

Abstract
BACKGROUND AND PURPOSE
Recent studies have suggested an essential role for aldehyde dehydrogenase 2 (ALDH2) in the bioactivation of organic nitrates such as glyceryl trinitrate (GTN). In the present study, we utilized an in vivo GTN tolerance model to further investigate the role of ALDH2 in GTN bioactivation and tolerance.
EXPERIMENTAL APPROACH
We assessed changes in aortic ALDH activity, and in ALDH2 protein expression in various rat blood vessels (aorta, vena cava, femoral artery and femoral vein) during continuous GTN exposure (0.4 mg·h−1 for 6, 12, 24 or 48 h) or after a 1-, 3- or 5-day drug-free period following a 48 h exposure to GTN, in relation to changes in vasodilator responses to GTN and in vascular GTN biotransformation.
KEY RESULTS
A decrease was observed in both ALDH2 protein expression (80% in tolerant veins and 30% in tolerant arteries after 48 h exposure to GTN) and aortic ALDH activity, concomitant with decreased vasodilator responses to GTN and decreased aortic GTN biotransformation. However, after a 24 h drug-free period following 48 h of GTN exposure, vasodilator responses to GTN and aortic GTN biotransformation activity had returned to control values, whereas vascular ALDH2 expression and aortic ALDH activity were still significantly depressed, and remained so for 3–5 days following cessation of GTN exposure.
CONCLUSIONS AND IMPLICATIONS
The dissociation of reduced ALDH activity and ALDH2 expression from the duration of the impaired vasodilator and biotransformation responses to GTN in nitrate-tolerant blood vessels, suggests that factors other than changes in ALDH2-mediated GTN bioactivation contribute to nitrate tolerance.
Keywords: glyceryl trinitrate, nitric oxide, aldehyde dehydrogenase, nitrate tolerance
Introduction
In the clinical setting, continuous exposure to nitrates such as glyceryl trinitrate (GTN) results in tolerance to their anti-anginal and haemodynamic effects. This has led to the use of elliptical dosage regimens to provide a nitrate-free interval, during which recovery of sensitivity to the vasodilator effects of the nitrate can occur. It is generally accepted that GTN is a pro-drug that requires bioactivation to yield NO or an ‘NO’-like species (termed mechanism-based biotransformation), resulting in activation of soluble guanylyl cyclase (sGC), increased cGMP accumulation and relaxation of vascular smooth muscle. Although the bioactive molecule derived from GTN is assumed to be NO, attempts to measure or visualize NO formation in blood vessels at pharmacologically relevant concentrations of GTN have been unsuccessful (Kleschyov et al., 2003; Nuñez et al., 2006; Miller et al., 2008), hence the use of the term ‘NO mimetic’ when referring to nitrates.
Tolerance to nitrates is widely acknowledged to be multifactorial in nature, and a number of proposed mechanisms of tolerance development have been advanced, some at the cellular level and others involving cardiovascular homeostatic mechanisms (see Fung et al., 2004; Münzel et al., 2005, for reviews). Although it is difficult to evaluate the relative role of various tolerance mechanisms, reduced mechanism-based biotransformation of GTN to the proximal activator of sGC appears to be of central importance, based on numerous studies showing both a reduction of GTN biotransformation and GTN-induced cGMP accumulation in GTN-tolerant tissues, concomitant with reduced vasodilator responses to GTN (Brien et al., 1988; Bennett et al., 1989; Slack et al., 1989; Sage et al., 2000; Chen et al., 2002). Importantly, only limited impairment of vasodilator responses to NO donors such as sodium nitroprusside or NONOates are observed in GTN-tolerant tissues and animals, suggesting that NO-dependent vasodilator mechanisms remain relatively intact, despite the development of GTN tolerance (Ratz et al., 2000b; 2002; Chen et al., 2002; MacPherson et al., 2006).
Although the vascular enzyme(s) that mediate the mechanism-based biotransformation of GTN to the proximal activator of sGC have yet to be identified with certainty, a number of enzymes and proteins have been shown to mediate GTN denitration to glyceryl dinitrates (GDNs) and inorganic nitrite anion (NO2-) (reviewed in Thatcher et al., 2004). These enzymes include cytochrome P450 (McDonald and Bennett 1990), NADPH cytochrome-P450 reductase (McGuire et al., 1998), cytosolic (Tsuchida et al., 1990; Nigam et al., 1996) and microsomal glutathione transferases (Ji et al., 2009) among others. However, one of these, namely aldehyde dehydrogenase 2 (ALDH2), has been the focus of a number of recent studies suggesting that this enzyme mediates the mechanism-based biotransformation of GTN, and that inactivation of ALDH2 during GTN biotransformation is the basis for tolerance development (reviewed in Daiber et al., 2008). Indeed, studies from Mayer's group have provided evidence for NO formation during incubation of GTN with purified ALDH2 (and ALDH1) (Beretta et al., 2008a; Wenzl et al., 2009a).
In 2002, Chen et al. proposed ALDH2 as an enzyme that bioactivates GTN, resulting in vascular smooth muscle relaxation. This finding was further supported in studies using Aldh2−/− mice, which showed impairment in drug-induced cGMP accumulation and vasodilator responses to GTN, but not to NO-donors such as sodium nitroprusside (Chen et al., 2005). This was taken as strong evidence that ALDH2 plays an important role in the bioactivation of GTN. Other studies have reported inhibition of GTN-induced relaxation in aorta by the ALDH inhibitors chloral hydrate, cyanamide and daidzin (Chen et al., 2002; DiFabio et al., 2003; de la Lande et al., 2004; Sydow et al., 2004; Kollau et al., 2005). These inhibitors had no effect on sodium nitroprusside-induced relaxation, suggesting that they inhibited GTN bioactivation. However, DiFabio et al. (2003) and de la Lande et al. (2004) showed that cyanamide, chloral hydrate and the ALDH substrate, propionaldehyde, were equally effective inhibitors of GTN-induced vasodilatation in both GTN-tolerant and non-tolerant blood vessels, indicating that these inhibitors have non-specific effects. Clinically, it has been shown that individuals who are heterozygous or homozygous for the ALDH2 point mutation Glu504Lys (ALDH2*1, ALDH2*2) exhibit reduced vasodilator responses to GTN (Mackenzie et al., 2005; Li et al., 2006). It is important to note that even though both of these clinical studies concluded that ALDH2 is involved in the bioactivation of GTN, they suggested the involvement of other mechanisms. Several concerns have been raised with respect to the ALDH2 bioactivation hypothesis, more specifically to the generation of NO from NO2- (Difabio et al., 2003) and to the regeneration of the inactivated ALDH2 enzyme formed during GTN treatment (Beretta et al., 2008b).
Previous studies have shown that 1,2-GDN is the predominant GTN metabolite formed in vascular tissues, and furthermore, that formation of 1,2-GDN is diminished in GTN-tolerant tissues. These findings suggest that ALDH2 plays a primary role in GTN tolerance as ALDH2 specifically catalyses the formation of 1,2-GDN, and ALDH2 activity is reduced during GTN tolerance. Recently, Hink et al. (2007) investigated whether inhibition of ALDH2 contributed to GTN tolerance in human blood vessels. They found that long-term GTN treatment resulted in GTN tolerance and endothelial dysfunction. Furthermore, GTN tolerance was associated with the inhibition and down-regulation of vascular ALDH2. The down-regulation of ALDH2 observed by Hink et al. (2007) could be of importance as the ALDH2 isoform has a low Km value (c. 1.0 µM) relative to other ALDH isoforms, and as a result mediates almost all hepatic aldehyde oxidation (Vasilou et al., 2000). Thus, the down-regulation of ALDH2 during nitrate tolerance could be important in the toxicology of a number of aldehydes and to the metabolism of alcohol. A number of studies have examined the expression of ALDH2 in GTN-tolerant blood vessels (Hink et al., 2007; Szöcs et al., 2007; Wenzel et al., 2007), and although these studies all showed a decrease in ALDH2 expression, this was only evaluated at a single time point. In the present study, we utilized an in vivo GTN tolerance model and examined ALDH2 protein and activity levels at various time points during the development and reversal of nitrate tolerance. We found that vascular ALDH2 protein and activity levels during exposure to GTN were not correlated with vasodilator responses to GTN or with vascular GTN biotransformation, suggesting the dissociation between ALDH2 inactivation and GTN tolerance.
Methods
Test system
All procedures for animal experimentation were undertaken in accordance with the principles and guidelines of the Canadian Council on Animal Care and were approved by the Queen's University Animal Care Committee. Animals were maintained under a 12 h light/dark cycle, with free access to food and water. Rats (male Sprague–Dawley, 250–350 g, Charles River Laboratories, Montreal, QC, Canada) were randomly assigned into GTN-treated or sham groups with each group consisting of 4–10 animals. GTN-treated rats were exposed to a continuous source of GTN via the subdermal implantation of two 0.2 mg·h−1 transdermal GTN patches (Ratz et al., 2000a) for 6, 12, 24 or 48 h, or for 48 h followed by removal of the patches for 1, 3 or 5 days. Surgery was performed under halothane anaesthesia, with the surgical plane of anaesthesia monitored by toe pinch and the depth and character of respiration. A 1-cm transverse incision was made in the upper dorsal region of the animal and the skin separated from the underlying fascia by blunt dissection. Two transdermal patches were inserted back to back into the resulting subdermal space. The site was sutured closed and disinfected with 10% providone-iodine solution. Animals were administered buprenorphine (0.05 mg·kg−1 s.c.) preoperatively, and then every 12 h as needed. For the groups of rats subjected to 48 h of GTN exposure, the site was re-opened after 24 h and both patches were replaced. Animals in the recovery groups also received GTN-treatment for 48 h. However, the GTN patches were removed after the initial 48 h GTN-treatment period for 1, 3 or 5 days prior to death. Animals in the sham groups received identical treatments; however, sham (drug-free) patches were used instead. Animals were killed at various time points and the aorta, vena cava, femoral arteries and femoral veins were removed for functional or biochemical analysis.
Isolated blood vessel relaxation responses
Isolated rings of aorta, femoral artery or femoral vein from GTN-tolerant or control animals were prepared for isometric tension measurements and were equilibrated for 1 h at an optimal resting tension of 2.5 mN for femoral veins, 5 mN for femoral arteries and 9.8 mN for aorta (MacPherson et al., 2006). The vascular preparations were contracted submaximally with phenylephrine (0.2–5 µM), and after the induced tone had stabilized, cumulative concentration-response curves were obtained for GTN (0.1 nM–30 µM) or 1,1-diethyl-2-hydroxy-2-nitrosohydrazine (DEA/NO) (0.1 nM–10 µM). In one series of experiments, aortic rings from control animals were submaximally contracted with phenylephrine and exposed to 0.1 mM daidzin or diluent (DMSO; final concentration, 0.1%) for 15 min prior to obtaining concentration-response curves for GTN.
Immunoblot analysis of ALDH2
Aorta, vena cava and femoral arteries and veins were homogenized in lysis buffer [50 mM Tris-HCl pH 7.4, 1 mM EDTA, 0.1 mM PMSF, 1 mM dithiothreitol, 1% Triton X-100 and protease inhibitors (Roche Diagnostics, Mannheim, Germany)] and centrifuged at 480× g for 10 min. Proteins in the supernatant fraction were separated by SDS-PAGE on 10% gels and transferred electrophoretically to PVDF membranes. Blots were probed with a rabbit polyclonal antibody to human ALDH2, and immunoreactive bands visualized by enhanced chemiluminescence. Membranes were then stripped and reprobed with a mouse monoclonal anti-β-actin antibody, and both immunoreactive bands quantified by optical densitometry using ImageJ software (version 1.43). To control for variability in optical density measurements between gels due to differences in protein loading, gel transfer time and time of film exposure, samples of all four blood vessel types from each treated animal and its matched control were loaded onto the same gel for comparison (see example in Figure 1), and densitometry values for ALDH2 normalized to β-actin. The normalized density value for each blood vessel type from treated animals was expressed as a percentage of its matched control.
Figure 1.
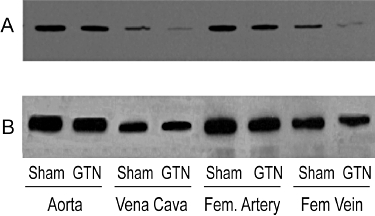
Immunoblot analysis of vascular ALDH2 expression after exposure of animals to 0.4 mg·h−1r GTN for 48 h. Blood vessels were excised from GTN-treated (GTN) and sham animals (Sham) and homogenized. Proteins (8 µg) from the 480×g supernatant fraction were resolved on 10% SDS gels under reducing conditions and transferred to PVDF membranes. The blot was probed with a rabbit polyclonal antibody to human ALDH2 (A), then stripped and re-probed with a mouse monoclonal antibody to β actin (B).
ALDH activity
Aortae were homogenized in buffer containing 0.25 M sucrose, 5 mM Tris-HCl pH 7.2, 0.5 mM EDTA and 0.1 mM PMSF and centrifuged at 480× g for 10 min. Protein content of the supernatant fraction was determined and deoxycholate (2.5 mg·mg−1 protein) was added to the homogenates to solubilize membrane-bound proteins. Total ALDH activity was measured as the change in absorbance at 340 nm during incubation of 100 µg aortic homogenate protein with 1 mM NAD+ in 50 mM sodium pyrophosphate buffer, pH 8.8, containing 2 µM rotenone (to inhibit NADH consumption by complex 1 of the electron transfer chain), 1 mM 4-methylpyrazone (to inhibit alcohol dehydrogenase) and substrate (5 mM propionaldehyde) (Tottmar et al., 1973; Loomis and Brien, 1983). Specific activity was calculated using the molar extinction coefficient for NADH of 6306 M−1 cm−1.
GTN Biotransformation
Rat aortic biotransformation of GTN to glyceryl-1,2-dinitrate (1,2-GDN) and glyceryl-1,3-dinitrate (1,3-GDN) was assessed essentially as described previously (Bennett et al., 1992). Briefly, aortas prepared from sham-treated animals or from animals treated with 0.4 mg·h−1 GTN for 48 h or treated for 48 h followed by a 1-day GTN-free period, were divided in half and placed into individual tubes containing 1 mL of Krebs' solution at 37°C aerated with 95% O2–5% CO2. Tissues were exposed to 0.2 µM phenylephrine for 5 min and to either diluent or 0.1 mM daidzin for an additional 10 min. This concentration of daidzin inhibited low Km ALDH activity (i.e. ALDH2) in rat liver mitochondrial fractions by almost 90% (from 15.2 ± 2.3 to 1.7 ± 0.9 nmol·min−1·mg−1 protein using 50 µM propionaldehyde as substrate, n = 3). Tissues were then incubated with 2 µM GTN for 1 min and frozen between liquid nitrogen precooled clamps. The 1,2-GDN and 1,3-GDN metabolites of GTN were extracted from the tissues as described (Bennett et al., 1992) and quantified by megabore capillary column gas-liquid chromatography (McDonald and Bennett, 1990).
Data analysis
Relaxation responses to GTN were measured as the percentage decrease in phenylephrine-induced tone. EC50 values for relaxation were determined from the concentration-response curves using a sigmoidal dose–response curve-fitting algorithm. Due to inhomogeneity of variance, statistical analysis for the relaxation experiments was performed using logarithmically transformed data. The differences in EC50 values for relaxation and aortic ALDH activity in blood vessels from sham- and GTN-treated animals were compared using Student's t-test for unpaired data. One-sample Student's t-tests were used to compare the normalized mean values for vascular ALDH2 protein expression from each treatment group with their matched controls. All data are expressed as the mean ± SD. A P-value of <0.05 was considered statistically significant.
Drugs and solutions
Transdermal GTN patches were obtained as Transderm-Nitro (0.2 mg·h−1) from Novartis Pharmaceuticals (Dorval, QC, Canada). Drug-free patches were produced by soaking patches for at least 2 days in 95% ethanol (patches were allowed to air dry for at least 15 min before implantation). Removal of GTN from the patches by this procedure was confirmed by the absence of GTN or GTN metabolites in the plasma of rats implanted with these sham patches (Ratz et al., 2002). GTN was obtained as a solution (TRIDIL®, 5 mg·mL−1) in ethanol, propylene glycol and water (1:1:1.33) from Sabex Inc. (Boucherville, QC, Canada). Halothane for inhalational anaesthesia was obtained from Halocarbon Laboratories (River Edge, NJ, USA), 1,1-diethyl-2-hydroxy-2-nitrosohydrazine (DEA/NO) was obtained from Calbiochem (La Jolla, CA, USA) and daidzin was obtained from LC Laboratories (Woburn, MA, USA). Chemiluminescence reagents were from Kirkegaard and Perry Laboratories (Gaithersburg, MA, USA). The rabbit anti-human ALDH2 antiserum was a gift from Dr V. Vasiliou (University of Colorado Health Science Center, Denver, CO, USA) and mouse monoclonal anti-β-actin antibody was obtained from Sigma (St. Louis, MO, USA). All other chemicals were of reagent grade and were obtained from a variety of commercial sources.
Results
ALDH2 protein expression
ALDH2 immunoreactive bands were observed with mobilities slightly less than the 50 kDa marker, and with the same mobility as purified recombinant human ALDH2, similar to the findings of Difabio et al. (2003) and Hink et al. (2007). The relative expression of ALDH2 was approximately twofold to threefold greater in arterial compared to venous blood vessels, and changes in the relative expression of ALDH2 in the four blood vessel types over the time-course of GTN tolerance development and reversal revealed vessel-specific differences. ALDH2 protein levels decreased in the aorta and femoral artery by approximately 30% after GTN treatment for 12, 24 or 48 h (Figures 2D and 3). In the vena cava and femoral vein, 48 h of GTN treatment resulted in an 80% reduction of ALDH2 protein (Figures 4 and 5). In all four blood vessel types, ALDH2 protein levels were still significantly reduced in animals that had been treated with GTN for 48 h, followed by a 1-day or 3-day nitrate-free period. A nitrate-free period of 5 days following 48 h of GTN treatment resulted in ALDH2 protein returning to control levels only in the aorta; in the other blood vessel types, ALDH2 protein was still reduced by 30–50%.
Figure 2.
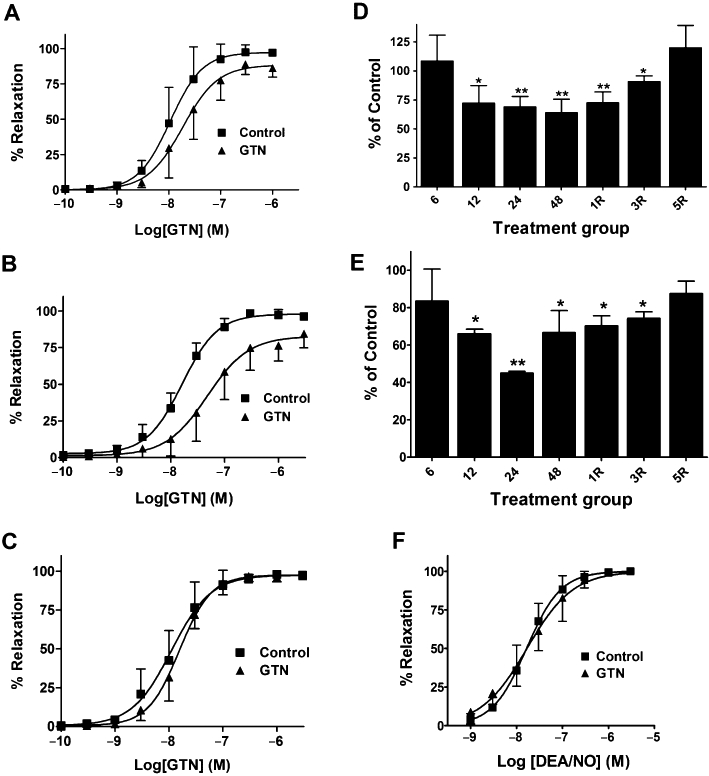
Effect of in vivo GTN exposure on GTN-induced relaxation of isolated aorta, and on ALDH activity and ALDH2 protein expression. (A–C) Isolated aortic rings were prepared from rats treated with 0.4 mg·h−1 GTN (GTN) or vehicle (Control) for (A) 24 h (B) 48 h GTN and (C) 48 h followed by a 1-day GTN-free period. Aortic rings were contracted submaximally with phenylephrine and cumulative concentration-response curves for GTN were obtained (n = 7–10). The EC50 values and maximal relaxation were significantly different from Control for the 24 h and 48 h GTN treatment groups (P < 0.05, Student's t-test for unpaired data). (D and E) Rats were treated with 0.4 mg·h−1 GTN (GTN) or vehicle for 6, 12, 24 and 48 h as well as 48 h GTN treatment followed by a 1-, 3- or 5-day GTN-free period (1R, 3R, 5R). (D) ALDH2 protein was determined by immunoblot analysis and immunoreactive bands were quantified by densitometry. ALDH2 protein levels in aortae (normalized to β-actin) from GTN-treated animals are expressed as a percentage of the levels in matching control animals. Data are presented as the mean percentage of control ± SD (n = 4) and were analysed using Student's one-sample t-test. *P < 0.05, **P < 0.01, significant decrease compared with Control. (E) Total ALDH activity from control and GTN-treated aortas was assessed in samples containing 100 µg of aortic protein and using 5 mM propionaldehyde as substrate. Data are presented as the mean percentage of control ± SD (n = 4) and were analysed using Student's t-test for unpaired data. *P < 0.05, **P < 0.01, significant decrease compared with Control. (F) Cumulative concentration-response curves for DEA/NO in aortic rings obtained from rats treated with 0.4 mg·h−1 GTN for 48 h (n = 6).
Figure 3.
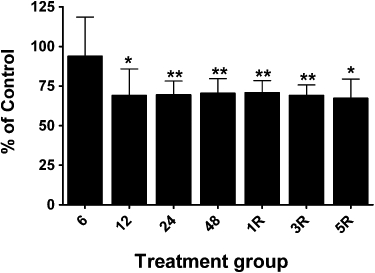
Changes in ALDH2 protein levels in femoral arteries from GTN-treated animals. Rats were treated with 0.4 mg·h−1 GTN for 6, 12, 24 or 48 h (6, 12, 24, 48) or for 48 h followed by a 1-, 3- or 5-day GTN-free period (1R, 3R, 5R). ALDH2 protein was determined by immunoblot analysis and immunoreactive bands were quantified by densitometry. ALDH2 protein levels in aortae (normalized to β-actin) from GTN-treated animals are expressed as a percentage of the levels in matching control animals. Data are presented as the mean percentage of control ± SD (n = 4) and were analysed using Student's one-sample t-test. *P < 0.05, **P < 0.01, significant decrease compared with Control.
Figure 4.
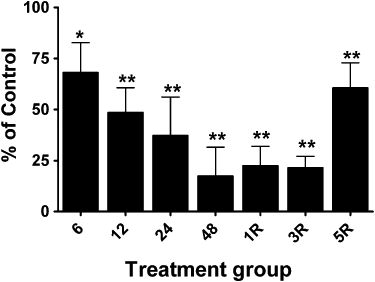
Changes in ALDH2 protein levels in vena cava from GTN-treated animals. Rats were treated with 0.4 mg·h−1 GTN for 6, 12, 24 or 48 h (6, 12, 24, 48) or for 48 h followed by a 1-, 3- or 5-day GTN-free period (1R, 3R, 5R). ALDH2 protein was determined by immunoblot analysis and immunoreactive bands were quantified by densitometry. ALDH2 protein levels in vena cava (normalized to β-actin) from GTN-treated animals are expressed as a percentage of the levels in matching control animals. Data are presented as the mean percentage of control ± SD (n = 4) and were analysed using Student's one-sample t-test. *P < 0.05, **P < 0.01, significant decrease compared with Control.
Figure 5.
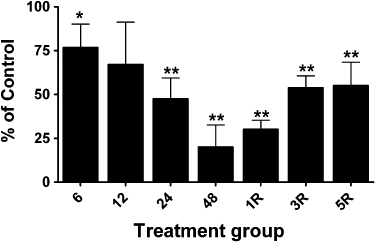
Changes in ALDH2 protein levels in femoral veins from GTN-treated animals. Rats were treated with 0.4 mg·h−1 GTN for 6, 12, 24 or 48 h (6, 12, 24, 48) or for 48 h followed by a 1-, 3- or 5-day GTN-free period (1R, 3R, 5R). ALDH2 protein was determined by immunoblot analysis and immunoreactive bands were quantified by densitometry. ALDH2 protein levels in aortae (normalized to β-actin) from GTN-treated animals are expressed as a percentage of the levels in matching control animals. Data are presented as the mean percentage of control ± SD (n = 4) and were analysed using Student's one-sample t-test. *P < 0.05, **P < 0.01, significant decrease compared with Control.
ALDH activity
ALDH2 has a low Km value for substrate relative to other ALDH isoforms and thus low and high substrate concentrations can be used to differentiate ALDH2 activity from other ALDH activities present in a particular sample. However, the ALDH2 content in aorta was not sufficient to yield reliable rates of NADH formation at a low substrate concentration (50 µM propionaldehyde). Therefore, total ALDH activity in whole cell homogenates of the rat aorta was determined at a high substrate concentration (5 mM propionaldehyde) to assess whether ALDH activity was altered during chronic GTN treatment. Furthermore, due to the low protein yields from the vena cava, femoral artery and femoral vein, total ALDH activity could not be measured accurately. In the aorta, there was a significant decrease in ALDH activity after a 12-h GTN exposure and a maximal decrease of approximately 55% after a 24-h exposure time (Figure 2E). ALDH activity remained significantly decreased after a 1- or 3-day nitrate-free period, only returning to control levels after a 5-day drug-free period.
GTN biotransformation
Consistent with previous studies, rat aortic GTN biotransformation was characterized by a highly selective formation of 1,2-GDN (1,2-GDN/1,3-GDN ratio ∼5) (Figure 6A). In aortae from GTN-tolerant animals, there was a significant decrease in GTN biotransformation, attributable to a selective decrease in 1,2-GDN formation. A nitrate-free period of 1 day after the induction of GTN-tolerance resulted in an increase in GTN biotransformation and selective 1,2-GDN formation back to control levels. Pretreatment of rat aortae with the relatively selective ALDH2 inhibitor, daidzin, did not significantly decrease GTN biotransformation, although a modest decrease in 1,2-GDN formation was observed. Daidzin mediated a similar inhibitory effect on 1,2-GDN formation under the three experimental conditions. Furthermore, daidzin-mediated inhibition of 1,2-GDN formation was markedly less than the decrease in 1,2-GDN formation observed in GTN-tolerant tissues in the absence of the inhibitor.
Figure 6.
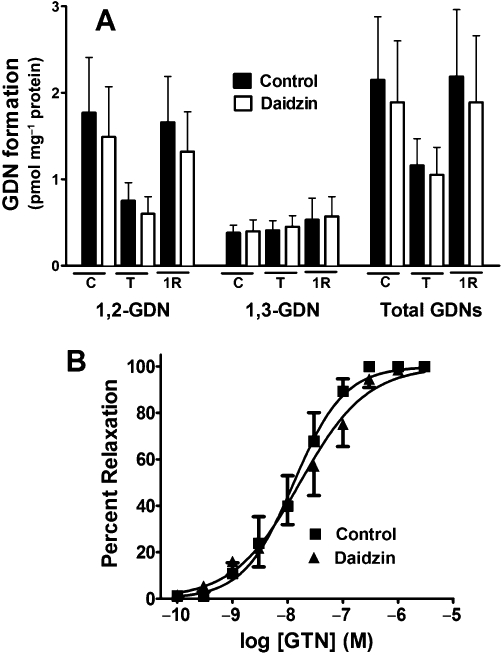
Effect of daidzin on GTN biotransformation and GTN-induced relaxation in rat aorta. (A) Segments of rat aorta prepared from sham-treated animals (C), or from animals treated with 0.4 mg·h−1 GTN for 48 h (T) or treated for 48 h followed by a 1-day GTN-free period (1R), were incubated with 2 µM GTN for 1 min in the presence or absence of the ALDH inhibitor daidzin (0.1 mM) and the formation of 1,2-GDN and 1,3-GDN quantitated. Data are presented as the mean ± SD (n = 8–10) and were analysed using two-way ANOVA. 1,2-GDN formation and total GDN formation were significantly decreased in tolerant aortae and daidzin-treated tolerant aortae compared with all other treatment groups (P < 0.05). (B) Aortic rings from control animals were contracted submaximally with phenylephrine and cumulative concentration-response curves for GTN were obtained in the presence or absence of 0.1 mM daidzin (n = 5).
Relaxation responses to GTN
No significant differences were observed in the relaxation responses of aortae from GTN-treated and sham animals for the 6 and 12 h treatment groups (Table 1A). After 24 h of GTN-treatment, there was a significant increase (approximately twofold) in the EC50 values for relaxation of aortae from GTN-treated animals compared to the untreated controls (Figure 2A), and an even greater relative increase (approximately threefold) in the EC50 values after a 48-h exposure to GTN (Figure 2B). In addition, there was a significant decrease in the maximal relaxation response to GTN at these time points. However, after a 1-, 2- or 4-day nitrate-free interval following a 48 h exposure to GTN, there were no differences in the concentration–response curves between the GTN-treated and control animals (Figure 2C, Table 1A). The induction of GTN tolerance did not significantly affect the responses of aortic rings to the NO donor DEA/NO, indicating that the GTN tolerance protocol used did not result in vascular tolerance to NO per se (Figure 2F). We also assessed the effect of 0.1 mM daidzin on GTN-induced relaxation of rat aorta and found no difference in the EC50 value or maximal relaxation response (Figure 6B).
Table 1.
EC50 and maximum relaxation values for GTN-induced relaxation in aorta (A), femoral artery (B) and femoral vein (C)
| Time point | Control EC50 (nM) | Treated EC50 (nM) | Control % relaxation | Treated % relaxation |
|---|---|---|---|---|
| A | ||||
| 6 h | 13 ± 7.2 | 15 ± 10 | 97.0 ± 4.3 | 99.5 ± 0.9 |
| 12 h | 19 ± 7.6 | 23 ± 7.1 | 99.6 ± 0.9 | 97.9 ± 3.6 |
| 24 h | 16 ± 14 | 26 ± 20* | 97.4 ± 5.1 | 89.0 ± 7.3** |
| 48 h | 25 ± 18 | 67 ± 46** | 97.2 ± 3.9 | 84.6 ± 9.6** |
| 1 day GTN-free | 15 ± 8.8 | 16 ± 5.0 | 97.5 ± 0.7 | 97.0 ± 2.6 |
| 2 day GTN-free | 21 ± 9.0 | 16 ± 7.1 | 99.1 ± 1.4 | 96.8 ± 2.6 |
| 4 day GTN-free | 27 ± 14 | 29 ± 13 | 98.7 ± 2.0 | 97.8 ± 3.8 |
| B | ||||
| 6 h | 2.4 ± 1.3 | 2.7 ± 1.2 | 98.6 ± 1.2 | 99.4 ± 1.0 |
| 12 h | 3.5 ± 1.2 | 3.1 ± 1.6 | 98.7 ± 1.1 | 98.7 ± 2.3 |
| 24 h | 1.8 ± 0.8 | 4.5 ± 1.1* | 98.0 ± 1.4 | 98.0 ± 1.4 |
| 48 h | 1.9 ± 1.1 | 6.3 ± 1.0** | 98.7 ± 1.2 | 93.2 ± 3.1** |
| 1 day GTN-free | 2.8 ± 0.9 | 3.5 ± 1.3 | 99.5 ± 1.0 | 98.9 ± 1.4 |
| 3 day GTN-free | 2.0 ± 1.2 | 2.5 ± 1.7 | 99.4 ± 1.0 | 97.2 ± 1.2 |
| 5 day GTN-free | 2.4 ± 1.8 | 3.2 ± 1.7 | 98.8 ± 1.0 | 99.3 ± 1.2 |
| C | ||||
| 6 h | 9.0 ± 1.9 | 8.7 ± 2.1 | 95.9 ± 0.6 | 96.9 ± 2.7 |
| 12 h | 6.1 ± 1.5 | 5.9 ± 1.2 | 96.0 ± 0.9 | 98.8 ± 2.0 |
| 24 h | 5.0 ± 1.0 | 11 ± 1.7* | 97.5 ± 2.6 | 94.9 ± 1.9 |
| 48 h | 5.2 ± 1.1 | 24 ± 7.0* | 96.7 ± 1.9 | 94.6 ± 0.9* |
| 1 day GTN-free | 6.4 ± 2.2 | 7.6 ± 2.3 | 99.2 ± 1.7 | 97.2 ± 1.9 |
| 3 day GTN-free | 5.9 ± 1.7 | 5.0 ± 1.9 | 95.5 ± 0.6 | 98.8 ± 2.1 |
| 5 day GTN-free | 4.9 ± 1.8 | 6.3 ± 1.3 | 98.8 ± 2.1 | 94.2 ± 1.6* |
Rats were treated with 0.4 mg·h−1 GTN (Treated) or vehicle (Control) for 6, 12, 24, and 48 h as well as 48 h GTN treatment followed by a 1 to 5 day drug-free period. Isolated rings of aorta, femoral artery and vein were prepared and cumulative concentration-response curves obtained. Values represent mean ± SD (n = 4–10).
P < 0.05 versus Control, Student's t-test for unpaired data.
P < 0.01 versus Control, Student's t-test for unpaired data.
A time-course analysis of GTN tolerance development and reversal was also conducted using isolated femoral arteries and veins from GTN-treated animals (Table 1B and C). Similar to the findings in aortic preparations, there were no differences in the EC50 values for relaxation between GTN-treated and sham animals after 6 or 12 h GTN exposure, and with both vessel types, complete reversal of tolerance occurred within 1 day, following cessation of GTN exposure. Also consistent with the findings in aorta, there was an approximate twofold increase in EC50 in both vessel types after 24 h treatment with GTN. After 48 h GTN treatment, the EC50 values for relaxation increased approximately threefold and fivefold in femoral artery and vein, respectively. The magnitudes of the rightward shifts are consistent with previous studies performed in our laboratory (Difabio et al., 2003; MacPherson et al., 2006). Hysteresis plots of the fold shift in EC50 values for relaxation as a function of ALDH2 status at the various time points in the study revealed marked hysteresis, thus highlighting the dissociation between the two variables (Figure 7). For example, examination of the hysteresis plot of ALDH2 expression in femoral vein (Figure 7E) indicates that at a number of time points, the EC50 value for relaxation was unaltered despite the fact that ALDH2 expression levels varied between 30 and 80% of control. Furthermore, an ALDH2 expression level of approximately 30% of control was associated with both a fivefold shift and no change in the EC50 value for relaxation. In contrast, the hysteresis plot of ALDH2 expression versus ALDH activity, with the exception of one data point, indicated a very good linear correlation between activity and expression (Figure 7C).
Figure 7.
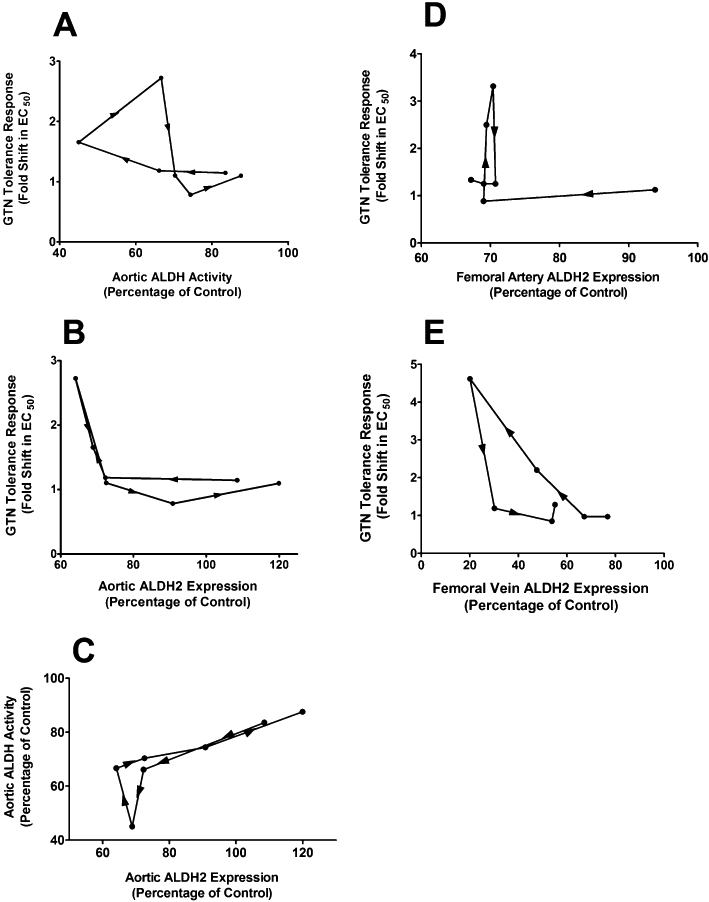
Hysteresis plots of ALDH2 status and fold shift in EC50 values for GTN-induced relaxation of various blood vessels. The fold shift in EC50 values for relaxation of aorta (A–B), femoral artery (D) and femoral vein (E) is plotted as a function of either ALDH activity (as a percentage of control) (A) or ALDH2 expression (as a percentage of control) (B–E). (C) Plot showing hysteresis of aortic ALDH activity as a function of aortic ALDH2 expression. Data are plotted as the mean percentage of control vs. mean fold shift in EC50 values. (n = 4–10).
Discussion
The role of ALDH2 in GTN bioactivation has been widely studied in animal models as well as in humans, but no study to date has examined the time-course effects of GTN exposure on ALDH2 activity and expression. Accordingly, we investigated the association between ALDH2 activity and protein expression and both the ex vivo functional responses to GTN and vascular GTN biotransformation, after various periods of chronic GTN exposure. We reasoned that if inactivation/loss of ALDH2 is the basis for GTN tolerance, then there should be an association between decreased ALDH2 activity/protein and the development of GTN tolerance. Furthermore, the decrease in ALDH2 activity and protein expression should correlate with functional responses in GTN-treated blood vessels. The major findings of the current study are that both the vascular biotransformation of GTN and the vasodilator responses to GTN during tolerance development and reversal do not correlate with GTN-induced changes in vascular ALDH2 protein expression and activity.
Although limitations in the sensitivity of the ALDH assay in aortic homogenates at low substrate concentration (selective for ALDH2) prevented us from attributing decreases in aortic ALDH activity, specifically to decreases in ALDH2 activity, the changes in aortic ALDH2 protein and total aortic ALDH activity were reasonably well correlated (Figure 2D and E). Furthermore, the hysteresis plot of aortic ALDH activity versus aortic ALDH2 protein levels did not indicate dissociation between the two variables (Figure 7C), suggesting that the decrease in total ALDH activity was primarily due to decreases in ALDH2.
As an alternative to the spectrophotometric assay of ALDH activity, we measured GDN formation in the presence and absence of the selective ALDH2 inhibitor, daidzin, in aortae from control and GTN-tolerant (48 h) animals, and from animals treated with GTN for 48 h followed by a 1-day GTN-free period (Figure 6A), and used this as a measure of ALDH2 activity. However, as only a relatively minor component of aortic GTN biotransformation was daidzin-inhibitable, this approach could not be used. Furthermore, to the extent that daidzin inhibition can be used as an index of ALDH2-mediated GTN biotransformation, it would appear that this enzyme does not contribute substantially to the overall biotransformation of GTN in rat aorta. In contrast, the flavoprotein inhibitor, diphenyleneiodonium sulphate, reduced 1,2-GDN formation from GTN by 60–75% under the same experimental conditions (McGuire et al., 1994), suggesting a dominant role for flavoproteins (e.g. the cytochrome P450-cytochrome P450 reductase system) in vascular GTN biotransformation. Consistent with the modest effect of daidzin on aortic GTN biotransformation, GTN-induced relaxation of rat aorta was unaffected at the concentration of daidzin employed (0.1 mM). Experiments were attempted using a higher daidzin concentration (0.3 mM), but this concentration of daidzin caused a 50–75% relaxation of phenylephrine-contracted tissues, indicating effects in addition to ALDH2 inhibition. Indeed, soy isoflavones such as daidzin and genestin were first characterized as phytoestrogens, and 17 β-oestradiol and selective oestrogen receptor modulators such as raloxifene, induce vascular relaxation via a non-genomic, oestrogen receptor-dependent activation of NO synthase and increased NO. The vasodilator effect of daidzin we observed is probably mediated by the same mechanism, as it was completely inhibited by pretreatment of tissues with the NO synthase inhibitor, L-NAME (Y. D'Souza and B.M. Bennet, unpubl. obs.).
In the absence of daidzin, there was a selective decrease in 1,2-GDN formation after incubation of GTN-tolerant tissues with GTN, whereas GTN biotransformation activity was completely restored following a 1-day recovery period, concomitant with the restoration of the vasodilator response to GTN (Figure 2C). This is in contrast to the ALDH2 expression (Figure 2D) and ALDH activity (Figure 2E) data, which show that after a 1-day recovery period, both expression and activity were still depressed to the same extent as observed at the 48 h time point. Together, these findings suggest dissociation between GTN biotransformation and vasodilator activity on the one hand, and changes in vascular ALDH2 protein expression and activity on the other.
The studies performed demonstrated that ALDH inactivation did not parallel the time-course for the development of GTN tolerance, but rather, that ALDH activity remained depressed long after relaxation responses to GTN had returned to control values. ALDH activity in aorta and ALDH2 protein expression in all of the blood vessels examined decreased in response to GTN treatment, with some variations in the extent of the decrease over time. After the induction of GTN tolerance, a nitrate-free interval of up to 5 days did not result in ALDH2 protein expression returning to control levels in the vena cava, femoral artery and vein, despite the fact that the functional responses to GTN returned to control values after a 1-day nitrate-free interval (Table 1). These data are inconsistent with the ALDH2 inactivation model of GTN tolerance, which would predict a close association between ALDH2 activity and protein levels and vascular responses to GTN. Rather, the data indicate that during tolerance, not only does ALDH activity and ALDH2 protein expression not correlate with the time-dependent onset of GTN tolerance and recovery from tolerance, but that vascular ALDH2 protein expression remains depressed long after the cessation of exposure to GTN. There have been other studies showing decreased ALDH2 protein expression during GTN tolerance (Hink et al., 2007; Szöcs et al., 2007; Wenzel et al., 2007), but in these studies, ALDH2 protein levels were only evaluated in GTN-tolerant blood vessels at a single time point, and were not evaluated during a nitrate-free interval following tolerance induction.
The dissociation between ALDH2 expression and GTN responsiveness is further illustrated by the plots of the EC50 values for relaxation by GTN as a function of ALDH2 status, which revealed significant hysteresis (Figure 7). At similar expression levels of ALDH2, we observed significant variations in the EC50 values for GTN-induced relaxation, and at a number of time points, the EC50 value for relaxation was unaltered despite the fact that ALDH2 expression levels varied considerably. Furthermore, we observed a twofold to threefold lower ALDH2 expression level in venous tissue compared to arterial tissue. This finding, taken with the observation of greater GTN biotransformation and cGMP accumulation in venous tissue compared to arterial tissue (Kawamoto et al., 1990), together with the well-known venoselective relaxation properties of nitrates, is further evidence of a dissociation between ALDH2 and the vasodilator actions of nitrates. However, it is entirely possible that different blood vessel types rely on different bioactivation processes, and that tolerance mechanisms could differ as well. For example, we have shown that increased phosphodiesterase 5 activity occurs in the venous, but not arterial circulation of GTN-tolerant animals, and that the phosphodiesterase inhibitor, zaprinast, selectively reverses the blunted venodilator response to GTN in GTN-tolerant animals (MacPherson et al., 2006), suggesting that increased phosphodiesterase 5 activity in the venous circulation contributes to the altered haemodynamic response to GTN following chronic GTN exposure.
The biotransformation of GTN by ALDH2 is proposed to result in ALDH2 inactivation after one catalytic cycle, due to oxidation of critical sulphhydryl groups in the active site of the enzyme (Chen et al., 2002), and this inactivation has been proposed to be the major factor in the development of GTN tolerance (Münzel et al., 2005). Thus, if continuous GTN biotransformation by ALDH2 is to occur, it would be necessary to regenerate free sulphhydryl groups in the active site. Wenzel et al. (2007) showed that dihydrolipoic acid could partially restore ALDH2 activity in GTN-treated blood vessels, but required relatively high concentrations (0.1 mM) of this reducing agent. More recently, Beretta et al. (2008b) showed that dihydrolipoic acid and dithiothreitol were able to restore ALDH2 activity. However, they concluded that these reducing agents only mediated partial reactivation of the enzyme, and concluded that significant irreversible inactivation of ALDH2 occurred during GTN tolerance. The decrease in ALDH2 protein levels shown in the current study would be consistent with a GTN-induced irreversible modification of the enzyme, provided that irreversibly inactivated enzyme is targeted for removal from the enzyme pool.
A number of studies support the notion that at least a portion of the bioactivation of GTN is mediated by ALDH2, including studies showing reduced vasodilator responses to GTN in Aldh2–/– mice (Chen et al., 2005; Wenzel et al., 2008), and in humans having the Glu504Lys ALDH2 loss of function mutation (Mackenzie et al., 2005; Li et al., 2006). Clearly, there are other mechanisms of GTN-induced vasodilatation in addition to those that may be mediated by ALDH2-mediated bioactivation, as vasodilatation still occurs in Aldh2–/– mice and in humans with the Glu504Lys mutation. Indeed, it has been suggested that high potency nitrates such as GTN and pentaerythrityl tetranitrate (PETN) are bioactivated by ALDH2, and that a low affinity pathway bioactivates both high potency nitrates and low potency nitrates such as ISDN (Daiber et al., 2004), in keeping with earlier studies suggesting low and high affinity pathways for GTN biotransformation (Bennett et al., 1989). Perhaps not so clear is whether inactivation of ALDH2 is the basis for GTN tolerance. In the study of Chen et al. (2005), in vitro exposure of aortae from Aldh2–/– mice with high concentrations of GTN did not result in further attenuation of the vasodilator response to GTN, and in aortae from GTN-tolerant guinea-pigs (Wenzl et al., 2009b), there was no further inhibitory effect of the ALDH2 inhibitor daidzin (although there was a further inhibition of vascular GTN biotransformation). However, in one study (Wenzel et al., 2008), in vivo infusion of GTN into Aldh2–/– mice did shift GTN concentration–response curves to the right, and in our own preliminary experiments in Aldh2–/– mice using an in vivo GTN tolerance protocol similar to that used in the current study, we observed a fourfold shift in the EC50 values for GTN-induced relaxation of aorta with little change in the vasodilator response to ISDN (unpublished data).
Two other aspects of GTN tolerance do not seem to be adequately explained by the ALDH2 hypothesis. Firstly, both GTN and PETN are proposed to be bioactivated by ALDH2 by the same chemical reaction pathway involving oxidation of critical SH groups in the active site, concomitant with inactivation of the enzyme (tolerance). However, PETN is a more potent vasodilator than GTN, while at the same time being a poorer substrate and causing less enzyme inactivation than GTN. Furthermore, unlike GTN, tolerance to PETN does not appear to occur, whereas GTN-tolerant animals exhibit reduced vasodilator responses to both GTN and PETN (Fink and Bassenge, 1997; Daiber et al., 2004; Griesberger et al., 2011; Oelze et al., 2011). Thus, two structurally similar compounds containing the same functional groups appear to interact with the enzyme in different ways, inconsistent with a proposed common mechanism of action. Secondly, the haemodynamic and vasodilator effects of GTN in both the arterial and venous circulation have been shown to be reduced in subjects pretreated with ISDN (Schelling and Lasagna, 1967; Zelis and Mason, 1975; Manyari et al., 1985). These findings of cross-tolerance between ISDN and GTN are inconsistent with the ALDH2 hypothesis, in which low potency nitrates such as ISDN are not considered to be ALDH2 substrates (or inactivators), and therefore ought not to affect the efficacy of GTN, especially at the low concentrations of the drug associated with clinical use.
During continuous GTN exposure, ALDH2 is thought to be inactivated either directly by reacting with GTN, or indirectly through the GTN-mediated generation of reactive oxygen species (Daiber et al., 2008). The down-regulation of other thiol-dependent enzymes during chronic GTN treatment that may mediate GTN biotransformation has not been reported, but would be of obvious interest. The biotransformation data obtained in the current study (Figure 6A) indicates that GTN biotransformation enzymes other than ALDH2 are inhibited during chronic GTN exposure. Nevertheless, the down-regulation of ALDH2 could be of importance because of the role of ALDH2 in the detoxification of a number of toxic aldehydes. Studies have shown that individuals with the ALDH2*2 polymorphism (Glu504Lys), and the corresponding decrease in ALDH2 activity, have an increased risk of colorectal cancer (Gao et al., 2008) as well as an increased risk of oesophageal and liver cancer (Seitz and Meier, 2007). Furthermore, Ohsawa et al. (2008) have shown that toxic aldehydes accumulate in ALDH2 transgenic mice lacking ALDH2 activity, resulting in a shorter lifespan, cognitive impairment and increased age-dependent neurodegeneration. Other studies have shown that ALDH2 deficiency renders mice more susceptible to exogenously triggered oxidative stress; Aldh2−/− mice were more susceptible to nitroglycerin-, acetaldehyde- and doxorubicin-induced cardiovascular damage (Wenzel et al., 2008). Thus, the inactivation of ALDH2 during GTN tolerance could be of pathological relevance, and warrants further investigation.
In summary, we have shown that during the development of GTN tolerance and during tolerance reversal, the changes in GTN-mediated vasodilator responses of several types of blood vessels and in vascular GTN biotransformation do not correlate with changes in ALDH activity or ALDH2 protein expression, suggesting that factors other than impaired ALDH2-mediated GTN bioactivation underlie nitrate tolerance.
Acknowledgments
This work was supported by grant from the Canadian Institutes of Health Research (MOP 81175). The authors wish to thank Mrs Diane Anderson for technical assistance.
Glossary
Abbreviations
- ALDH
aldehyde dehydrogenase
- DEA/NO
1,1-diethyl-2-hydroxy-2-nitrosohydrazine
- GTN
glyceryl trinitrate
- GDN
glyceryl dinitrate
- sGC
soluble GC
- PETN
pentaerythrityl tetranitrate
Conflicts of interest
None.
References
- Bennett BM, Leitman DC, Schröder H, Kawamoto JH, Nakatsu K, Murad F. Relationship between biotransformation of glyceryl trinitrate and cyclic GMP accumulation in various cultured cell lines. J Pharmacol Exp Ther. 1989;250:316–323. [PubMed] [Google Scholar]
- Bennett BM, McDonald BJ, St James MJ. Hepatic cytochrome P450-mediated activation of rat aortic guanylyl cyclase by glyceryl trinitrate. J Pharmacol Exp Ther. 1992;261:716–723. [PubMed] [Google Scholar]
- Beretta M, Gruber K, Kollau A, Russwurm M, Koesling D, Goessler W, et al. Bioactivation of nitroglycerin by purified mitochondrial and cytosolic aldehyde dehydrogenases. J Biol Chem. 2008a;283:17873–17880. doi: 10.1074/jbc.M801182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beretta M, Sottler A, Schmidt K, Mayer B, Gorren AC. Partially irreversible inactivation of mitochondrial aldehyde dehydrogenase by nitroglycerin. J Biol Chem. 2008b;283:30735–30744. doi: 10.1074/jbc.M804001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien JF, McLaughlin BE, Kobus SM, Kawamoto JH, Nakatsu K, Marks GS. Mechanism of glyceryl trinitrate-induced vasodilation. I. Relationship between drug biotransformation, tissue cyclic GMP elevation and relaxation of rabbit aorta. J Pharmacol Exp Ther. 1988;244:322–327. [PubMed] [Google Scholar]
- Chen Z, Zhang J, Stamler JS. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2002;99:8306–8311. doi: 10.1073/pnas.122225199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhang J, Foster MW, Stamler JS. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, Oelze M, Coldewey M, Bachschmid M, Wenzel P, Sydow K, et al. Oxidative stress and mitochondrial aldehyde dehydrogenase activity: a comparison of pentaerythritol tetranitrate with other organic nitrates. Mol Pharmacol. 2004;66:1372–1382. doi: 10.1124/mol.104.002600. [DOI] [PubMed] [Google Scholar]
- Daiber A, Wenzel P, Oelze M, Münzel T. New insights into bioactivation of organic nitrates, nitrate tolerance and cross-tolerance. Clin Res Cardiol. 2008;97:12–20. doi: 10.1007/s00392-007-0588-7. [DOI] [PubMed] [Google Scholar]
- De la Lande IS, Stepien JM, Philpott AC, Hughes PA, Stafford I, Horowitz JD. Aldehyde dehydrogenase, nitric oxide synthase and superoxide in ex vivo nitrate tolerance in rat aorta. Eur J Pharmacol. 2004;496:141–149. doi: 10.1016/j.ejphar.2004.06.010. [DOI] [PubMed] [Google Scholar]
- DiFabio J, Ji Y, Vasiliou V, Thatcher RJ, Bennett BM. Role of mitochondrial aldehyde dehydrogenase in nitrate tolerance. Mol Pharmacol. 2003;64:1109–1116. doi: 10.1124/mol.64.5.1109. [DOI] [PubMed] [Google Scholar]
- Fink B, Bassenge E. Unspected, tolerance-devoid vasomotor and platelet actions of pentaerythrityl tetranitrate. J Cardiovasc Pharmacol. 1997;30:831–836. doi: 10.1097/00005344-199712000-00020. [DOI] [PubMed] [Google Scholar]
- Fung HL. Biochemical mechanism of nitroglycerin action and tolerance: is this old mystery solved? Annu Rev Pharmacol Toxicol. 2004;44:67–85. doi: 10.1146/annurev.pharmtox.44.101802.121646. [DOI] [PubMed] [Google Scholar]
- Gao CM, Takezaki T, Wu JZ, Zhang XM, Cao HX, Ding JH, et al. Polymorphisms of alcohol dehydrogenase 2 and aldehyde dehydrogenase 2 and colorectal cancer risk in Chinese males. World J Gastroenterol. 2008;14:5078–5083. doi: 10.3748/wjg.14.5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesberger M, Kollau A, Wölkart G, Wenzl MV, Beretta M, Russwurm M, et al. Bioactivation of pentaerythrityl tetranitrate by mitochondrial aldehyde dehydrogenase. Mol Pharmacol. 2011;79:541–548. doi: 10.1124/mol.110.069138. [DOI] [PubMed] [Google Scholar]
- Hink U, Daiber A, Kayhan N, Trischler J, Kraatz C, Oelze M, et al. Oxidative inhibition of the mitochondrial aldehyde dehydrogenase promotes nitroglycerin tolerance in human blood vessels. J Am Coll Cardiol. 2007;50:2226–2232. doi: 10.1016/j.jacc.2007.08.031. [DOI] [PubMed] [Google Scholar]
- Ji Y, Anderson DJ, Bennett BM. Role of microsomal glutathione transferase 1 in the mechanism-based biotransformation of glyceryl trinitrate in LLC-PK1 cells. Biochem Pharmacol. 2009;77:1702–1708. doi: 10.1016/j.bcp.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Kawamoto JH, McLaughlin BE, Brien JF, Marks GS, Nakatsu K. Biotransformation of glyceryl trinitrate and elevation of cyclic GMP precede glyceryl trinitrate-induced vasodilation. J Cardiovasc Pharmacol. 1990;15:714–719. doi: 10.1097/00005344-199005000-00005. [DOI] [PubMed] [Google Scholar]
- Kleschyov AL, Oelze M, Daiber A, Huang Y, Mollnau H, Schulz E, et al. Does nitric oxide mediate the vasodilator activity of nitroglycerin? Circ Res. 2003;93:104–112. doi: 10.1161/01.RES.0000100067.62876.50. [DOI] [PubMed] [Google Scholar]
- Kollau A, Hofer A, Russwurm M, Koesling D, Keung WM, Schmidt K, et al. Contribution of aldehyde dehydrogenase to mitochondrial bioactivation of nitroglycerin: evidence for the activation of purified soluble guanylate cyclase through direct formation of nitric oxide. Biochem J. 2005;385:769–777. doi: 10.1042/BJ20041354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang D, Jin W, Shao C, Yan P, Xu C, et al. Mitochondrial aldehyde dehydrogenase-2 (ALDH2) Glu504Lys polymorphism contributes to the variation in efficacy of sublingual nitroglycerin. J Clin Invest. 2006;116:506–511. doi: 10.1172/JCI26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis CW, Brien JF. Inhibition of hepatic aldehyde dehydrogenases in the rat by calcium carbimide (calcium cyanamide) Can J Physiol Pharmacol. 1983;61:1025–1034. doi: 10.1139/y83-153. [DOI] [PubMed] [Google Scholar]
- Mackenzie IS, Maki-Petaja KM, McEniery CM, Bao YP, Wallace SM, Cheriyan J, et al. Aldehyde dehydrogenase 2 plays a role in the bioactivation of nitroglycerin in humans. Arterioscler Thromb Vasc Biol. 2005;25:1891–1895. doi: 10.1161/01.ATV.0000179599.71086.89. [DOI] [PubMed] [Google Scholar]
- MacPherson JD, Gillespie T, Maurice DH, Bennett BM. Inhibition of phosphodiesterase type 5 selectively reverses nitrate tolerance in venous circulation. J Pharmacol Exp Ther. 2006;317:188–195. doi: 10.1124/jpet.105.094763. [DOI] [PubMed] [Google Scholar]
- Manyari MD, Smith ER, Spragg J. Isosorbide dinitrate and glyceryl trinitrate: demonstration of cross tolerance in the capacitance vessels. Am J Cardiol. 1985;55:927–931. doi: 10.1016/0002-9149(85)90719-2. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Bennett BM. Cytochrome P-450 mediated biotransformation of organic nitrates. Can J Physiol Pharmacol. 1990;68:1552–1557. doi: 10.1139/y90-236. [DOI] [PubMed] [Google Scholar]
- McGuire JJ, Anderson DJ, Bennett BM. Inhibition of the biotransformation and pharmacological actions of glyceryl trinitrate by the flavoprotein inhibitor, diphenyleneiodonium sulfate. J Pharmacol Exp Ther. 1994;271:708–714. [PubMed] [Google Scholar]
- McGuire JJ, Anderson DJ, McDonald BJ, Narayanasami R, Bennett BM. Inhibition of NADPH-cytochrome P450 reductase and glyceryl trinitrate biotransformation by diphenyleneiodonium sulfate. Biochem Pharmacol. 1998;56:881–893. doi: 10.1016/s0006-2952(98)00216-0. [DOI] [PubMed] [Google Scholar]
- Miller MR, Grant S, Wadsworth RM. Selective arterial dilatation by glyceryl trinitrate is not associated with nitric oxide formation in vitro. J Vasc Res. 2008;45:375–385. doi: 10.1159/000121407. [DOI] [PubMed] [Google Scholar]
- Münzel T, Daiber A, Mülsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- Nigam R, Anderson DJ, Bennett BM. Isoform-specific biotransformation of glyceryl trinitrate by rat aortic glutathione S-transferases. J Pharmacol Exp Ther. 1996;279:1527–1534. [PubMed] [Google Scholar]
- Nuñez C, Esplugues JV, Rocha M, Bosca I, Ibiza S, Herance JR, et al. Complex I dysfunction and tolerance to nitroglycerin: an approach based on mitochondrial-targeted antioxidants. Circ Res. 2006;99:1067–1075. doi: 10.1161/01.RES.0000250430.62775.99. [DOI] [PubMed] [Google Scholar]
- Oelze M, Knorr M, Schell R, Kamuf J, Pautz A, Art J, et al. Regulation of human mitochondrial aldehyde dehydrogenase (ALDH-2) activity by electrophiles in vitro. J Biol Chem. 2011;286:8843–8900. doi: 10.1074/jbc.M110.190017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa I, Nishimaki K, Murakami Y, Suzuki Y, Ishikawa M, Ohta S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J Neurosci. 2008;28:6239–6249. doi: 10.1523/JNEUROSCI.4956-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratz JD, Fisher AB, Rees-Milton KJ, Adams MA, Bennett BM. Endothelin receptor antagonism does not prevent the development of in vivo glyceryl trinitrate tolerance in the rat. J Pharmacol Exp Ther. 2000a;285:578–585. [PubMed] [Google Scholar]
- Ratz JD, McGuire JJ, Anderson DJ, Bennett BM. Effects of the flavoprotein inhibitor, diphenyleneiodonium sulfate, on ex vivo organic nitrate tolerance in the rat. J Pharmacol Exp Ther. 2000b;293:569–577. [PubMed] [Google Scholar]
- Ratz JD, Adams MA, Bennett BM. Effect of in vivo nitrate tolerance on the hypersensitivity to NO-donors after NO-synthase blockade. Can J Physiol Pharmacol. 2002;80:1106–1118. doi: 10.1139/y02-141. [DOI] [PubMed] [Google Scholar]
- Sage PR, de la Lande IS, Stafford I, Bennett CL, Phillipov G, Stubberdfield J, et al. Nitroglycerin tolerance in human vessels. Evidence for impaired nitroglycerin bioconversion. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- Schelling JL, Lasagna L. A study of cross-tolerance to circulatory effects of organic nitrates. Clin Pharmacol Ther. 1967;8:256–260. doi: 10.1002/cpt196782256. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Meier P. The role of acetaldehyde in upper digestive tract cancer in alcoholics. Transl Res. 2007;149:293–297. doi: 10.1016/j.trsl.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Slack CJ, McLaughlin BE, Brien JF, Marks GS, Nakatsu K. Biotransformation of glyceryl trinitrate and isosorbide dinitrate in vascular smooth muscle made tolerant to organic nitrates. Can J Physiol Pharmacol. 1989;67:1381–1385. doi: 10.1139/y89-221. [DOI] [PubMed] [Google Scholar]
- Sydow K, Daiber A, Oezle M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113:482–489. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szöcs K, Lassègue B, Wenzel P, Wendt M, Daiber A, Oelze M, et al. Increased superoxide production in nitrate tolerance is associated with NAD(P)H oxidase and aldehyde dehydrogenase 2 downregulation. J Mol Cell Cardiol. 2007;42:1111–1118. doi: 10.1016/j.yjmcc.2007.03.904. [DOI] [PubMed] [Google Scholar]
- Thatcher GRJ, Nicolescu AC, Bennett BM, Toader V. Nitrates and NO release: contemporary aspects in biological and medicinal chemistry. Free Radic Biol Med. 2004;37:1122–1143. doi: 10.1016/j.freeradbiomed.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Tottmar SOC, Pettersson H, Kiessling K-H. The subcellular distribution and properties of aldehyde dehydrogenases in rat liver. Biochem J. 1973;135:577–586. doi: 10.1042/bj1350577a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida S, Maki J, Sato K. Purification and characterization of glutathione transferases with an activity towards nitroglycerin from human aorta and heart: multiplicity of the human class mu forms. J Biol Chem. 1990;265:7150–7157. [PubMed] [Google Scholar]
- Vasilou V, Pappa A, Petersen DR. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem Biol Interact. 2000;129:1–19. doi: 10.1016/s0009-2797(00)00211-8. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Hink U, Oelze M, Schuppan S, Schaeuble K, Schildknecht S, et al. Role of reduced lipoic acid in the redox regulation of mitochondrial aldehyde dehydrogenase (ALDH-2) activity: implications for mitochondrial oxidative stress and nitrate tolerance. J Biol Chem. 2007;282:792–799. doi: 10.1074/jbc.M606477200. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Muller J, Zurmeyer S, Schuhmacher S, Schulz E, Oelze M, et al. ALDH-2 deficiency increases cardiovascular oxidative stress-evidence for indirect antioxidative properties. Biochem Biophys Res Commun. 2008;367:137–143. doi: 10.1016/j.bbrc.2007.12.089. [DOI] [PubMed] [Google Scholar]
- Wenzl MV, Beretta M, Gorren ACF, Zeller A, Baral PK, Gruber K, et al. Role of the general base Glu-268 in nitroglycerin bioactivation and superoxide formation by aldehyde dehydrogenase-2. J Biol Chem. 2009a;284:19878–19886. doi: 10.1074/jbc.M109.005652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzl MV, Wölkart G, Stessel H, Beretta M, Schmidt K, Mayer B. Different effects of ascorbate deprivation and classical vascular nitrate tolerance on aldehyde dehydrogenase-catalysed bioactivation of nitroglycerin. Br J Pharmacol. 2009b;156:1248–1255. doi: 10.1111/j.1476-5381.2009.00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelis R, Mason DT. Isosorbide dinitrate. Effect on the vasodilator response to nitroglycerin. J Am Med Assoc. 1975;234:166–170. doi: 10.1001/jama.234.2.166. [DOI] [PubMed] [Google Scholar]