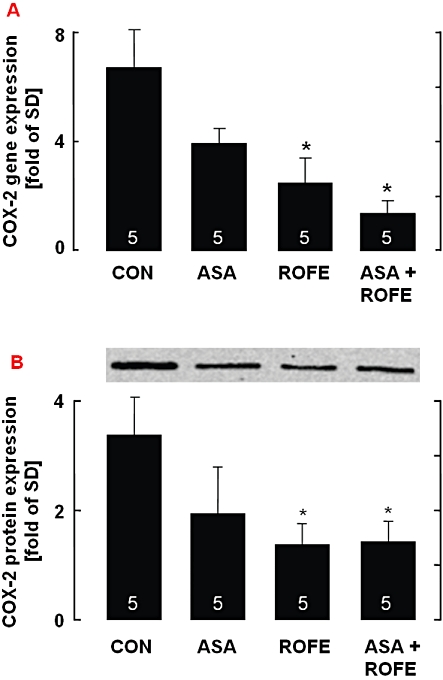
(A) Expression of COX-2 mRNA in the aorta of rabbits fed an atherogenic diet and its modification by ASA (5 mg·kg−1 per os, bid), rofecoxib (25 mg·kg−1 per os, bid) or a combination of both. For each aorta, the ratio of COX-2 versus GAPDH gene expression was determined, and the relative increase in comparison with control animals on standard diet was calculated and set to one. (B) Western blot analysis of COX-2 protein expression in the aorta of rabbits fed an atherogenic diet and its modification by ASA (5 mg·kg−1 per os, bid), rofecoxib (25 mg·kg−1 per os, bid) or a combination of both; the results were obtained by using fluorescent secondary antibodies and the Odyssey Infrared Imaging System. The data are mean ± SEM of the number of experiments indicated in the columns; *P < 0.05 (treatment vs. CON).
Immunohistochemistry
Cholesterol feeding resulted in a strong up-regulation of COX-2, thrombomodulin and PAI-1 protein expression in sections of the aortic root (Figure 5). In agreement with the mRNA levels, rofecoxib had no effect on the protein expression in rabbits on standard diet (not shown). Treatment of cholesterol-fed animals with rofecoxib, ASA or the combination of both strongly reduced the elevated COX-2 expression. The expression levels of TM and PAI-1 were only marginally reduced by the different treatments. Due to the limitations of immunohistochemistry and the method used for quantification, further experiments are needed to establish the changes in protein expression.
Figure 5.
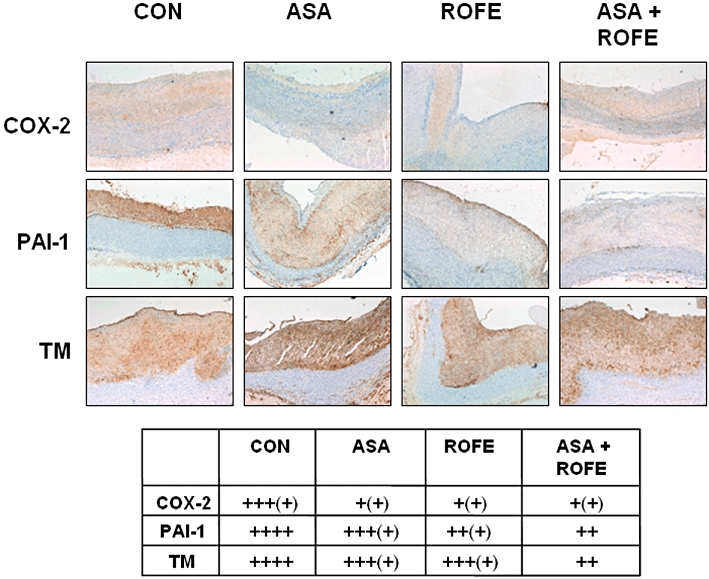
Immunohistochemical staining for COX-2, TM and PAI-1 in sections of the aortic arch of rabbits fed standard diet (SD, n = 11) or atherogenic (CON, n = 13) diet for 12 weeks are shown. COX-2 expression is reduced by ROFE (n = 7), ASA (n = 8) and the combination of both (ASA + ROFE, n = 5) compared with animals fed an atherogenic diet alone. A semiquantitative scale: no staining (−), mild (+), moderate (++), strong (+++) and intensive (++++) staining was used in order to evaluate staining intensity of the sections by five independent observers in a blinded fashion (see table).
Discussion
The biological significance of the different COX isoforms and their products for interactions between blood and the vessel wall is still a matter of discussion. This is particularly true for atherosclerosis. Some studies on the role of cyclooxygenases in atherogenesis revealed that COX-1 is important, particularly in early stages of lesion development, and that selective inhibition of COX-1 reduces the progression of atherosclerotic vessel injury (Pratico et al., 2001; Belton et al., 2003; Prasad and Lee, 2003; Egan et al., 2005). However, it is also clear that (reversible) COX-1 inhibition ultimately will not prevent the atherosclerotic process, as seen for example in a subgroup analysis of the physicians' health study. The role of COX-2 in atherosclerosis is more complex (FitzGerald, 2003). COX-2 inhibitors have been found to retard (Burleigh et al., 2002; 2005;), to accelerate (Rott et al., 2003) or not to modify (Olesen et al., 2002; Bea et al., 2003; Belton et al., 2003) atherogenesis in hypercholesterolaemic mice. Thus, while the process of atherogenesis clearly involves enhanced COX-2-dependent prostaglandin production (Belton et al., 2003), its biological function is difficult to predict. Coxibs do not directly inhibit platelet function and therefore might cause a thrombus-prone state, specifically on the background of reduced generation of anti-platelet/vasodilator prostaglandins by the vessel wall.
Our study confirms that hypercholesterolaemia not only causes marked aortic plaque formation but also a prothrombotic state. This becomes particularly evident after vascular injury. All animals with vessel wall injury exhibited thrombus formation, as seen from cyclic flow variations or formation of occluding thrombi. Treatment with rofecoxib did not change this but rather resulted in a slightly improved perfusion. ASA significantly reduced both aortic plaque size and thrombus formation after vessel injury, whereas rofecoxib only slightly attenuated the formation of atherosclerotic plaques. The anti-atherosclerotic effect of ASA is generally attributed to its anti-platelet action but might also involve an improvement of (Husain et al., 1998) or a protective effect on (Kharbanda et al., 2002) inflammation-induced endothelial dysfunction in atherosclerotic vessels as well as a decrease in the oxidative stress in hypercholesterolaemia (Prasad and Lee, 2003). A similar tendency, that is reduced aortic plaque formation and reduced thrombogenicity, was also seen with rofecoxib. This fits well into the overall concept of an anti-inflammatory/anti-atherosclerotic action of coxibs.
The most striking and unexpected effect, however, was the significant attenuation of the ASA protection from thrombotic events by rofecoxib cotreatment. This was seen in terms of both plaque formation and thrombogenicity. To our knowledge, this is the first demonstration of this type of drug interaction, which is most likely due to the changes in prostaglandin and thromboxane formation. In this context, it has to be realized that a long-term treatment was used, which, as seen from the transcriptional effects on COX-2 mRNA and protein expression, was associated with up-regulated enzyme levels.
In contrast to conventional non-selective NSAIDs, selective COX-2 inhibitors do not interact with the anti-platelet effects of ASA (Catella-Lawson et al., 2001) and also do not inhibit COX-1-derived platelet-dependent thromboxane formation (Belton et al., 2000; Van Hecken et al., 2000). This was confirmed in the present investigation and we also showed that ASA markedly reduced serum thromboxane formation. Interestingly, this inhibition was incomplete, suggesting the contribution of COX-2-derived prostaglandin endoperoxides from non-platelet sources, such as monocytes/macrophages or the atherosclerotic vessel wall.
Additionally, platelet turnover is extremely rapid in cholesterol-fed rabbits. Platelet half-life is reduced from approximately 40 h at standard chow to less than 30 h in animals receiving a cholesterol- and lipid-enriched diet (Barrett and Butler, 1983; Butler et al., 1987). In the present study we investigated the consequences of an interaction between ASA and coxibs during long-term treatment, rabbits received the last dose of the test substances 12 h before the beginning of the experiments. Thus, the incomplete inhibition of serum thromboxane formation can possibly be explained by the rapid platelet turnover with a partial regeneration of platelet thromboxane synthesis capacity by newly formed platelets.
The inhibition of the protective ASA effects on plaque formation and cyclic flow reductions by simultaneously applied rofecoxib was paralleled by a significant increase in thromboxane generation in these rabbits, whereas in ASA-treated animals, platelet-dependent thromboxane formation was significantly reduced.
The increase in serum thromboxane by co-administration of rofecoxib is most likely caused by the increase in atherosclerotic lesion size. The amount of TXA2 synthesized by slices of the aorta from cholesterol-fed rabbits was correlated with the degree of coverage with aortic lesions and was increased more than 10-fold as compared with controls (Wang et al., 1991). In normal mice, approximately 20–25% of total thromboxane A2 is synthesized by the vessel wall and monocytes/macrophages and is not produced by platelets (Babaev et al., 2006). This is especially important, because, in contrast to platelets, thromboxane synthesis in the vessel wall is not inhibited by low-dose ASA.
In healthy blood vessels, COX-1 is expressed constitutively without detectable COX-2, and the COX-1 in the vessel wall is more sensitive to inhibition by ASA and other NSAIDs than platelet COX-1 (Mitchell et al., 2006). However, because of the rapid pre-systemic acetylation and inhibition of platelet COX-1 by ASA and the low systemic bioavailability, the inhibitory effect of ASA on COX-1 in the vessel wall is low (Pedersen and FitzGerald, 1984; Mitchell et al., 2006).
It has been hypothesized that PGI2 modulates the platelet–vessel wall interactions in vivo and specifically controls the response to TXA2 in tissue haemostasis (Cheng et al., 2002). This control may become even more important if endogenous PGI2 production is increased, for example in patients at advanced stages of atherosclerosis, where platelet-dependent generation of TXA2 is also significantly enhanced (FitzGerald et al., 1984; Belton et al., 2000). Treatment of these patients with a COX-2 selective inhibitor will reduce PGI2 generation but leaves the enhanced thromboxane generation unimpaired. This depression of PGI2 generation without coincidental inhibition of TXA2 formation may result in an impaired haemostasis with an elevated thrombotic risk via (i) a decreased generation of antithrombotic (PGI2) and antihypertensive (PGE2) factors; and (ii) an unchanged or even elevated formation of prothrombotic and vasoconstrictive factors. These mechanisms may contribute to the cardiovascular side effects of coxibs observed in clinical trials (Cheng et al., 2002) and also the antagonistic action of rofecoxib on the anti-atherosclerotic and antithrombotic effect of ASA demonstrated in our study with hypercholesterolaemic rabbits. Using the coronary thrombosis model of the dog, Hennan et al. (2001) previously showed that the antithrombotic efficacy of ASA mediated by COX-1 inhibition is attenuated in the presence of celecoxib, suggesting that the actions of ASA might depend on the maintenance of an endothelial COX-2-derived biosynthesis of prostacyclin. We have shown that this is also valid for long-term treatment and the antithrombotic effect of ASA in atherosclerotic animals at increased vascular risk.
Acknowledgments
This work was supported in part by the Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM), the Deutsche Forschungsgemeinschaft (DFG) and the Forschungskommission der Medizinischen Fakultät der Heinrich-Heine-Universität Düsseldorf.
Glossary
Abbreviations
- APPROVe
adenomatous polyp prevention on VIOXX
- ASA
acetylsalicylic acid
- CABG
coronary artery bypass graft
- CFVs
cyclic flow variations
- CON
control diet
- NSAID
nonsteroidal anti-inflammatory drugs
- PAI-1
plasminogen activator inhibitor-1
- ROFE
rofecoxib
- SD
standard diet
- TF
tissue factor
- TM
thrombomodulin
- TXA2
thromboxane A2
Conflict of interest
None.
References
- Babaev VR, Ding L, Reese J, Morrow JD, Breyer MD, Dey SK, et al. Cyclooxygenase-1 deficiency in bone marrow cells increases early atherosclerosis in apolipoprotein E- and low-density lipoprotein receptor-null mice. Circulation. 2006;113:108–117. doi: 10.1161/CIRCULATIONAHA.105.591537. [DOI] [PubMed] [Google Scholar]
- Barrett PA, Butler KD. Shortening of platelet survival by induced hypercholesterolaemia in rabbits and its prolongation by anagrelide. Thromb Haemost. 1983;50:656–659. [PubMed] [Google Scholar]
- Bea F, Blessing E, Bennett BJ, Kuo CC, Campbell LA, Kreuzer J, et al. Chronic inhibition of cyclooxygenase-2 does not alter plaque composition in a mouse model of advanced unstable atherosclerosis. Cardiovasc Res. 2003;60:198–204. doi: 10.1016/s0008-6363(03)00464-4. [DOI] [PubMed] [Google Scholar]
- Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;102:840–845. doi: 10.1161/01.cir.102.8.840. [DOI] [PubMed] [Google Scholar]
- Belton OA, Duffy A, Toomey S, Fitzgerald DJ. Cyclooxygenase isoforms and platelet vessel wall interactions in the apolipoprotein E knockout mouse model of atherosclerosis. Circulation. 2003;108:3017–3023. doi: 10.1161/01.CIR.0000104565.78013.AD. [DOI] [PubMed] [Google Scholar]
- Burleigh ME, Babaev VR, Oates JA, Harris RC, Gautam S, Riendeau D, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation. 2002;105:1816–1823. doi: 10.1161/01.cir.0000014927.74465.7f. [DOI] [PubMed] [Google Scholar]
- Burleigh ME, Babaev VR, Yancey PG, Major AS, McCaleb JL, Oates JA, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in ApoE-deficient and C57BL/6 mice. J Mol Cell Cardiol. 2005;39:443–452. doi: 10.1016/j.yjmcc.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Butler KD, Butler PA, Shand RA, Ambler J, Wallis RB. Prolongation of platelet survival in hypercholesterolaemic rabbits by CGS 12970 (3-methyl-2-(3-pyridyl)-1 indoleoctanoic acid) and dazoxiben. Thromb Res. 1987;45:751–761. doi: 10.1016/0049-3848(87)90085-5. [DOI] [PubMed] [Google Scholar]
- Catella-Lawson F, Reilly MP, Kapoor SC, Cucchiara AJ, DeMarco S, Tournier B, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809–1817. doi: 10.1056/NEJMoa003199. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Austin SC, Rocca B, Koller BH, Coffman TM, Grosser T, et al. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science. 2002;296:539–541. doi: 10.1126/science.1068711. [DOI] [PubMed] [Google Scholar]
- Egan KM, Wang M, Fries S, Lucitt MB, Zukas AM, Pure E, et al. Cyclooxygenases, thromboxane, and atherosclerosis: plaque destabilization by cyclooxygenase-2 inhibition combined with thromboxane receptor antagonism. Circulation. 2005;111:334–342. doi: 10.1161/01.CIR.0000153386.95356.78. [DOI] [PubMed] [Google Scholar]
- FitzGerald GA. COX-2 and beyond: approaches to prostaglandin inhibition in human disease. Nat Rev Drug Discov. 2003;2:879–890. doi: 10.1038/nrd1225. [DOI] [PubMed] [Google Scholar]
- Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- FitzGerald GA, Smith B, Pedersen AK, Brash AR. Increased prostacyclin biosynthesis in patients with severe atherosclerosis and platelet activation. N Engl J Med. 1984;310:1065–1068. doi: 10.1056/NEJM198404263101701. [DOI] [PubMed] [Google Scholar]
- Folts J. An in vivo model of experimental arterial stenosis, intimal damage, and periodic thrombosis. Circulation. 1991;83(6) Suppl:IV3–I14. [PubMed] [Google Scholar]
- Folts JD, Crowell EB, Jr, Rowe GG. Platelet aggregation in partially obstructed vessels and its elimination with aspirin. Circulation. 1976;54:365–370. doi: 10.1161/01.cir.54.3.365. [DOI] [PubMed] [Google Scholar]
- Hennan JK, Huang J, Barrett TD, Driscoll EM, Willens DE, Park AM, et al. Effects of selective cyclooxygenase-2 inhibition on vascular responses and thrombosis in canine coronary arteries. Circulation. 2001;104:820–825. doi: 10.1161/hc3301.092790. [DOI] [PubMed] [Google Scholar]
- Hohlfeld T, Zimmermann N, Weber AA, Jessen G, Weber H, Schrör K, et al. Pyrazolinone analgesics prevent the antiplatelet effect of aspirin and preserve human platelet thromboxane synthesis. J Thromb Haemost. 2008;6:166–173. doi: 10.1111/j.1538-7836.2007.02800.x. [DOI] [PubMed] [Google Scholar]
- Husain S, Andrews NP, Mulcahy D, Panza JA, Quyyumi AA. Aspirin improves endothelial dysfunction in atherosclerosis. Circulation. 1998;97:716–720. doi: 10.1161/01.cir.97.8.716. [DOI] [PubMed] [Google Scholar]
- Juni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021–2029. doi: 10.1016/S0140-6736(04)17514-4. [DOI] [PubMed] [Google Scholar]
- Kharbanda RK, Walton B, Allen M, Klein N, Hingorani AD, MacAllister RJ, et al. Prevention of inflammation-induced endothelial dysfunction: a novel vasculo-protective action of aspirin. Circulation. 2002;105:2600–2604. doi: 10.1161/01.cir.0000017863.52347.6c. [DOI] [PubMed] [Google Scholar]
- Livio M, Del Maschio A, Cerletti C, de Gaetano G. Indomethacin prevents the long-lasting inhibitory effect of aspirin on human platelet cyclo-oxygenase activity. Prostaglandins. 1982;23:787–796. doi: 10.1016/0090-6980(82)90123-x. [DOI] [PubMed] [Google Scholar]
- McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, Lucas R, Vojnovic I, Hasan K, Pepper JR, Warner TD. Stronger inhibition by nonsteroid anti-inflammatory drugs of cyclooxygenase-1 in endothelial cells than platelets offers an explanation for increased risk of thrombotic events. FASEB J. 2006;20:2468–2475. doi: 10.1096/fj.06-6615com. [DOI] [PubMed] [Google Scholar]
- Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, et al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med. 2005;352:1081–1091. doi: 10.1056/NEJMoa050330. [DOI] [PubMed] [Google Scholar]
- Olesen M, Kwong E, Meztli A, Kontny F, Seljeflot I, Arnesen H, et al. No effect of cyclooxygenase inhibition on plaque size in atherosclerosis-prone mice. Scand Cardiovasc J. 2002;36:362–367. doi: 10.1080/140174302762659094. [DOI] [PubMed] [Google Scholar]
- Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987;68:231–240. doi: 10.1016/0021-9150(87)90202-4. [DOI] [PubMed] [Google Scholar]
- Patrignani P, Panara MR, Greco A, Fusco O, Natoli C, Lacobelli S, et al. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J Pharmacol Exp Ther. 1994;271:1705–1712. [PubMed] [Google Scholar]
- Pedersen AK, FitzGerald GA. Dose-related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N Engl J Med. 1984;311:1206–1211. doi: 10.1056/NEJM198411083111902. [DOI] [PubMed] [Google Scholar]
- Prasad K, Lee P. Suppression of oxidative stress as a mechanism of reduction of hypercholesterolemic atherosclerosis by aspirin. J Cardiovasc Pharmacol Ther. 2003;8:61–69. doi: 10.1177/107424840300800i109. [DOI] [PubMed] [Google Scholar]
- Pratico D, Tillmann C, Zhang ZB, Li H, FitzGerald GA. Acceleration of atherogenesis by COX-1-dependent prostanoid formation in low density lipoprotein receptor knockout mice. Proc Natl Acad Sci USA. 2001;98:3358–3363. doi: 10.1073/pnas.061607398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao GH, Johnson GG, Reddy KR, White JG. Ibuprofen protects platelet cyclooxygenase from irreversible inhibition by aspirin. Arteriosclerosis. 1983;3:383–388. doi: 10.1161/01.atv.3.4.383. [DOI] [PubMed] [Google Scholar]
- Rimarachin JA, Jacobson JA, Szabo P, Maclouf J, Creminon C, Weksler BB. Regulation of cyclooxygenase-2 expression in aortic smooth muscle cells. Arterioscler Thromb. 1994;14:1021–1031. doi: 10.1161/01.atv.14.7.1021. [DOI] [PubMed] [Google Scholar]
- Rott D, Zhu J, Burnett MS, Zhou YF, Zalles-Ganley A, Ogunmakinwa J, et al. Effects of MF-tricyclic, a selective cyclooxygenase-2 inhibitor, on atherosclerosis progression and susceptibility to cytomegalovirus replication in apolipoprotein-E knockout mice. J Am Coll Cardiol. 2003;41:1812–1819. doi: 10.1016/s0735-1097(03)00304-8. [DOI] [PubMed] [Google Scholar]
- Schrör K, Seidel H. Blood-vessel wall arachidonate metabolism and its pharmacological modification in a new in vitro assay. Naunyn Schmiedebergs Arch Pharmacol. 1988;337:177–182. doi: 10.1007/BF00169246. [DOI] [PubMed] [Google Scholar]
- Van Hecken A, Schwartz JI, Depre M, De Lepeleire I, Dallob A, Tanaka W, et al. Comparative inhibitory activity of rofecoxib, meloxicam, diclofenac, ibuprofen, and naproxen on COX-2 versus COX-1 in healthy volunteers. J Clin Pharmacol. 2000;40:1109–1120. [PubMed] [Google Scholar]
- Wang T, Falardeau P, Powell WS. Synthesis of prostaglandins and thromboxane B2 by cholesterol-fed rabbits. Arterioscler Thromb. 1991;11:501–508. doi: 10.1161/01.atv.11.3.501. [DOI] [PubMed] [Google Scholar]