

Abstract
BACKGROUND AND PURPOSE
Infections with respiratory viruses induce exacerbations of asthma, increase acetylcholine release and potentiate vagally mediated bronchoconstriction by blocking inhibitory M2 muscarinic receptors on parasympathetic neurons. Here we test whether virus-induced M2 receptor dysfunction and airway hyperresponsiveness are tumour necrosis factor-alpha (TNF-α) dependent.
EXPERIMENTAL APPROACH
Guinea pigs were pretreated with etanercept or phosphate-buffered saline 24 h before intranasal infection with parainfluenza. Four days later, pulmonary inflation pressure, heart rate and blood pressure were measured. M2 receptor function was assessed by the potentiation by gallamine (an M2 receptor antagonist) of bronchoconstriction caused by electrical stimulation of the vagus nerves and measured as increased pulmonary inflation pressure. Human airway epithelial cells were infected with influenza and TNF-α concentration in supernatant was measured before supernatant was applied to human neuroblastoma cells. M2 receptor expression in these neuroblastoma cells was measured by qRT-PCR.
KEY RESULTS
Influenza-infected animals were hyperresponsive to vagal stimulation but not to intravenous ACh. Gallamine did not potentiate vagally induced bronchoconstriction in virus-infected animals, indicating M2 receptor dysfunction. Etanercept prevented virus-induced airway hyperresponsiveness and M2 receptor dysfunction, without changing lung viral titres. Etanercept caused a non-significant decrease in total cells, macrophages and neutrophils in bronchoalveolar lavage. Influenza infection significantly increased TNF-α release from isolated epithelial cells, sufficient to decrease M2 receptors in neuroblastoma cells. This ability of supernatants from infected epithelial cells to inhibit M2 receptor expression was blocked by etanercept.
CONCLUSIONS AND IMPLICATIONS
TNF-α is a key mediator of virus-induced M2 muscarinic receptor dysfunction and airway hyperresponsiveness.
Keywords: TNF-α, virus, asthma, airway hyperreactivity, etanercept
Introduction
Viruses cause 80–85% of asthma exacerbations in children (Johnston et al., 1995) and are also associated with the majority of asthma exacerbations in adults (Nicholson et al., 1993; Johnston et al., 1996). Virus-induced airway hyperresponsiveness can be mediated by increased vagal reflexes in humans (Empey et al., 1976) and in animals (Fryer and Jacoby, 1991; Jacoby and Fryer, 1991). Parasympathetic nerves normally control airway tone (Caulfield and Birdsall, 1998) and cause bronchoconstriction by releasing acetylcholine (ACh) onto M3 muscarinic receptors (Alexander et al., 2009) on airway smooth muscle. Release of ACh is limited by the negative feedback provided by inhibitory M2 muscarinic receptors on airway parasympathetic nerves. Decreased neuronal M2 muscarinic receptor function leads to increased ACh release and airway hyperresponsiveness (Fryer and Maclagan, 1984). Virus infection reduces M2 muscarinic receptor function (Fryer and Jacoby, 1991) through a mechanism potentially shared by different respiratory RNA viruses because double-stranded RNA (dsRNA), produced as a replicative intermediate during infection, also causes M2 muscarinic receptor dysfunction and airway hyperresponsiveness (Bowerfind et al., 2002). However, the signalling mechanism following virus infection that leads to M2 muscarinic receptor dysfunction is not completely understood.
The multifunctional cytokine TNF-α plays a key role in development of airway hyperresponsiveness in asthma (Gosset et al., 1991; Ying et al., 1991; Broide et al., 1992; Bradding et al., 1994; Hallsworth et al., 1994; Thomas, 2001; Berry et al., 2006). TNF-α expression, TNF-α receptor I and TNF-α converting enzyme are all increased in airway lavage from patients with asthma compared with normal subjects (Thomas, 2001; Berry et al., 2006; Cazzola et al., 2006). In patients with severe and steroid-resistant asthma, anti-TNF-α therapy decreases airway hyperresponsiveness and reduces frequency of asthma exacerbations (Howarth et al., 2005; Berry et al., 2006). In antigen sensitized and challenged animals, we have shown that TNF-α causes M2 muscarinic receptor dysfunction by increasing inter-cellular adhesion molecule 1 (ICAM-1) expression on parasympathetic nerves. Increased ICAM-1 in turn facilitates migration and adhesion of inflammatory cells onto parasympathetic nerves (Nie et al., 2007). Inflammatory cell adhesion to nerves, particularly eosinophil adhesion, is associated with airway hyperresponsiveness in humans and animal models (Costello et al., 1997; Fryer et al., 1999; Sawatzky et al., 2002). We have also shown that TNF-α directly reduces M2 muscarinic receptor expression on airway nerves (Nie et al., 2009) and M2 muscarinic receptor function in antigen-challenged guinea pigs can be protected by anti-TNF-α therapy (Nie et al., 2009). TNF-α expression is also increased in airways with influenza and parainfluenza virus infection (Uhl et al., 1998). Here we test whether viruses initiate TNF-α signalling which inhibits M2 muscarinic receptor function on airway nerves leading to airway hyperresponsiveness.
Method
Animals
All animal care and experimental procedures were in accordance with standards established by the US Animal Welfare Acts set forth in the National Institutes of Health guidelines and were approved by the Oregon Health and Science University Animal Care and Use Committee. Specific pathogen-free female Dunkin-Hartley guinea pigs (300–350 g; Elm Hill, Chelmsford, MA, USA) were shipped in filtered crates and housed in high-efficiency particulate-filtered air.
Viral infection and titration
Parainfluenza type 1 (Sendai virus, VR-105; ATCC) was grown in rhesus monkey kidney cell monolayers and titred as previously described (Fryer and Jacoby, 1991). Human influenza A/Port Chalmers/72 (H3N2) was grown in Rhesus monkey kidney cells, and the virus in the lysate was purified by sucrose gradient centrifugation. For control experiments, influenza viruses were inactivated by exposure to ultraviolet light for 15 min.
Pathogen-free guinea pigs were treated with etanercept (3 mg·kg−1 i.p, Immunex) or PBS. The dose of etanercept was chosen based upon previous animal (Kivilcim et al., 2007; Nie et al., 2009) and clinical studies (Takei et al., 2001). Etanercept is a human dimeric fusion protein consisting of p75 TNF-α receptor linked to the Fc of IgG1 (Lesslauer et al., 1991; Peppel et al., 1991; Suffredini et al., 1995; Ritchlin et al., 2003; Wang et al., 2006; Baumann et al., 2007). Twenty-four hours after etanercept or PBS treatment, animals were anesthetized with ketamine (30 mg·kg−1 i.m.) and xylazine (5 mg·kg−1 i.m.) and infected with parainfluenza virus type 1 (Sendai virus, ATCC-VR 105, 105 TCID50 per animal, intranasal) or PBS (vehicle control). TCID50 is defined as the amount of virus required to infect 50% of monolayers of cultured rhesus monkey kidney cells (Jacoby et al., 2000). At the end of each experiment, viral content in the lungs was determined by real time RT-PCR (primer pairs: sense: ATG CGG CTG ATC TTC TCA CT, antisense: CTT TGC CAC GAC ATT AGG GT). Based on the standard curve, viral titres are presented as TCID50 per mg tissue.
Vagal nerve activity and M2 receptor function
Four days after infection, guinea pigs were anesthetized with urethane (1.9 g·kg−1 i.p.). Heart rate and blood pressure were measured via a carotid artery cannula. Both jugular veins were cannulated for administration of drugs. After vagotomy, distal ends of both vagus nerves were placed on shielded electrodes immersed in mineral oil. Body temperature was maintained at 37°C on a heating blanket. Animals were paralysed with succinylcholine (10 µg·kg−1·min−1 i.v.) and ventilated via a tracheal cannula (tidal volume 1 mL per 100 g body weight at 100 breaths·min−1; Harvard Apparatus, South Natick, MA, USA). All animals were treated with guanethidine (5 mg·kg−1, i.v.) 20 min before physiological measurements to deplete noradrenaline as previously described (Fryer and Maclagan, 1984).
Pulmonary inflation pressure (Ppi) was measured via a pressure transducer on a sidearm of the tracheal cannula, as previously described (Nie et al., 2009). Electrical stimulation of both vagus nerves (2–25 Hz, 0.2 ms pulse duration, 10 V, and 5 s pulse train at 40 s intervals) caused bronchoconstriction measured as an increase in Ppi. The function of post-junctional M3 receptors on airway smooth muscle was assessed by measuring bronchoconstriction in response to exogenous ACh (1–10 µg·kg−1 i.v.) in vagotomized animals.
Neuronal M2 muscarinic receptor function was measured by the ability of a selective M2 antagonist, gallamine (0.1–10 mg·kg−1 i.v.), to potentiate vagally induced bronchoconstriction in a dose-dependent manner. For these experiments, both vagi were electrically stimulated at 15 Hz, 0.2 ms pulse duration, 5–15 V, and 3 s pulses per train. The voltage was chosen in the absence of gallamine to increase Ppi 10–15 mmH2O above baseline. The effect of gallamine on vagally induced bronchoconstriction was measured as a ratio of bronchoconstriction in the presence of gallamine to bronchoconstriction in the absence of gallamine. Voltages were not significantly different between groups.
Atropine was applied to block the bronchoconstriction in randomly selected animals to confirm the vagal-mediated bronchoconstriction was mediated via muscarinic receptors.
Lung lavage
At the end of the experiment, the lungs were lavaged five times with 10 mL aliquots of warm PBS via the tracheal cannula. Cells were counted and 500–1000 cells were applied to cytospin preparations. In total, 200 cells were counted for cell differentials, using cytospin slides stained with Diff-Quik (American Scientific Products, McGaw, IL, USA).
Primary culture and infection of human tracheal epithelial cell, M2 muscarinic receptor measurement in neuroblastoma cells
Airway epithelial cells were isolated from human tracheas (provided by the Pacific Northwest Transplant Bank) as previously described (Fryer et al., 2006; Nie et al., 2007) and grown in serum free (LHC8, Invitrogen) medium for 1 week to near confluence in Collagen Type 4 (Sigma c5533 – 5 mg) coated six-well tissue culture plates. Cultures were infected with Human influenza A/Port Chalmers/72 (H3N2) influenza virus (5.62 × 103 TCID50 per ml) for 2 h. UV inactivated H3N2 virus and LHC8 medium were used as controls. The medium was then removed and fresh serum-free LHC8 medium was replaced. Infected cells were incubated at 34°C under 5% CO2 in humidified air. Four days later, supernatants were collected and TNF-α concentration was measured by elisa (R&D Systems Human TNF-α DuoSet elisa Development Kit, Cat# DY210). The supernatants with or without etanercept (1 µg·mL−1) from every condition were subsequently applied to SK-N-SH human neuroblastoma cells (ATCC) for 24 h. RNA from treated SK-N-SH cells was isolated using RNeasy Mini Kit (74106; QIAGEN) and reverse transcribed using SuperScript III (18080-051; Invitrogen Corp) with random hexamer primers. Quantitative PCR was carried out in triplicate at 60°C annealing temperature over 45 cycles using Applied Biosystems SYBR Green PCR kit (4309155; Applied Biosystems). PCR products were quantified using the Applied Biosystems 7500 Real-Time PCR System. Oligonucleotide PCR primer pairs were designed from published sequences as follows: human M2 muscarinic receptor: sense, TTA AAG TCA ACC GCC ACC TC, antisense, CAA AGG TCA CAC ACC ACA GG; 18S rRNA: sense, GTAACCCGTTGAACCCCATT, antisense, CCATCCAATCGGTAGTAGCG. Threshold cycle number was measured and relative expression of M2 muscarinic receptor mRNA was normalized to 18S rRNA.
Statistical analysis
All data were expressed as means ± SE. Frequency, gallamine and ACh responses were analysed by two-way anovas for repeated measures. Baseline parameters for Ppi, heart rate, blood pressure, TNF-α concentration and M2 muscarinic receptor mRNA expression were evaluated by one-way anova for analysis of variance. Bronchoalveolar lavage (BAL) cell counts were evaluated by two-way anova. Viral titre in lung tissue and total number of cells in BAL was analysed by t-test. A P-value of <0.05 was considered significant. All statistical analyses were made with the software package Graphpad Prism5 (Version 5.01, Graphpad Software).
Materials
ACh chloride, atropine, guanethidine, succinylcholine chloride, urethane and human TNF-α (T0157 Sigma) were purchased from Sigma (St. Louis, MO, USA). Rhesus monkey kidney cells were purchased from Viromed (Minneapolis, MN, USA). Dulbecco's modified Eagle's medium (DMEM) and LHC8 medium were purchased from Invitrogen (Carlsbad, California). Etanercept (Immunex) was purchased from OHSU pharmacy.
Results
Baseline responses
There was no statistically significant difference in any of the baseline parameters for Ppi, heart rate and blood pressure among groups (Table 1).
Table 1.
Baseline parameters of the experimental groups of guinea pigs
| Treatment group | Ppi (mmH2O) | Heart rate (beat•min−1) | Systolic BP (mmHg) | Diastolic BP (mmHg) |
|---|---|---|---|---|
| Control (n = 5) | 114 ± 2.5 | 296 ± 4 | 32.5 ± 4.9 | 17.5 ± 6.9 |
| Virus (n = 5) | 147 ± 18.9 | 306 ± 19 | 43.5 ± 4.9 | 30 ± 5.6 |
| Virus + etanercept (n = 5) | 122 ± 5.8 | 285 ± 4 | 44 ± 2.0 | 25 ± 5.0 |
Ppi, pulmonary inflation pressure.
Virus caused airway hyperresponsiveness
Electrical stimulation of both vagus nerves produced frequency-dependent bronchoconstriction measured as a rapid and reversible increase in Ppi (Figure 1). Vagally induced bronchoconstriction was significantly increased in virus-infected animals compared with vehicle controls (PBS), indicating airway hyperreactivity. This potentiation was prevented by pretreatment with etanercept. In our previous study, etanercept did not affect airway responses to vagal stimulation in control guinea pigs (Nie et al., 2009).
Figure 1.
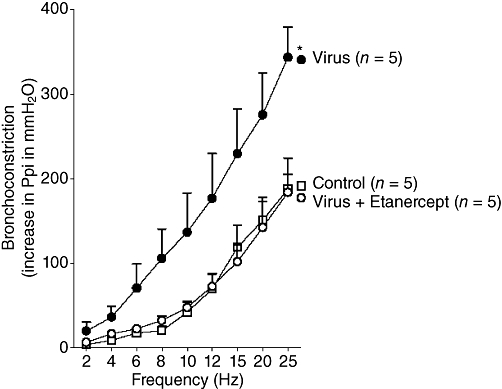
Etanercept prevents virus-induced airway hyperreactivity. Electrical stimulation of both vagus nerves produces frequency-dependent bronchoconstriction measured as an increase in pulmonary inflation pressure. Bronchoconstriction induced by vagal stimulation is potentiated in virus-infected guinea pigs compared with controls and this potentiation is blocked by etanercept. n = 5 in each group and points are mean ± SEM. *The entire frequency response is significantly different from controls, using anova.
Airway smooth muscle M3 muscarinic receptor function
Neither virus infection nor etanercept changed M3 muscarinic receptor function (Figure 2) as there were no significant differences in ACh dose–response curves among control, virus-infected and etanercept-treated virus-infected animals, that were all vagotomized to prevent any reflex.
Figure 2.
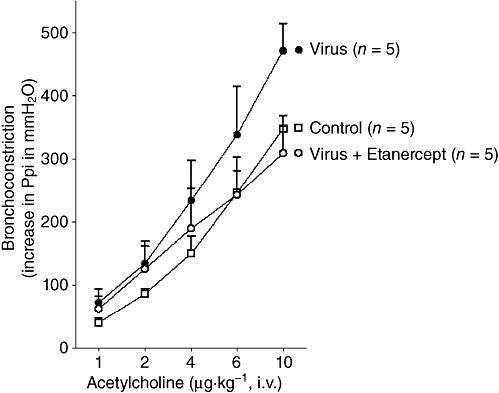
Etanercept does not change M3 muscarinic receptor function on smooth muscle in virus-infected guinea pigs. ACh-induced bronchoconstriction, measured as an increase in pulmonary inflation pressure, is not changed by virus infection or by etanercept, compared with controls. n = 5 in each group and points are mean ± SEM.
Neuronal M2 muscarinic receptor function
Gallamine, a M2 muscarinic receptor antagonist, potentiated vagally induced bronchoconstriction in a dose-dependent manner in control guinea pigs (Figure 3) demonstrating normal M2 muscarinic receptor function. In virus-infected guinea pigs, the ability of gallamine to potentiate vagally induced bronchoconstriction was significantly reduced compared with controls, indicating virus-induced dysfunction of neuronal M2 muscarinic receptors. Etanercept pretreatment protected M2 receptor function in virus-infected guinea pigs.
Figure 3.
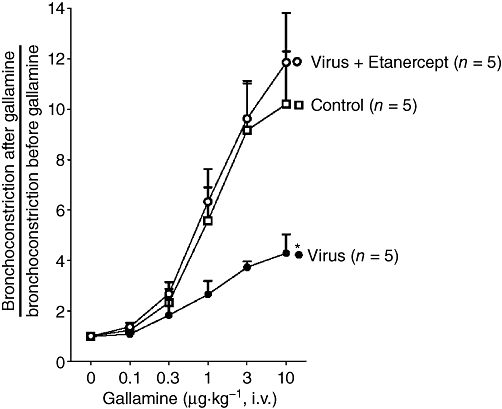
Etanercept protects neuronal M2 muscarinic receptor function in virus-infected guinea pigs. In controls, gallamine potentiates vagally induced bronchoconstriction by inhibiting M2 muscarinic receptor function. Virus infection significantly reduces gallamine-induced potentiation of vagally induced bronchoconstriction and this is blocked by etanercept. Points are mean ± SEM. *The dose–response curve is significantly different from control animals and etanercept-treated virus-infected animals, using anova.
Cardiac M2 muscarinic receptor function
Bradycardia was induced by either electrical stimulation of both vagus nerves or by intravenous injection of ACh; both of which decreased heart rate via stimulation of cardiac M2 muscarinic receptors (Figure 4). Bradycardia was not changed by virus infection or by etanercept.
Figure 4.
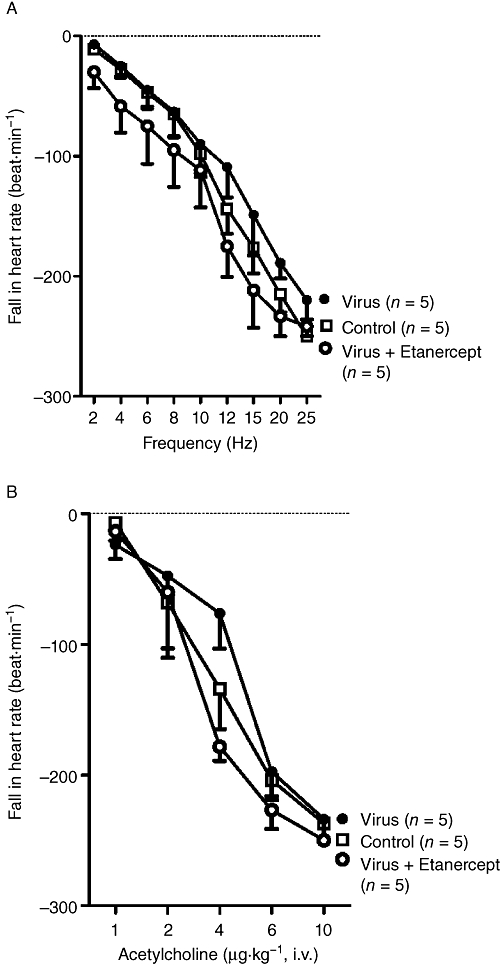
Etanercept does not change M2 muscarinic receptor function in the heart. Bradycardia induced either by electrical stimulation of both vagus nerves (A) or intravenous ACh (B) was not changed by virus infection, or by etanercept in virus-infected animals.
Inflammation
The average number of total cells in BAL from vehicle control animals (mock infection with PBS) was 17 × 106 cells, higher than uninfected control (no PBS and no virus infection, the average of total cell number was 5.6 × 106 cells), which was done within the same time period and published previously (Nie et al., 2009). Total cells, macrophages and neutrophils in BAL were slightly, but not significantly, decreased in virus-infected animals, compared with vehicle control (Figure 5). In our previous study, etanercept pretreatment did not change the total inflammatory cells in BAL from control guinea pigs [no PBS and no virus infection (Nie et al., 2009)]. Similarly, pretreatment with etanercept in virus-infected guinea pigs did not cause a significant decrease in total cells in BAL, compared with that of virus-infected animals (Figure 5A, P = 0.134). This non-significant decrease was made up predominantly of macrophages (P = 0.095) and neutrophils (P = 0.279, Figure 5B).
Figure 5.
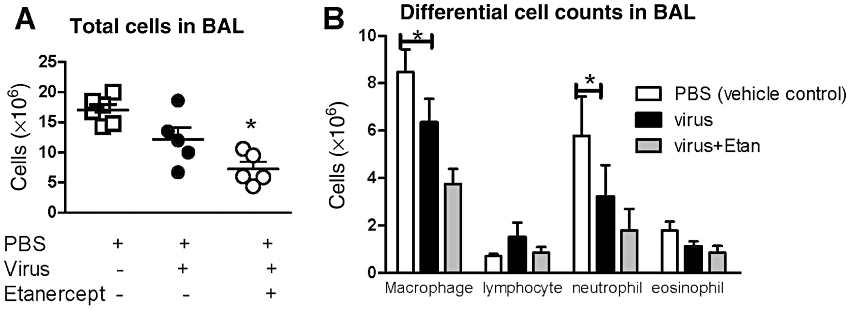
Etanercept (Etan) treatment in virus-infected animals does not significantly reduce total cells (A), macrophages and neutrophils (B) in bronchoalveolar lavage (BAL), compared with virus-infected animals. n = 5 in each group and points are mean ± SEM. * significant difference between vehicle control and etanercept-treated, virus-infected animals (P < 0.05, using anova with Bonferroni's correction).
Viral titres in the lungs of infected guinea pigs
Virus was recovered from all infected guinea pigs demonstrating that they all had active infections. The average viral titres recovered from infected guinea pig lung was 40.4 TCID50 per mg, which were not significantly different to that from etanercept pretreated virus-infected lung (79.3 TCID50 per mg, t-test P = 0.233).
TNF-α release from human trachea epithelial cells
Non-infected primary cultures of human airway epithelial cells express and release baseline TNF-α protein as measured by elisa. Each donor had different baseline expression of TNF-α and the average of baseline TNF-α expression was 10 pg·mL−1. Influenza virus infection increased TNF-α concentration in the supernatants threefold over uninfected controls (Figure 6) and the average of TNF-α expression induced by virus was 30 pg·mL−1. UV-inactivated influenza virus did not increase TNF-α protein in the supernatant as compared with baseline (Figure 6).
Figure 6.
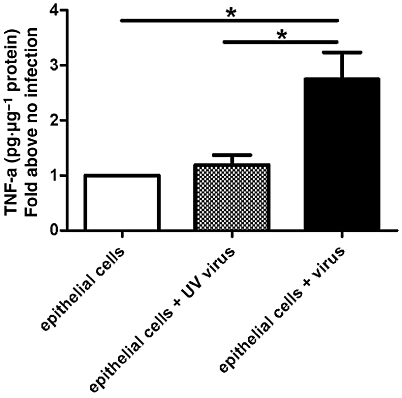
-Human tracheal epithelial cells infected with influenza virus release TNF-α. Four days post infection, the TNF-α concentration in the supernatant of infected cells is measured by elisa and significantly higher than in uninfected controls.
TNF-α reduces M2 receptor mRNA in neuroblastoma cells
Supernatants from virus-infected human tracheal epithelial cells in primary culture significantly reduced M2 muscarinic receptor mRNA in SK-N-SH cells (Figure 7). This inhibitory effect was completely blocked by adding etanercept to the supernatant (Figure 7). These findings demonstrate that virus-infected human airway epithelial cells produce physiologically significant levels of TNF-α. Supernatants from uninfected epithelial cells in culture and supernatants from epithelial cells exposed to UV-inactivated virus slightly decreased M2 receptor expression, but not significantly, compared with control media. This decrease was also blocked by etanercept (Figure 7). This confirmed our previous data in Figure 6 that there was a basal release of TNF-α in uninfected airway epithelial cells.
Figure 7.
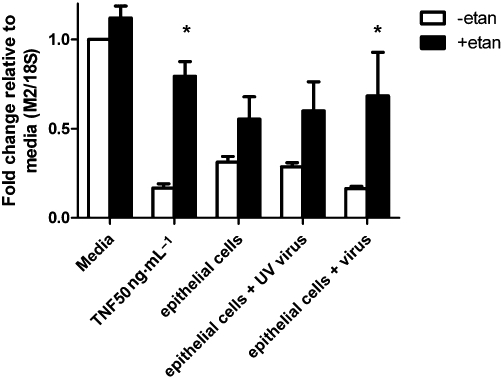
Reduction of M2 muscarinic receptor mRNA in neuroblastoma cells is prevented by etanercept. M2 receptor mRNA in neuroblastoma cells are measured by real time RT-PCR and expressed as fold change from control media. The supernatant from virus-infected cells significantly reduces M2 receptor mRNA in neuroblastoma cells and this reduction is blocked by etanercept (etan). *P < 0.05,significant difference, (two-way anova with Bonferroni's post-test).
The difference between the reduction in mRNA for M2 receptors caused by supernatant with virus infection and that caused by supernatant from uninfected epithelial cells was significant (P = 0.0129) (Figure 7). Additionally, the difference in reduction in mRNA for M2 receptors caused by supernatant with virus infection versus UV-inactivated virus was also significant (P = 0.009) (Figure 7). This indicates active virus infection contributes to the further reduction of expression of M2 receptor mRNA.
Discussion
Here we have shown that virus-induced airway hyperresponsiveness and M2 muscarinic receptor dysfunction are blocked by etanercept and thus are likely to be TNF-α dependent. Etanercept caused a non-significant decrease in macrophages, neutrophils and total cells in BAL (Figure 5) but did not affect viral titre in lung.
Neuronal M2 muscarinic receptor dysfunction is a common mechanism underlying airway hyperresponsiveness in human asthma patients (Minette et al., 1989), antigen challenged animals (Fryer and Jacoby, 1992), virus-infected animals (Fryer and Jacoby, 1991), ozone exposed animals (Gambone et al., 1994) and animals exposed to organophosphate pesticides (Lein and Fryer, 2005). We have shown that M2 muscarinic receptor dysfunction is mediated by a variety of mechanisms and that preventing M2 muscarinic receptor dysfunction prevents airway hyperresponsiveness. In antigen challenged animals, eosinophils are recruited to airway nerves, where they degranulate and release major basic protein (MBP), which blocks the function of M2 receptors acting as an M2 receptor antagonist (Jacoby et al., 1993). In contrast, in virus-infected animals, depleting macrophages (a major source of TNF-α) protects M2 receptors and prevents virus-induced airway hyperresponsiveness (Lee et al., 2004). In vitro, inflammatory cytokines such as TNF-α directly inhibit M2 muscarinic receptor mRNA expression (Nie et al., 2009).
TNF-α concentration in BAL is increased in virus-infected patients (Calhoun et al., 1994; McNamara et al., 2004) and animals (You et al., 2006). A major source of TNF-α production in airway diseases are macrophages. Depleting macrophages by pretreating with liposome-encapsulated clodronate prevented both vagally mediated airway hyperresponsiveness and M2 receptor dysfunction in virus-infected guinea pigs (Lee et al., 2004). Here, we show that there was no statistically significant decrease of macrophages in BAL from etanercept-treated virus-infected animals (Figure 5). This indicates the protective effect of etanercept on M2 receptor function is not due to reducing macrophages. However, our data (Figure 6) and another study (Matsukura et al., 1998) have shown that airway epithelial cells, the primary site of airway virus infection, are another important source of TNF-α after virus infection. Although virus-infected epithelial cells produce many cytokines in addition to TNF-α (Noah and Becker, 1993; Bryan et al., 2005), blocking TNF-α with etanercept completely reversed the decrease in neuronal M2 muscarinic receptor expression in response to treatment with epithelial cell supernatants. This indicates that among the pro-inflammatory cytokines released from virus-infected epithelial cells, TNF-α was a predominant inhibitor of neuronal M2 muscarinic receptor expression.
Baseline inflation pressure, blood pressure and heart rate were not different among control, virus-infected and etanercept-treated virus-infected animals (Table 1). The M2 muscarinic receptor function and airway hyperresponsiveness were significantly different between virus-infected and etanercept-treated virus-infected animals (Figures 1 and 2), but viral titres and BAL inflammatory cell numbers were not significantly different between these two groups. This indicates titres of virus and inflammatory cell numbers in BAL may not be critical in the effect of etanercept on virus-induced airway hyperresponsiveness.
Asthma is a heterogeneous disease with diverse environmental causes. A synergistic interaction between allergen sensitization, allergen exposure and viral infection has been shown in adult asthmatic subjects during acute exacerbations (Green et al., 2002; Murray et al., 2006). Individuals who are sensitized, exposed to allergen and virus-infected have significantly increased risk of admission for exacerbations. Our data help explain this synergistic interaction. We have shown both virus-induced (Fryer and Jacoby, 1991) and antigen challenge-induced (Fryer and Wills-Karp, 1991; Fryer and Jacoby, 1992) airway hyperresponsiveness is due to dysfunction of M2 muscarinic receptors. In antigen sensitized and challenged animals, M2 muscarinic receptors are blocked by MBP release by eosinophils in contact with the parasympathetic nerves (Fryer and Wills-Karp, 1991; Fryer and Jacoby, 1992). These airway eosinophils are redistributed away from the nerves by etanercept, suggesting a prominent role for TNF-α (Nie et al., 2009). Here, we show virus-induced dysfunction of M2 muscarinic receptor was through a TNF-α dependent pathway too, as etanercept completely protected M2 receptor function (Figure 3) in virus-infected guinea pigs. Thus, TNF-α is a common factor in the molecular mechanisms of virus infection and antigen challenge-induced airway hyperresponsiveness, although the mechanisms in these two models are different. Our previous studies have shown that TNF-α reduces neuronal M2 muscarinic receptor expression by increasing the rate of M2 mRNA degradation (Nie et al., 2009).
The results of the current study suggest TNF-α mediated M2 muscarinic receptor dysfunction is central in virus induced vagally mediated airway hyperresponsiveness. Our results provide evidence supporting anti-TNF-α therapy in treating and preventing virus-induced asthma exacerbation.
Acknowledgments
We thank the Pacific Northwest Transplant Bank (Portland, OR) for human organ donor tracheas. We thank Elizabeth Bivins-Smith for preparing the H3N2 virus.
Glossary
Abbreviations
- BAL
bronchoalveolar lavage
- MBP
major basic protein
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann B, Wagner M, Aleksic T, von Wichert G, Weber CK, Adler G, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest. 2007;117:1502–1513. doi: 10.1172/JCI30876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, et al. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- Bowerfind WM, Fryer AD, Jacoby DB. Double-stranded RNA causes airway hyperreactivity and neuronal M2 muscarinic receptor dysfunction. J Appl Physiol. 2002;92:1417–1422. doi: 10.1152/japplphysiol.00934.2001. [DOI] [PubMed] [Google Scholar]
- Bradding P, Roberts JA, Britten KM, Montefort S, Djukanovic R, Mueller R. Interleukin-4, -5, and -6 and tumor necrosis factor-alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. 1994;10:471–480. doi: 10.1165/ajrcmb.10.5.8179909. [DOI] [PubMed] [Google Scholar]
- Broide DH, Lotz M, Cuomo AJ, Coburn DA, Federman EC, Wasserman SI. Cytokines in symptomatic asthma airways. J Allergy Clin Immunol. 1992;89:958–967. doi: 10.1016/0091-6749(92)90218-q. [DOI] [PubMed] [Google Scholar]
- Bryan DL, Hart P, Forsyth K, Gibson R. Modulation of respiratory syncytial virus-induced prostaglandin E2 production by n-3 long-chain polyunsaturated fatty acids in human respiratory epithelium. Lipids. 2005;40:1007–1011. doi: 10.1007/s11745-005-1463-4. [DOI] [PubMed] [Google Scholar]
- Calhoun WJ, Dick EC, Schwartz LB, Busse WW. A common cold virus, rhinovirus 16, potentiates airway inflammation after segmental antigen bronchoprovocation in allergic subjects. J Clin Invest. 1994;94:2200–2208. doi: 10.1172/JCI117581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Cazzola M, Polosa R, Erzurum SC, Berry MA, Hargadon B, Shelley M, et al. Anti-TNF-alpha and Th1 cytokine-directed therapies for the treatment of asthma. Curr Opin Allergy Clin Immunol. 2006;6:43–50. doi: 10.1097/01.all.0000199798.10047.74. [DOI] [PubMed] [Google Scholar]
- Costello RW, Schofield BH, Kephart GM, Gleich GJ, Jacoby DB, Fryer AD. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am J Physiol. 1997;273(Pt 1):L93–103. doi: 10.1152/ajplung.1997.273.1.L93. [DOI] [PubMed] [Google Scholar]
- Empey DW, Laitinen LA, Jacobs L, Gold WM, Nadel JA. Mechanisms of bronchial hyperreactivity in normal subjects after upper respiratory tract infection. Am Rev Respir Dis. 1976;113:131–139. doi: 10.1164/arrd.1976.113.2.131. [DOI] [PubMed] [Google Scholar]
- Fryer AD, Jacoby DB. Parainfluenza virus infection damages inhibitory M2 muscarinic receptors on pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol. 1991;102:267–271. doi: 10.1111/j.1476-5381.1991.tb12164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer AD, Jacoby DB. Function of pulmonary M2 muscarinic receptors in antigen-challenged guinea pigs is restored by heparin and poly-L-glutamate. J Clin Invest. 1992;90:2292–2298. doi: 10.1172/JCI116116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer AD, Maclagan J. Muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br J Pharmacol. 1984;83:973–978. doi: 10.1111/j.1476-5381.1984.tb16539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer AD, Wills-Karp M. Dysfunction of M2-muscarinic receptors in pulmonary parasympathetic nerves after antigen challenge. J Appl Physiol. 1991;71:2255–2261. doi: 10.1152/jappl.1991.71.6.2255. [DOI] [PubMed] [Google Scholar]
- Fryer AD, Adamko DJ, Yost BL, Jacoby DB. Effects of inflammatory cells on neuronal M2 muscarinic receptor function in the lung. Life Sci. 1999;64:449–455. doi: 10.1016/s0024-3205(98)00587-6. [DOI] [PubMed] [Google Scholar]
- Fryer AD, Stein LH, Nie Z, Curtis DE, Evans CM, Hodgson ST, et al. Neuronal eotaxin and the effects of ccr3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J Clin Invest. 2006;116:228–236. doi: 10.1172/JCI25423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambone LM, Elbon CL, Fryer AD. Ozone-induced loss of neuronal M2 muscarinic receptor function is prevented by cyclophosphamide. J Appl Physiol. 1994;77:1492–1499. doi: 10.1152/jappl.1994.77.3.1492. [DOI] [PubMed] [Google Scholar]
- Gosset P, Tsicopoulos A, Wallaert B, Vannimenus C, Joseph M, Tonnel AB, et al. Increased secretion of tumor necrosis factor alpha and interleukin-6 by alveolar macrophages consecutive to the development of the late asthmatic reaction. J Allergy Clin Immunol. 1991;88:561–571. doi: 10.1016/0091-6749(91)90149-i. [DOI] [PubMed] [Google Scholar]
- Green RM, Custovic A, Sanderson G, Hunter J, Johnston SL, Woodcock A. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ. 2002;324:763. doi: 10.1136/bmj.324.7340.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallsworth MP, Soh CP, Lane SJ, Arm JP, Lee TH. Selective enhancement of GM-CSF, TNF-alpha, IL-1 beta and IL-8 production by monocytes and macrophages of asthmatic subjects. Eur Respir J. 1994;7:1096–1102. [PubMed] [Google Scholar]
- Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, et al. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–1018. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby DB, Fryer AD. Virus-induced airway hyperresponsiveness–possible involvement of neural mechanisms. Am Rev Respir Dis. 1991;144:1422–1423. doi: 10.1164/ajrccm/144.6.1422a. [DOI] [PubMed] [Google Scholar]
- Jacoby DB, Gleich GJ, Fryer AD. Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor. J Clin Invest. 1993;91:1314–1318. doi: 10.1172/JCI116331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby DB, Yost BL, Elwood T, Fryer AD. Effects of neurokinin receptor antagonists in virus-infected airways. Am J Physiol Lung Cell Mol Physiol. 2000;279:L59–L65. doi: 10.1152/ajplung.2000.279.1.L59. [DOI] [PubMed] [Google Scholar]
- Johnston SL, Pattemore PK, Sanderson G, Smith S, Lampe F, Josephs L, et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ. 1995;310:1225–1229. doi: 10.1136/bmj.310.6989.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SL, Pattemore PK, Sanderson G, Smith S, Campbell MJ, Josephs LK, et al. The relationship between upper respiratory infections and hospital admissions for asthma: a time-trend analysis. Am J Respir Crit Care Med. 1996;154(Pt 1):654–660. doi: 10.1164/ajrccm.154.3.8810601. [DOI] [PubMed] [Google Scholar]
- Kivilcim M, Peyman GA, Kazi AA, Dellacroce J, Ghobrial RN, Monzano R. Intravitreal toxicity of high-dose etanercept. J Ocul Pharmacol Ther. 2007;23:57–62. doi: 10.1089/jop.2006.0083. [DOI] [PubMed] [Google Scholar]
- Lee AM, Fryer AD, van Rooijen N, Jacoby DB. Role of macrophages in virus-induced airway hyperresponsiveness and neuronal M2 muscarinic receptor dysfunction. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1255–L1259. doi: 10.1152/ajplung.00451.2003. [DOI] [PubMed] [Google Scholar]
- Lein PJ, Fryer AD. Organophosphorus insecticides induce airway hyperreactivity by decreasing neuronal M2 muscarinic receptor function independent of acetylcholinesterase inhibition. Toxicol Sci. 2005;83:166–176. doi: 10.1093/toxsci/kfi001. [DOI] [PubMed] [Google Scholar]
- Lesslauer W, Tabuchi H, Gentz R, Brockhaus M, Schlaeger EJ, Grau G, et al. Recombinant soluble tumor necrosis factor receptor proteins protect mice from lipopolysaccharide-induced lethality. Eur J Immunol. 1991;21:2883–2886. doi: 10.1002/eji.1830211134. [DOI] [PubMed] [Google Scholar]
- McNamara PS, Flanagan BF, Selby AM, Hart CA, Smyth RL. Pro- and anti-inflammatory responses in respiratory syncytial virus bronchiolitis. Eur Respir J. 2004;23:106–112. doi: 10.1183/09031936.03.00048103. [DOI] [PubMed] [Google Scholar]
- Matsukura S, Kokubu F, Kubo H, Tomita T, Tokunaga H, Kadokura M, et al. Expression of RANTES by normal airway epithelial cells after influenza virus A infection. Am J Respir Cell Mol Biol. 1998;18:255–264. doi: 10.1165/ajrcmb.18.2.2822. [DOI] [PubMed] [Google Scholar]
- Minette PA, Lammers JW, Dixon CM, McCusker MT, Barnes PJ. A muscarinic agonist inhibits reflex bronchoconstriction in normal but not in asthmatic subjects. J Appl Physiol. 1989;67:2461–2465. doi: 10.1152/jappl.1989.67.6.2461. [DOI] [PubMed] [Google Scholar]
- Murray CS, Poletti G, Kebadze T, Morris J, Woodcock A, Johnston SL, et al. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. 2006;61:376–382. doi: 10.1136/thx.2005.042523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson KG, Kent J, Ireland DC. Respiratory viruses and exacerbations of asthma in adults. BMJ. 1993;307:982–986. doi: 10.1136/bmj.307.6910.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Jacoby DB, Fryer AD. Etanercept prevents airway hyperresponsiveness by protecting neuronal M2 muscarinic receptors in antigen challenged guinea pigs. Br J Pharmacol. 2009;156:201–210. doi: 10.1111/j.1476-5381.2008.00045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Nelson CS, Jacoby DB, Fryer AD. Expression and regulation of intercellular adhesion molecule-1 on airway parasympathetic nerves. J Allergy Clin Immunol. 2007;119:1415–1422. doi: 10.1016/j.jaci.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Noah TL, Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am J Physiol. 1993;265(Pt 1):L472–L478. doi: 10.1152/ajplung.1993.265.5.L472. [DOI] [PubMed] [Google Scholar]
- Peppel K, Crawford D, Beutler B. A tumor necrosis factor (TNF) receptor-IgG heavy chain chimeric protein as a bivalent antagonist of TNF activity. J Exp Med. 1991;174:1483–1489. doi: 10.1084/jem.174.6.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003;111:821–831. doi: 10.1172/JCI16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawatzky DA, Kingham PJ, Court E, Kumaravel B, Fryer AD, Jacoby DB, et al. Eosinophil adhesion to cholinergic nerves via ICAM-1 and VCAM-1 and associated eosinophil degranulation. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1279–L1288. doi: 10.1152/ajplung.00279.2001. [DOI] [PubMed] [Google Scholar]
- Suffredini AF, Reda D, Banks SM, Tropea M, Agosti JM, Miller R. Effects of recombinant dimeric TNF receptor on human inflammatory responses following intravenous endotoxin administration. J Immunol. 1995;155:5038–5045. [PubMed] [Google Scholar]
- Takei S, Groh D, Bernstein B, Shaham B, Gallagher K, Reiff A. Safety and efficacy of high dose etanercept in treatment of juvenile rheumatoid arthritis. J Rheumatol. 2001;28:1677–1680. [PubMed] [Google Scholar]
- Thomas PS. Tumour necrosis factor-alpha: the role of this multifunctional cytokine in asthma. Immunol Cell Biol. 2001;79:132–140. doi: 10.1046/j.1440-1711.2001.00980.x. [DOI] [PubMed] [Google Scholar]
- Uhl EW, Moldawer LL, Busse WW, Jack TJ, Castleman WL. Increased tumor necrosis factor-alpha (TNF-alpha) gene expression in parainfluenza type 1 (Sendai) virus-induced bronchiolar fibrosis. Am J Pathol. 1998;152:513–522. [PMC free article] [PubMed] [Google Scholar]
- Wang H, Peters T, Kess D, Sindrilaru A, Oreshkova T, Van Rooijen N, et al. Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J Clin Invest. 2006;116:2105–2114. doi: 10.1172/JCI27180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Robinson DS, Varney V, Meng Q, Tsicopoulos A, Moqbel R, et al. TNF alpha mRNA expression in allergic inflammation. Clin Exp Allergy. 1991;21:745–750. doi: 10.1111/j.1365-2222.1991.tb03205.x. [DOI] [PubMed] [Google Scholar]
- You D, Becnel D, Wang K, Ripple M, Daly M, Cormier SA. Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir Res. 2006;7:107. doi: 10.1186/1465-9921-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]