

Abstract
Suppression therapy is a treatment strategy for genetic diseases caused by nonsense mutations. This therapeutic approach utilizes pharmacological agents that suppress translation termination at in-frame premature termination codons (PTCs) to restore translation of a full-length, functional polypeptide. The efficiency of various classes of compounds to suppress PTCs in mammalian cells is discussed along with the current limitations of this therapy. We also elaborate on approaches to improve the efficiency of suppression that include methods to enhance the effectiveness of current suppression drugs, and the design or discovery of new, more effective suppression agents. Finally, we discuss the role of nonsense-mediated mRNA decay (NMD) in limiting the effectiveness of suppression therapy, and describe tactics that may allow the efficiency of NMD to be modulated in order to enhance suppression therapy.
Keywords: Suppression therapy, nonsense mutations, premature termination codon, PTC, translation termination, suppression agents, nonsense-mediated mRNA decay, NMD
Introduction
Premature termination codons (PTCs) can arise from various types of mutations in germ or somatic cells. Genetic lesions that result in a PTC include: 1) single base pair substitutions that change a sense codon to an in-frame PTC, commonly known as nonsense mutations; 2) insertion or deletion mutations that alter the ribosomal reading frame, causing translating ribosomes to encounter a PTC; 3) an insertion mutation that maintains the proper distal reading frame but introduces an in-frame PTC; 4) mutations that lead to mRNA splicing defects that cause retention of an intron that alters the reading frame, leading translating ribosomes to encounter a PTC; and 5) mRNA splicing defects that cause retention of an intron that contains an in-frame PTC but does not alter the distal ribosomal reading frame. Overall, these various classes of PTCs are associated with one-third of all genetic disorders, including many types of cancer 1.
The introduction of a PTC can have two important consequences on gene expression. First, a PTC will terminate mRNA translation prior to completion of a full-length polypeptide, leading to production of truncated proteins that are often non-functional and/or unstable. In addition, PTC-containing mRNAs are also frequently unstable, resulting in a severe reduction in steady-state mRNA abundance. Together, these two PTC-induced events reduce the level of functional protein produced to such an extent that a severe disease state results. For the purpose of this review, we will limit our focus to mutations that introduce PTCs that retain the proper distal reading frame. In recent years, a novel therapeutic approach called suppression therapy has been developed that utilizes low molecular weight compounds to induce the translation machinery to recode a PTC into a sense codon. Suppression of translation termination at a PTC allows translation elongation to continue in the correct reading frame so that synthesis of a full-length polypeptide can be completed and protein function can be restored.
Mechanism of premature termination codon suppression
The recognition of stop codons greatly differs from the recognition of sense codons. During translation elongation, the movement of a sense codon into the ribosomal A site is followed by sampling of aminoacyl tRNAs until a cognate aminoacyl tRNA binds. The term “cognate” refers to a tRNA whose anticodon can base pair with all three nucleotides of the A site codon. Following a proofreading step to confirm the proper codon-anticodon interaction, the cognate aminoacyl tRNA becomes accommodated into the A site 2. Peptide bond formation then occurs, resulting in the addition of the amino acid carried by the cognate aminoacyl tRNA onto the carboxy-terminus of the growing polypeptide chain. After the peptidyl-tRNA is translocated back into the ribosomal P site, aminoacyl tRNA selection continues at the next codon. The three stop codons (UAA, UAG, and UGA) are not decoded by aminoacyl tRNAs, but rather, are recognized by proteins called eukaryotic release factors (eRFs) (Figure 1). There are two release factors in eukaryotes, eRF1 and eRF3, which together function as a termination complex 3. eRF1, which has a three-dimensional structure that resembles the size and shape of an aminoacyl tRNA, recognizes and binds to all three stop codons in the ribosomal A site and mediates release of the nascent polypeptide from the ribosome 4. eRF3 is a GTPase that assists eRF1 in both stop codon recognition and polypeptide release 5,6.
Figure 1. Translation termination.

During translation termination, eRF1 and eRF3 bind the pre-termination complex with a stop codon located in the ribosomal A site. eRF1 mediates the initial recognition of the stop codon. GTP hydrolysis by eRF3 finalizes stop codon recognition and facilitates polypeptide chain release.
When a stop codon enters the ribosomal A site, the sampling process is initiated just as it does at a sense codon. Near-cognate aminoacyl tRNAs with anticodons that are complementary to two of the three nucleotides of a stop codon can compete with the release factors for A site binding. Normally, stop codon recognition by the eRF1/3 complex efficiently out-competes near-cognate aminoacyl tRNAs and efficient polypeptide chain release occurs. On occasion, aminoacyl tRNAs that are near-cognate to a stop codon become accommodated in the ribosomal A site and their amino acid is incorporated into the polypeptide 7 (Figure 2). This process recodes a stop codon into a sense codon. This recoding or “readthrough” event suppresses translation termination and allows translation elongation to continue in the correct reading frame until the normal stop codon is encountered. Under basal conditions, the recoding of a PTC into a sense codon occurs in less than 1% of translation events 8,9, while suppression of a normal stop codon occurs at a frequency of <0.1% 10,11.
Figure 2. Suppression of translation termination.

During the suppression of a premature stop codon, a near-cognate aminoacyl tRNA pairs at two of the three bases of the stop codon. The amino acid carried by the near-cognate tRNA is added to the polypeptide chain and translation resumes in the proper reading frame until the natural stop codon is reached.
The goal of suppression therapy is to enhance the ability of near-cognate aminoacyl tRNAs to compete with the release factor complex for binding PTCs in the ribosomal A site. By increasing the frequency that PTCs are recoded into sense codons, enough full-length, functional protein may be restored to provide a therapeutic benefit to patients that carry PTCs. However, one concern raised about suppression therapy is its effect on global translation efficiency. For example, does increasing suppression at PTCs also increase suppression at normal stop codons located at the end of open reading frames (ORFs)? If so, readthrough of normal stop codons could be predicted to increase the expression of misfolded proteins with C-terminal extensions due to continued translation into the 3′ untranslated region (UTR) of an mRNA. Subsequently, the presence of a large increase in misfolded proteins would be expected to induce an unfolded protein response. Analysis of the inducible form of Hsp70, which is elevated when unfolded proteins are present such as during heat shock conditions, revealed only a slight increase in Hsp70 abundance at high suppression drug concentrations 12. In addition, investigations of suppression agents in mammalian cell culture indicate that overall translation rates are unaffected by these compounds at doses that induce PTC readthrough 13,14. From these results, it appears that PTC suppression does not cause large effects on either global translation or normal stop codon recognition.
These observations indicate that suppression agents are more effective in suppressing termination at PTCs than natural stop codons, suggesting that there are differences in termination at these two types of stop codons. Consistent with this hypothesis, ribosomal toe-printing experiments found that the toe-print observed at a PTC is much more pronounced than the toe-print at a normal stop codon 15. This indicates that a much longer ribosomal pause occurs at PTCs than at normal stop codons, suggesting that the kinetics of translation termination are less efficient at PTCs than at natural stop codons. It has been shown that the eRF3 release factor normally interacts with poly(A) binding protein (PABP) associated with the poly(A) tail at the 3′ end of mRNAs, which increases the efficiency of translation termination 16,17 (Figure 3). At natural stop codons located at the end of ORFs, the release factors are placed in close proximity to PABP, which facilitates this interaction. In contrast, the spatial distance between the PTC-bound release factors and PABP diminishes the interaction between eRF3 and PABP, rendering the termination reaction less efficient 18. Ribosomal pausing has been shown to induce many forms of ribosomal recoding including frameshifting, ribosome hopping, selenocysteine incorporation, etc. 19. Similarly, it is thought that the inefficient kinetics of termination observed at PTCs makes them more susceptible to suppression therapy than normal stop codons.
Figure 3. Comparison of translation termination at natural stop codons vs. premature stop codons.

A) Termination at natural stop codons. Termination efficiency at natural stop codons located near the 3′ end of mRNAs is enhanced by interactions between eRF3 and PABP. B) Termination at premature stop codons. eRF3 and PABP cannot interact efficiently because of the spatial distance between premature termination codons and the poly (A) tail of the mRNA where PABP binds, thus reducing the efficiency of termination. The absence of this interaction is thought to result in ribosome pausing at the premature stop codon, making them more susceptible to suppression.
Other factors are also likely to prevent the suppression of a natural stop codon and translation into the 3′ UTR of an mRNA. First, the stop codon and nucleotide sequence surrounding a stop codon greatly influence the suppression level 8,9,14,20. Sequence analysis of mammalian cDNAs showed the most efficient termination sequences that are least susceptible to suppression are frequently found at the end of ORFs of highly expressed genes 10. In addition, multiple in-frame stop codons are frequently found at the end of many ORFs, which significantly reduces the possibility that translation elongation can continue into the 3′ UTR 21. Finally, mRNA turnover pathways have been identified that target mRNAs for degradation when translation continues into the 3′ UTR. These RNA turnover pathways include nonstop mRNA decay, which degrades mRNAs that do not contain any stop codons such that translation continues to the end of the poly(A) tail 22; and ribosome-extension mediated RNA decay, that degrades mRNAs when translation continues into the 3′ UTR until another stop codon is encountered in the 3′ UTR 23. While natural substrates for these mRNA degradation pathways could be rare or may occur in a tissue-specific manner, these pathways also likely function as surveillance mechanisms to eliminate mRNAs that encode aberrant proteins.
Various amino acids can be incorporated into a polypeptide at the site of a PTC. The amino acids inserted in place of a PTC during suppression can be donated by various near-cognate aminoacyl tRNAs that base pair with two nucleotides in any position of the stop codon (with the 1st and 2nd nucleotides, the 2nd and 3rd nucleotides, or the 1st and 3rd nucleotides) 7. Many factors probably influence the frequency that a particular amino acid is inserted in place of a PTC during suppression, including the nucleotide sequence of the PTC and the abundance of the various near-cognate aminoacyl tRNAs. Unless a PTC is located at a residue where a specific amino acid is needed for protein function, such as in a catalytic pocket, PTC suppression would be expected to restore at least a fraction of functional protein that might provide a therapeutic benefit for many disorders.
Drugs that suppress disease-causing premature termination codons
Numerous low molecular weight compounds have been shown to increase the frequency that termination is suppressed at PTCs. The best characterized of these drugs are the aminoglycosides 24. Aminoglycosides generally consist of two to three aminosugars joined to a 2-deoxystreptamine ring by glycosidic linkages. Aminoglycosides have traditionally been used as antibiotics, where they bind to a region of the 16S ribosomal RNA in the bacterial ribosome called the decoding center 25. This leads to an inhibition of translation initiation at high doses and a reduction of translational fidelity at low doses. The decoding center normally carries out a proofreading function that monitors base pairing between codons and anticodons in the ribosomal A site to ensure that a cognate aminoacyl tRNA is accommodated in the A site. By binding to the decoding center, aminoglycosides are able to reduce the proofreading ability of the ribosome and increase the misincorporation of near-cognate aminoacyl tRNAs into the ribosomal A site at both sense codons and stop codons, resulting in translational misreading and inhibition of protein synthesis. The decoding center is generally well-conserved between prokaryotes and eukaryotes; however, differences at two nucleotides prevent aminoglycosides from binding as tightly to the eukaryotic ribosome 26. The reduced binding affinity of aminoglycosides for the eukaryotic decoding center allows their use as antibacterial agents without blocking protein synthesis in eukaryotes. Only a subset of aminoglycosides has been shown to bind weakly to the eukaryotic ribosome. Studies in eukaryotic cells have found that aminoglycosides that bind to the eukaryotic ribosome do not appear to induce significant misreading at sense codons, but can induce low levels of misreading at PTCs 27. Various aminoglycosides, including gentamicin, amikacin, paromomycin, G418 (geneticin), lividomycin, tobramycin, and streptomycin have been shown to suppress disease-causing PTCs in mammalian cells and partially restore protein function to various extents for more than twenty different disease models in vitro, and eight different disease models in vivo (for reviews see 28-30). Several pilot clinical trials with patients carrying nonsense mutations with cystic fibrosis 31-34, Duchenne muscular dystrophy 35,36, hemophilia A and B 37, and Hailey-Hailey disease 38 have shown the partial restoration of full-length, functional protein to a variable extent when administered gentamicin.
While these results are promising, there are several obstacles that must be overcome before aminoglycosides can be used long-term in the suppression of nonsense mutations. First, the efficiency of suppressing PTCs is greatly influenced by the identity of the stop codon and the surrounding mRNA sequence 9,39. Various aminoglycosides show different abilities to suppress a PTC depending upon the sequence context. This suggests that screening compounds to identify those that best suppress a particular PTC in its natural sequence context is needed. Second, the use of aminoglycosides long-term is limited due to side effects that result in nephrotoxicity and ototoxicity.
One way aminoglycosides enter cells is through their interaction with megalin, a multi-ligand, endocytic receptor that is particularly enriched in the proximal tubules of the kidney and hair cells of the inner ear 40. Three distinct mechanisms have been proposed to explain the basis of aminoglycoside toxicity. First, endocytosed aminoglycosides become positively charged in the low pH environment of the lysosomal compartment and bind to the negatively charged phospholipids in the lysosomal membrane 41. This interferes with enzymatic reactions on these membranes such as phospholipase signaling, which induces pathological effects such as phospholipidosis. Second, the charged nature of aminoglycosides leads to the formation of reactive oxygen species and oxidative damage 42. Finally, aminoglycosides may induce mitochondrial dysfunction due to similarities between bacterial and mitochondrial ribosomes 43. This is a particular problem with individuals carrying specific polymorphisms in their mitochondrial ribosomal RNA that increase aminoglycoside binding to the mitochondrial ribosome and induce translational misreading or arrest.
Several approaches to reduce aminoglycoside toxicity while maintaining or improving their ability to suppress PTCs are being explored. First, co-administration of antioxidants that scavenge free radicals has been shown to protect cochlear hair cells from aminoglycoside-induced damage and may also aid in protection against nephrotoxicity 44. Another approach to protect against aminoglycoside-induced nephrotoxicity is to co-administer aminoglycosides with poly-L-aspartate or daptomycin 45,46. Both molecules sequester aminoglycosides away from lysosomal membrane phospholipids. In addition, by preventing the accumulation of aminoglycosides within the lysosomal membrane, poly-L-aspartate increases the cytoplasmic aminoglycoside concentration and enhances nonsense suppression. A recent study using a cystic fibrosis mouse model that carried a human CFTR transgene with a nonsense mutation showed that co-administration of poly-L-aspartate with gentamicin restored a higher level of functional CFTR protein than administration of gentamicin alone 47. In addition, co-administration of poly-L-aspartate prolonged the therapeutic effect of suppression therapy after the suppression drug was withdrawn. These results indicate that in addition to protecting against aminoglycoside-induced nephrotoxicity, poly-L-aspartate can also enhance the efficiency of aminoglycosides in PTC suppression.
Another approach that may reduce aminoglycoside toxicity while enhancing suppression therapy is to encapsulate aminoglycosides into liposomes 48,49. It was found that much of aminoglycoside toxicity associated with its use as an antibiotic could be circumvented by encapsulation in liposomes since this alters the route of aminoglycoside clearance from the kidneys to the liver. Current clinical protocols for aminoglycoside administration monitor serum peak and trough concentrations to maximize their antibiotic effects and minimize their toxic side effects. Liposome encapsulation of aminoglycosides has been shown to improve aminoglycoside pharmacokinetics by maintaining higher aminoglycoside serum levels for longer periods of time, which may be a more effective pharmacokinetic profile for the administration of aminoglycosides as suppression agents.
Gentamicin is the most commonly used aminoglycoside for suppression therapy. However, gentamicin preparations contain a mixture of related structures (congeners) that result from its synthesis as a natural product by Micromonospora species. Some of these congeners may increase potential toxicity and hinder its effectiveness in suppression therapy. During gentamicin synthesis, a mixture of five isoforms (C1, C1a, C2, C2a, and C2b) is generated in varying proportions (Figure 4). It has been shown that the various congeners have significantly different abilities to induce toxicity and to suppress PTCs. Such variability in different gentamicin preparations could potentially affect the efficiency of suppression therapy and the side effects observed. It was previously shown that the C2 isoform exhibits significantly less toxicity than the other isoforms in gentamicin preparations 50 and also efficiently suppresses PTCs (our unpublished results). This suggests that purification of the C2 isoform of gentamicin may significantly reduce toxicity while enhancing PTC suppression.
Figure 4. Structures of gentamicin congeners.

Since a significant aspect of aminoglycoside toxicity is due to off-target effects that are unrelated to their ability to suppress PTCs, attempts are also being made to separate specific structural components of aminoglycosides that facilitate nonsense suppression from other features responsible for off-target side effects. In so doing, novel aminoglycoside derivatives may be designed that show enhanced PTC suppression with reduced toxicity. In addition, current commercial aminoglycosides have been selected for clinical use based on their antibacterial properties. Since the similarity between the bacterial and mitochondrial translation machineries can contribute to one aspect of aminoglycoside toxicity, antibacterial properties of aminoglycosides are undesirable and unnecessary for their role in PTC suppression 43. Structural studies have recently allowed a better visualization of the differences in the aminoglycoside binding pocket between eukaryotes and prokaryotes, which may assist in the design of novel aminoglycoside derivatives that are specific for the eukaryotic ribosome for PTC suppression, while also reducing any potential off-target effects that induce toxicity 51.
Several novel aminoglycoside derivatives have been generated based on the logic outlined above and tested in mammalian cells (Figure 5). These include NB30, a derivative of paromomycin 52; NB54, which combines components of paromomycin and amikacin 53; and NB84, which is composed of structural elements from paromomycin, amikacin, and G418 54. These novel compounds show more than a 10-fold reduction in cellular toxicity compared to the classical aminoglycosides 53,54. In addition, each of the compounds was found to restore a significant amount of functional protein in mammalian cells carrying PTCs related to Usher syndrome 55, Rett syndrome 56, cystic fibrosis, and mucopolysaccharidosis I-Hurler (MPS I-H) (unpublished results). These designer drugs are currently being evaluated for their ability to suppress disease-causing PTCs in vivo.
Figure 5. Structures of conventional aminoglycosides and novel synthetic aminoglycosides.

The novel synthetic aminoglycoside derivatives shown contain structural components of conventional aminoglycosides (shaded and numbered A-C) that are predicted to enhance suppression nonsense mutations while decreasing their nonspecific interactions.
Additional, non-aminoglycoside compounds have also been found to suppress PTCs in mammalian cells. The dipeptide antibiotic negamycin (Figure 6A) was shown to suppress a nonsense mutation in a mouse model of Duchenne muscular dystrophy and restore approximately 10% of wild-type dystrophin protein levels 57. While gentamicin restored a comparable amount of dystrophin protein, negamycin was reported to be less toxic. The mechanism of negamycin-induced PTC suppression has not yet been elucidated, but negamycin was reported to bind to the decoding site of the eukaryotic ribosome and the nascent polypeptide exit tunnel of the bacterial ribosome 58,59. These results suggest that negamycin may target more than one site of the ribosome to induce suppression.
Figure 6. Structures of non-aminoglycoside compounds that suppress translation termination at premature termination codons.

Other compounds that are unrelated to aminoglycosides and do not function as antibiotics have been identified by high-throughput drug screens. In one such screen of a 34,000 small molecule library using an ELISA-based reporter platform, Du et al. identified twelve non-aminoglycoside compounds that suppressed PTCs 60. Two of these compounds, RT#13 and RT#14 were identified as lead compounds (Figure 6B, C). Upon further evaluation in lymphoblastoid and fibroblast cell lines derived from ataxia telangiectasia patients that carried nonsense mutations in the ATM gene, RT#13 and RT#14 were found to restore ATM kinase function when administered in micromolar amounts 61. In addition, these drugs were shown to suppress a nonsense mutation in the dystrophin gene in myoblasts derived from a mouse model of Duchenne muscular dystrophy 60. While the mechanism underlying the ability of these drugs to suppress PTCs is unknown, RT#13 and RT#14 were non-toxic in mammalian cells at concentrations that suppressed PTCs and did not alter global protein expression patterns, suggesting that these drugs show potential for further development.
Another compound, PTC124 (Figure 6D), was recently identified by PTC Therapeutics, Inc. by screening a library of 800,000 low molecular weight compounds using a luciferase-based reporter platform 62. Significantly, PTC124 (also known by the trade name Ataluren®) was found to suppress nonsense mutations as well or better than aminoglycosides at nanomolar concentrations in mammalian cells without toxic side effects. PTC124 did not alter global protein or mRNA profiles and did not possess antibacterial activity.
PTC124 was initially shown to suppress nonsense mutations associated with Duchenne muscular dystrophy and cystic fibrosis in mouse models 62,63. In the Duchenne muscular dystrophy mouse model, approximately 20-25% of normal dystrophin levels were restored in PTC124-treated mice. In addition, significant improvements in other muscular dystrophy disease markers such as an increased steady state level of the γ-sarcoglycan protein and a decrease in serum creatine kinase levels were also observed in PTC124-treated mice. In a Cftr knockout mouse that carried a human CFTR transgene with a nonsense mutation, PTC124 treatment restored up to 29% of wild-type CFTR cAMP-activated chloride channel activity in relevant mouse tissues. PTC124 treatment also resulted in the appearance of CFTR protein at the apical membrane in tissues of treated mice 63.
Given the promising results obtained in suppressing PTCs in animal models, PTC124 proceeded to Phase I clinical trials where it was deemed safe for potential therapeutic uses 64. Phase II clinical trials of PTC124 administered to both adult and pediatric cystic fibrosis patients that carry nonsense mutations have also been completed, with mixed results. Studies in Belgium and Israel found that PTC124 treatment restores measurable CFTR function in around 50% of treated patients 65-67. In contrast, another study in the U.S. did not observe any significant increase in CFTR function 68. While there are many possible explanations for this discrepancy, differences in the assay protocol used at different study sites were considered likely. After extensive efforts to standardize the assay conditions among all participating sites, PTC124 has now entered Phase III clinical trials for treatment of cystic fibrosis.
In phase II clinical trials for Duchenne and Becker muscular dystrophy patients, two doses of PTC124 were initially tested over a 48-week period. Evaluations of the trial indicated that no improvement was observed in patients administered the high dose of PTC124. However, patients given the lower dose showed a trend toward increased walk distances in a six minute walk distance test and a statistically significant decrease in progression of the disease 69 (and unpublished results). Phase II clinical trials are also underway for the treatment of hemophilia A and B, and methylmalonic acidemia in patients with nonsense mutations. Recently, additional experimental evidence has been obtained suggesting that PTC124 is effective in suppressing nonsense mutations associated with carnitine palmitoyltransferase 1A deficiency 70, peroxisome biogenesis disorders 71, limb girdle muscular dystrophy 72, Usher syndrome 73, and the lysosomal storage disease MPS I-Hurler (unpublished data).
Some controversy has surrounded the identification of PTC124 as a suppression agent. The high throughput platform used to screen compound libraries for PTC suppression agents utilized a firefly luciferase construct containing a PTC that prevented the synthesis of full-length firefly protein 62. The screening strategy was based on the logic that suppression of the PTC by a readthrough agent at significant levels would induce translation of full-length firefly protein that could be measured by an increase in luciferase activity. A recent study suggested that the identification of PTC124 as a suppression agent might have been an artifact derived from the platform that was used to perform the high-throughput screen 74. Those authors showed that PTC124 forms an adduct with ATP, a firefly substrate, during the reaction catalyzed by firefly luciferase 75. The PTC124-ATP complex binds to the firefly protein and stabilizes it, which increases the steady state luciferase activity. It was suggested that in the firefly-based high throughput drug screen, PTC124 was identified based on its ability to increase luciferase activity by stabilizing the residual firefly protein that was present from basal readthrough, rather than its ability to enhance suppression of the nonsense mutation. Rebuttals that argued against the conclusion of this study based on technical issues pertaining to the components of the luciferase assay systems and the concentrations of PTC124 used in the original screen have been published 76. Regardless of the role of off-target effects of PTC124 on firefly luciferase activity during its initial identification as a readthrough agent, subsequent in vivo studies using disease models that were independent of firefly luciferase-based assays have provided strong evidence that PTC124 suppresses termination at PTCs and restores functional protein 62,63. In addition, reports from clinical trials indicate that the level of suppression provided by PTC124 in vivo is sufficient to provide a therapeutic benefit in a significant portion of patients with cystic fibrosis 65-67.
Approaches to enhance suppression therapy
While pharmacological PTC suppression as a therapy for diseases caused by nonsense mutations is promising, there are still limitations that hinder the wide spread implementation of this approach to treat a variety of disorders. While the most promising suppression agent at the present time, PTC124, was found to be safe and offers a therapeutic benefit to many patients, not all patients respond equally well to PTC124 administration. For example, in a Phase II PTC124 clinical trial in cystic fibrosis patients, only 50% of the treated patients showed a restoration of CFTR protein function 65. This could possibly be due to differences in the ability of PTC124 to suppress different termination sequences. However, in many cases various patients with identical CFTR mutations showed very different response profiles to suppression therapy. Other factors could include technical differences in assaying CFTR protein function, as well as epigenetic factors and/or genetic modifiers that allow some patients to respond to suppression therapy more robustly than others.
One factor that possibly affects the response to suppression therapy in many patients is the efficiency of nonsense-mediated mRNA decay (NMD). NMD is an mRNA surveillance pathway that degrades mRNAs that contain a PTC 77. It has been proposed that the main purpose of NMD is to dispose of mRNAs that could encode aberrant, truncated polypeptides with the potential of having a dominant-negative phenotype. The NMD pathway reduces the efficiency of suppression therapy because it depletes the pool of PTC-containing transcripts available for translation and subsequent PTC suppression. Interestingly, a recent study reported that cystic fibrosis patients that responded most robustly to suppression therapy generally had a higher level of residual PTC-containing CFTR mRNA 78. In addition, when RNA silencing was used to reduce the abundance of several NMD factors in order to moderate NMD efficiency, the level of functional CFTR protein restored by suppression therapy was significantly increased. This suggests that strategies designed to reduce the efficiency of NMD may improve the therapeutic benefit afforded by suppression therapy.
The NMD pathway has been an active area of research in recent years and our understanding of the factors involved in NMD, as well as the function and mechanism of NMD, has greatly increased. However, for the purpose of this discussion and for brevity, a simplified model of mammalian NMD will be presented (Figure 7). For more detailed information about NMD, please see the following recent reviews 77,79,80. In mammalian cells, mRNAs become messenger ribonucleoprotein (mRNP) complexes in the nucleus. This includes binding of a nuclear cap-binding complex (CBC) composed of CBP20 and CBP80 to the 5′ methylguanosine cap; binding of the nuclear poly(A) binding protein (PABPN1) to the 3′ poly(A) tail; and the association of factors called the exon junction complex (EJC) approximately 20-24 nucleotides upstream of exon-exon junctions as a consequence of mRNA splicing. Several factors required for NMD are components of EJCs. EJCs remain associated with mRNA transcripts as they are exported from the nucleus to the cytoplasm. Upon entry into the cytoplasm and initiation of the first or “pioneer” round of translation, the ribosome displaces the EJC complexes from mRNAs as they are translated. For mRNAs without PTCs, the translating ribosome remodels the mRNP structure by removing the EJC complexes. In addition, the nuclear CBC is replaced with eIF4E and nuclear PABPN1 is replaced with cytoplasmic PAPB (PABPC1). These remodeling events stabilize the mRNA for subsequent rounds of translation. If a PTC is located in an mRNA that is at least 50-55 nucleotides upstream of the final exon-exon junction, the ribosome terminates translation of the mRNA prior to removal of all of the EJC complexes from the mRNA. This incomplete mRNP remodeling marks the mRNA as aberrant, leading to its subsequent degradation by the NMD pathway.
Figure 7. A simplified model of mammalian NMD.

The NMD pathway is found in all eukaryotes. Several NMD factors are highly conserved among diverse species, including UPF1, UPF2, and UPF3. UPF3 is a component of the EJC complex that is deposited onto the mRNA during nuclear mRNA splicing. UPF2 resides in the perinuclear space and binds to mRNA-bound UPF3 as mRNPs transit from the nucleus to the cytoplasm. If a PTC prohibits removal of distal EJCs from an mRNA during the pioneer round of translation, UPF1 and the SMG-1 kinase associate with the eRF1 and eRF3 release factors on the ribosomal pre-termination complex at the PTC. UPF1 then interacts with the UPF2/UPF3 proteins associated with the downstream EJC complex. This interaction induces UPF1 phosphorylation by SMG-1 and marks the mRNA as PTC-containing. In addition, an association between UPF1 and a component of the CBC, CBP80, enhances the efficiency of the NMD process. A complex composed of SMG5, SMG6, SMG7 and the PP2A phosphatase then dephosphorylates UPF1, and the mRNA is subsequently degraded. It is unclear from current NMD models if UPF1 dephosphorylation is a prerequisite for recruitment of the mRNA degradation machinery.
Several pharmacological agents have been shown to inhibit NMD. These include translation inhibitors such as cycloheximide, emetine, puromycin, and pateamine A. However, these drugs are not beneficial for therapeutic use as NMD inhibitors due to their toxicity. Since UPF1 undergoes repeated cycles of phosphorylation and dephosphorylation and this cycle is required for NMD to occur, this provides a pharmacological target for NMD inhibition 77. Several compounds have been identified that inhibit this process. The UPF1 kinase SMG1 is a member of the phosphoinositide-3-kinase related protein kinases (PIKKs) that also include ATM, ATR, and DNA-PKcs that regulate the cellular DNA damage response; and mTOR that regulates the cellular nutrition response 81,82. Kinase inhibitors such as caffeine and wortmannin have been shown to inhibit SMG1 and prevent UPF1 phosphorylation, resulting in partial NMD inhibition 83,84. However, these drugs are not specific for SMG1 and not only affect other PIKKs, but also affect some phosphoinositide 3-kinases (PI3Ks). However, a unique region of 1,000 amino acids in the SMG1 protein that is not found in other PIKKs or PI3Ks may provide a basis for the therapeutic development of molecules that specifically target SMG1 and inhibit its function 85.
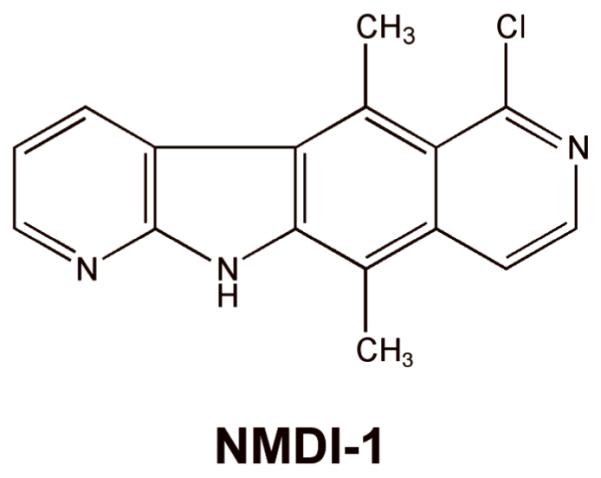
Through a high-throughput drug screen, another NMD inhibitor was recently discovered that also modulates the phosphorylation state of UPF1. This compound, called NMDI-1 (Figure 8), blocks the interaction between SMG5 and UPF1 86. This inhibits the dephosphorylation of UPF1, leading to the inhibition of NMD. Initial characterization of NMDI-1 suggests that it is relatively specific for NMD. It also did not induce toxicity in mammalian cells treated with concentrations much higher than the dose required to inhibit NMD 86. NMDI-1 or an NMDI-1 derivative may show promise as a pharmacological agent that could be moved into clinical use in order to partially inhibit NMD and enhance the efficacy of suppression therapy.
Figure 8. Structure of the NMD inhibitor, NMDI-1.
In addition, the partial inhibition of NMD may in itself be beneficial for some disorders. For example, it has been shown that NMD inhibition by SMG1 or UPF1 depletion, or by pharmacological inactivation of SMG1, could alleviate the Ullrich disease phenotype in human fibroblasts 83,84. This disorder is caused by a deficiency in the collagen VI alpha2 protein. Partial NMD inhibition was found to restore adequate levels of the truncated collagen protein to assemble with other collagen fibers, leading to partial restoration of a functional extracellular matrix. In addition, knockdown of UPF1 expression to inhibit NMD of a nonsense-containing hERG mRNA associated with an inherited form of cardiovascular dysfunction called long-QT syndrome was found to restore channel protein expression and function as a pore-forming subunit of the rapidly activating delayed rectifier potassium channel 87.
While NMD inhibition may prove to be a viable way to provide therapeutic benefits for some diseases, either by increasing the level of partially functional truncated polypeptides, or in combination with suppression therapy to restore full-length, functional proteins, caution must be taken. In addition to degrading mRNAs that contain PTCs, NMD also modulates the level of many endogenous transcripts, including those that contain upstream open reading frames, introns in their 3′ UTR, or abnormally long 3′ UTRs 77. Consistent with the importance of the NMD process and/or certain NMD factors, UPF1, UPF2, and SMG1 knockouts result in embryonic lethality in mice 88-90. However, partial inhibition of NMD by constitutive expression of a dominant negative UPF1 mutant protein allowed the production of viable mice 91. NMD also functions to eliminate nonproductive rearrangements during VDJ and T-cell receptor recombination during immunological system development 90,91. In addition, NMD factors are important for the maintenance of telomeric regions at the ends of chromosomes, play a role in DNA replication and repair, and safeguard genomic integrity 77,80. Thus, modulation of NMD must be monitored carefully to ensure that other NMD-mediated cellular functions are not detrimentally affected. Since the efficiency of NMD varies among individuals within the general population 30,78,92 it is likely that mild pharmacological inhibition of NMD efficiency may be possible without complications.
The success of suppression therapy to provide a therapeutic benefit in various individuals depends on many factors. One particularly important factor is the threshold of correction for a particular disorder that varies depending upon the function of the factor and the tissues where it is expressed. For example, for some disorders that result from an enzyme deficiency such as the lysosomal storage disease mucopolysaccharidosis type I-Hurler (MPS I-H), as little as 1% of wild-type enzymatic activity can significantly alleviate the disease phenotype 93. In other disorders, much more functional protein is required in order to provide therapeutic benefits. While the efficiency of suppression therapy may be improved by developing better suppression agents and combining suppression therapy with NMD inhibition, these efforts may still not restore enough functional protein to provide a therapeutic benefit for some disorders.
In some cases, additional therapeutic approaches may be combined with suppression therapy to reach a therapeutic threshold, particularly in certain tissues. For example, in the case of MPS I-H, administration of enzyme replacement therapy appears to be sufficient to alleviate the disease in many organs 94. However, since the enzyme cannot cross the blood-brain barrier, no correction can be achieved in neurological tissues. The ability of some suppression drugs to cross the blood-brain barrier raises the possibility that the combination of suppression therapy (with or without NMD inhibition) and enzyme replacement therapy may provide a therapeutic benefit in all affected tissues. Similarly, CF drugs called potentiators have been identified that allow the CFTR protein, which is a cAMP-activated chloride channel located on the apical surface of epithelial cells, to remain open for longer periods of time 95. These potentiator compounds, when combined with suppression therapy or suppression therapy together with NMD inhibition, may provide an enhanced therapeutic benefit by restoring greater CFTR protein function than either treatment alone. Approaches that enhance CFTR transcription also have the potential to enhance CFTR protein expression. For example, sodium butyrate has been shown to increase CFTR mRNA levels and, when combined with suppression therapy, may also provide another method to enhance the yield of functional CFTR protein 96.
Conclusion
According to the National Organization for Rare Disorders (NORD), roughly 7,000 genetic disorders are known to occur in humans. In the United States alone, approximately 30 million individuals are afflicted with a genetic disease. Since 11% of all reported mutations are in-frame nonsense mutations 97, potentially 3 million individuals in the US alone could be candidates for PTC suppression therapy. However, specific limits related to the molecular genetics of each disease-causing mutation must be considered. For example, the sequence of the nonsense mutation and the surrounding sequence context in the mRNA are important factors in the susceptibility of mutations to suppression. This affects the overall level of suppression that can be obtained, as well as the specific choice of readthrough compound that will provide the maximal level of suppression. Current methods to predict these variables are limited, so context effects must still be determined empirically in order to find the best readthrough agent to suppress a particular mutation in its natural sequence context. Given the significant differences in the susceptibility of different mutations, suppression therapy is an example of a therapeutic approach that will clearly benefit from increased utilization of individualized genome sequencing and personalized medicine.
Beyond the variable susceptibility of specific nonsense mutations in various genes to suppression, another important factor that must be considered is the threshold of restored protein function needed to provide a therapeutic benefit. In disorders where ten percent or less of WT activity could alleviate some or all of the disease phenotype, this approach may provide a viable treatment option. This level of restoration could possibly be achieved by suppression alone, by the combination of NMD inhibition with nonsense suppression, or by various other combinatorial approaches to amplify the amount of functional protein obtained. Given that a PTC suppression compound is currently undergoing clinical trials for several diseases, it is possible that this novel therapeutic approach could be available for a broad range of genetic disorders in the coming years.
Acknowledgements and Disclosure
Research in the authors’ labs is supported by grants from the NIH to DMB (5RO1 GM068854) and to DMB and KMK (5RO1 NS057412). DMB and KMK have proprietary and financial interests in aminoglycoside-mediated suppression of disease-causing PTCs. DMB is a paid consultant for PTC Therapeutics, Inc.
Contributor Information
Kim M. Keeling, Dept. of Microbiology, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL 35294-2170, USA
David M. Bedwell, Dept. of Microbiology, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL 35294-2170, USA.
References
- 1.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8(10):1893–900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 2.Rodnina MV, Gromadski KB, Kothe U, Wieden HJ. Recognition and selection of tRNA in translation. FEBS Lett. 2005;579(4):938–42. doi: 10.1016/j.febslet.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 3.Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, Philippe M. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995;14(16):4065–72. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D. The crystal structure of human eukaryotic release factor eRF1-- mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100(3):311–21. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 5.Frolova L, Le Goff X, Zhouravleva G, Davydova E, Philippe M, Kisselev L. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA. 1996;2(4):334–41. [PMC free article] [PubMed] [Google Scholar]
- 6.Salas-Marco J, Bedwell DM. GTP Hydrolysis by eRF3 Facilitates Stop Codon Decoding during Eukaryotic Translation Termination. Mol Cell Biol. 2004;24(17):7769–78. doi: 10.1128/MCB.24.17.7769-7778.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fearon K, McClendon V, Bonetti B, Bedwell DM. Premature translation termination mutations are efficiently suppressed in a highly conserved region of yeast Ste6p, a member of the ATP-binding cassette (ABC) transporter family. J Biol Chem. 1994;269(27):17802–8. [PubMed] [Google Scholar]
- 8.Cassan M, Rousset JP. UAG readthrough in mammalian cells: effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol Biol. 2001;2(1):3. doi: 10.1186/1471-2199-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6(7):1044–55. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCaughan KK, Brown CM, Dalphin ME, Berry MJ, Tate WP. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc Natl Acad Sci U S A. 1995;92(12):5431–5. doi: 10.1073/pnas.92.12.5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tate WP, Poole ES, Horsfield JA, Mannering SA, Brown CM, Moffat JG, Dalphin ME, McCaughan KK, Major LL, Wilson DN. Translational termination efficiency in both bacteria and mammals is regulated by the base following the stop codon. Biochem Cell Biol. 1995;73(11-12):1095–103. doi: 10.1139/o95-118. [DOI] [PubMed] [Google Scholar]
- 12.Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet. 2001;10(3):291–9. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- 13.Chernikov VG, Terekhov SM, Krokhina TB, Shishkin SS, Smirnova TD, Kalashnikova EA, Adnoral NV, Rebrov LB, Denisov-Nikol’skii YI, Bykov VA. Comparison of cytotoxicity of aminoglycoside antibiotics using a panel cellular biotest system. Bull Exp Biol Med. 2003;135(1):103–5. doi: 10.1023/a:1023474719042. [DOI] [PubMed] [Google Scholar]
- 14.Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med. 2002;80(6):367–76. doi: 10.1007/s00109-001-0317-z. [DOI] [PubMed] [Google Scholar]
- 15.Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432(7013):112–8. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- 16.Hoshino S, Imai M, Kobayashi T, Uchida N, Katada T. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3′-Poly(A) tail of mRNA. Direct association of erf3/GSPT with polyadenylate-binding protein. J Biol Chem. 1999;274(24):16677–80. doi: 10.1074/jbc.274.24.16677. [DOI] [PubMed] [Google Scholar]
- 17.Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE. Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J. 2008;27(5):736–47. doi: 10.1038/emboj.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh G, Rebbapragada I, Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6(4):e111. doi: 10.1371/journal.pbio.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atkins JF, Baranov PV, Fayet O, Herr AJ, Howard MT, Ivanov IP, Matsufuji S, Miller WA, Moore B, Prere MF. Overriding standard decoding: implications of recoding for ribosome function and enrichment of gene expression. Cold Spring Harb Symp Quant Biol. 2001;66:217–32. doi: 10.1101/sqb.2001.66.217. others. [DOI] [PubMed] [Google Scholar]
- 20.Namy O, Hatin I, Rousset JP. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001;2(9):787–93. doi: 10.1093/embo-reports/kve176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalphin ME, Stockwell PA, Tate WP, Brown CM. TransTerm, the translational signal database, extended to include full coding sequences and untranslated regions. Nucleic Acids Res. 1999;27(1):293–4. doi: 10.1093/nar/27.1.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frischmeyer PA, van Hoof A, O’Donnell K, Guerrerio AL, Parker R, Dietz HC. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295(5563):2258–61. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- 23.Kong J, Liebhaber SA. A cell type-restricted mRNA surveillance pathway triggered by ribosome extension into the 3′ untranslated region. Nat Struct Mol Biol. 2007;14(7):670–6. doi: 10.1038/nsmb1256. [DOI] [PubMed] [Google Scholar]
- 24.Tok JB, Bi L. Aminoglycoside and its derivatives as ligands to target the ribosome. Curr Top Med Chem. 2003;3(9):1001–19. doi: 10.2174/1568026033452131. [DOI] [PubMed] [Google Scholar]
- 25.Recht MI, Douthwaite S, Puglisi JD. Basis for prokaryotic specificity of action of aminoglycoside antibiotics. EMBO J. 1999;18(11):3133–8. doi: 10.1093/emboj/18.11.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynch SR, Puglisi JD. Structural origins of aminoglycoside specificity for prokaryotic ribosomes. J Mol Biol. 2001;306(5):1037–58. doi: 10.1006/jmbi.2000.4420. [DOI] [PubMed] [Google Scholar]
- 27.Salas-Marco J, Bedwell DM. Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J Mol Biol. 2005;348(4):801–15. doi: 10.1016/j.jmb.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 28.Dietz HC. New therapeutic approaches to mendelian disorders. N Engl J Med. 2011;363(9):852–63. doi: 10.1056/NEJMra0907180. [DOI] [PubMed] [Google Scholar]
- 29.Keeling KM, Bedwell DM. Recoding therapies for genetic diseases. In: Atkins JF, Gesteland RF, editors. Recoding: Expansion of Decoding Rules Enriches Gene Expression. Springer Publishing; New York: 2010. [Google Scholar]
- 30.Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24(11):552–63. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 31.Clancy JP, Bebok Z, Ruiz F, King C, Jones J, Walker L, Greer H, Hong J, Wing L, Macaluso M. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med. 2001;163(7):1683–92. doi: 10.1164/ajrccm.163.7.2004001. others. [DOI] [PubMed] [Google Scholar]
- 32.Clancy JP, Rowe SM, Bebok Z, Aitken ML, Gibson R, Zeitlin P, Berclaz P, Moss R, Knowles MR, Oster RA. No Detectable Improvements in CFTR by Nasal Aminoglycosides in CF Patients with Stop Mutations. Am J Respir Cell Mol Biol. 2007;37(1):57–66. doi: 10.1165/rcmb.2006-0173OC. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilschanski M, Famini C, Blau H, Rivlin J, Augarten A, Avital A, Kerem B, Kerem E. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med. 2000;161(3 Pt 1):860–5. doi: 10.1164/ajrccm.161.3.9904116. [DOI] [PubMed] [Google Scholar]
- 34.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349(15):1433–41. doi: 10.1056/NEJMoa022170. others. [DOI] [PubMed] [Google Scholar]
- 35.Dunant P, Walter MC, Karpati G, Lochmuller H. Gentamicin fails to increase dystrophin expression in dystrophin-deficient muscle. Muscle Nerve. 2003;27(5):624–7. doi: 10.1002/mus.10341. [DOI] [PubMed] [Google Scholar]
- 36.Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O, Comi LI. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003;22(1):15–21. [PubMed] [Google Scholar]
- 37.James PD, Raut S, Rivard GE, Poon MC, Warner M, McKenna S, Leggo J, Lillicrap D. Aminoglycoside suppression of nonsense mutations in severe hemophilia. Blood. 2005;106(9):3043–8. doi: 10.1182/blood-2005-03-1307. [DOI] [PubMed] [Google Scholar]
- 38.Kellermayer R, Szigeti R, Keeling KM, Bedekovics T, Bedwell DM. Aminoglycosides as potential pharmacogenetic agents in the treatment of Hailey-Hailey disease. J Invest Dermatol. 2006;126(1):229–31. doi: 10.1038/sj.jid.5700031. [DOI] [PubMed] [Google Scholar]
- 39.Martin R. On the relationship between preferred termination codon contexts and nonsense suppression in human cells. Nucleic Acids Res. 1994;22(1):15–9. doi: 10.1093/nar/22.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moestrup SK, Cui S, Vorum H, Bregengard C, Bjorn SE, Norris K, Gliemann J, Christensen EI. Evidence that epithelial glycoprotein 330/megalin mediates uptake of polybasic drugs. J Clin Invest. 1995;96(3):1404–13. doi: 10.1172/JCI118176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laurent G, Carlier MB, Rollman B, Van Hoof F, Tulkens P. Mechanism of aminoglycoside-induced lysosomal phospholipidosis: in vitro and in vivo studies with gentamicin and amikacin. Biochem Pharmacol. 1982;31(23):3861–70. doi: 10.1016/0006-2952(82)90303-3. [DOI] [PubMed] [Google Scholar]
- 42.Guthrie OW. Aminoglycoside induced ototoxicity. Toxicology. 2008;249(2-3):91–6. doi: 10.1016/j.tox.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Bottger EC. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci U S A. 2008;105(52):20888–93. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawamoto K, Sha SH, Minoda R, Izumikawa M, Kuriyama H, Schacht J, Raphael Y. Antioxidant gene therapy can protect hearing and hair cells from ototoxicity. Mol Ther. 2004;9(2):173–81. doi: 10.1016/j.ymthe.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 45.Gilbert DN, Wood CA, Kohlhepp SJ, Kohnen PW, Houghton DC, Finkbeiner HC, Lindsley J, Bennett WM. Polyaspartic acid prevents experimental aminoglycoside nephrotoxicity. J Infect Dis. 1989;159(5):945–53. doi: 10.1093/infdis/159.5.945. [DOI] [PubMed] [Google Scholar]
- 46.Thibault N, Grenier L, Simard M, Bergeron MG, Beauchamp D. Attenuation by daptomycin of gentamicin-induced experimental nephrotoxicity. Antimicrob Agents Chemother. 1994;38(5):1027–35. doi: 10.1128/aac.38.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du M, Keeling KM, Fan L, Liu X, Bedwell DM. Poly-L-aspartic Acid Enhances and Prolongs Gentamicin-mediated Suppression of the CFTR-G542X Mutation in a Cystic Fibrosis Mouse Model. J Biol Chem. 2009;284(11):6885–92. doi: 10.1074/jbc.M806728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fielding RM, Lewis RO, Moon-McDermott L. Altered tissue distribution and elimination of amikacin encapsulated in unilamellar, low-clearance liposomes (MiKasome) Pharm Res. 1998;15(11):1775–81. doi: 10.1023/a:1011925132473. [DOI] [PubMed] [Google Scholar]
- 49.Schiffelers R, Storm G, Bakker-Woudenberg I. Liposome-encapsulated aminoglycosides in pre-clinical and clinical studies. J Antimicrob Chemother. 2001;48(3):333–44. doi: 10.1093/jac/48.3.333. [DOI] [PubMed] [Google Scholar]
- 50.Sandoval RM, Reilly JP, Running W, Campos SB, Santos JR, Phillips CL, Molitoris BA. A non-nephrotoxic gentamicin congener that retains antimicrobial efficacy. J Am Soc Nephrol. 2006;17(10):2697–705. doi: 10.1681/ASN.2005101124. [DOI] [PubMed] [Google Scholar]
- 51.Ben-Shem A, Jenner L, Yusupova G, Yusupov M. Crystal structure of the eukaryotic ribosome. Science. 2010;330(6008):1203–9. doi: 10.1126/science.1194294. [DOI] [PubMed] [Google Scholar]
- 52.Nudelman I, Rebibo-Sabbah A, Shallom-Shezifi D, Hainrichson M, Stahl I, Ben-Yosef T, Baasov T. Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorg Med Chem Lett. 2006;16(24):6310–5. doi: 10.1016/j.bmcl.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T. Development of Novel Aminoglycoside (NB54) with Reduced Toxicity and Enhanced Suppression of Disease-Causing Premature Stop Mutations. J Med Chem. 2009;52:2836–45. doi: 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T. Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg Med Chem. 2010;18(11):3735–46. doi: 10.1016/j.bmc.2010.03.060. [DOI] [PubMed] [Google Scholar]
- 55.Rebibo-Sabbah A, Nudelman I, Ahmed ZM, Baasov T, Ben-Yosef T. In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Hum Genet. 2007;122(3-4):373–81. doi: 10.1007/s00439-007-0410-7. [DOI] [PubMed] [Google Scholar]
- 56.Brendel C, Belakhov V, Werner H, Wegener E, Gartner J, Nudelman I, Baasov T, Huppke P. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med. 2010 doi: 10.1007/s00109-010-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arakawa M, Shiozuka M, Nakayama Y, Hara T, Hamada M, Kondo S, Ikeda D, Takahashi Y, Sawa R, Nonomura Y. Negamycin restores dystrophin expression in skeletal and cardiac muscles of mdx mice. J Biochem (Tokyo) 2003;134(5):751–8. doi: 10.1093/jb/mvg203. others. [DOI] [PubMed] [Google Scholar]
- 58.Schroeder SJ, Blaha G, Moore PB. Negamycin binds to the wall of the nascent chain exit tunnel of the 50S ribosomal subunit. Antimicrob Agents Chemother. 2007;51(12):4462–5. doi: 10.1128/AAC.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uehara Y, Hori M, Umezawa H. Specific inhibition of the termination process of protein synthesis by negamycin. Biochim Biophys Acta. 1976;442(2):251–62. doi: 10.1016/0005-2787(76)90495-0. [DOI] [PubMed] [Google Scholar]
- 60.Du L, Damoiseaux R, Nahas S, Gao K, Hu H, Pollard JM, Goldstine J, Jung ME, Henning SM, Bertoni C. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J Exp Med. 2009;206(10):2285–97. doi: 10.1084/jem.20081940. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lai CH, Chun HH, Nahas SA, Mitui M, Gamo KM, Du L, Gatti RA. Correction of ATM gene function by aminoglycoside-induced read-through of premature termination codons. Proc Natl Acad Sci U S A. 2004;101(44):15676–81. doi: 10.1073/pnas.0405155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. others. [DOI] [PubMed] [Google Scholar]
- 63.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci U S A. 2008;105(6):2064–9. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007;47(4):430–44. doi: 10.1177/0091270006297140. others. [DOI] [PubMed] [Google Scholar]
- 65.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–27. doi: 10.1016/S0140-6736(08)61168-X. others. [DOI] [PubMed] [Google Scholar]
- 66.Sermet-Gaudelus I, Leal T, DeBoeck Cea. PTC124 induces CFTR full-length production and activity in children with nonsense-mutation-mediated CF. J Cyst Fibros. 2008;7:S22. [Google Scholar]
- 67.Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, Aviram M, Cohen M, Armoni S, Yaakov Y, Pugatch T. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011 doi: 10.1183/09031936.00120910. others. [DOI] [PubMed] [Google Scholar]
- 68.Clancy JP, Konstan MW, Rowe SMea. A phase II study of PTC124 in CF patients harboring premature stop mutations. Ped Pulmonol Suppl. 2006;41 Abstract 269. [Google Scholar]
- 69.Finkel RS. Read-through strategies for suppression of nonsense mutations in Duchenne/ Becker muscular dystrophy: aminoglycosides and ataluren (PTC124) J Child Neurol. 2010;25(9):1158–64. doi: 10.1177/0883073810371129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tan L, Narayan SB, Chen J, Meyers GD, Bennett MJ. PTC124 improves readthrough and increases enzymatic activity of the CPT1A R160X nonsense mutation. J Inherit Metab Dis. 2011 doi: 10.1007/s10545-010-9265-5. [DOI] [PubMed] [Google Scholar]
- 71.Dranchak PK, Di Pietro E, Snowden A, Oesch N, Braverman NE, Steinberg SJ, Hacia JG. Nonsense suppressor therapies rescue peroxisome lipid metabolism and assembly in cells from patients with specific PEX gene mutations. J Cell Biochem. 2011 doi: 10.1002/jcb.22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang B, Yang Z, Brisson BK, Feng H, Zhang Z, Welch EM, Peltz SW, Barton ER, Brown RH, Jr., Sweeney HL. Membrane blebbing as an assessment of functional rescue of dysferlin-deficient human myotubes via nonsense suppression. J Appl Physiol. 2010;109(3):901–5. doi: 10.1152/japplphysiol.01366.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goldmann T, Overlack N, Wolfrum U, Nagel-Wolfrum K. PTC124 mediated translational read-through of a nonsense mutation causing Usher type 1C. Hum Gene Ther. 2011 doi: 10.1089/hum.2010.067. [DOI] [PubMed] [Google Scholar]
- 74.Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci U S A. 2009;106(9):3585–90. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Auld DS, Lovell S, Thorne N, Lea WA, Maloney DJ, Shen M, Rai G, Battaile KP, Thomas CJ, Simeonov A. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc Natl Acad Sci U S A. 2010;107(11):4878–83. doi: 10.1073/pnas.0909141107. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Peltz SW, Welch EM, Jacobson A, Trotta CR, Naryshkin N, Sweeney HL, Bedwell DM. Nonsense suppression activity of PTC124 (ataluren) Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0901936106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Muhlemann O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci. 2010;67(5):677–700. doi: 10.1007/s00018-009-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Linde L, Boelz S, Nissim-Rafinia M, Oren YS, Wilschanski M, Yaacov Y, Virgilis D, Neu-Yilik G, Kulozik AE, Kerem E. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117(3):683–92. doi: 10.1172/JCI28523. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maquat LE, Tarn WY, Isken O. The pioneer round of translation: features and functions. Cell. 2010;142(3):368–74. doi: 10.1016/j.cell.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J. 2010;430(3):365–77. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- 81.Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst) 2009;8(9):1004–8. doi: 10.1016/j.dnarep.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ward S, Sotsios Y, Dowden J, Bruce I, Finan P. Therapeutic potential of phosphoinositide 3-kinase inhibitors. Chem Biol. 2003;10(3):207–13. doi: 10.1016/s1074-5521(03)00048-6. [DOI] [PubMed] [Google Scholar]
- 83.Usuki F, Yamashita A, Higuchi I, Ohnishi T, Shiraishi T, Osame M, Ohno S. Inhibition of nonsense-mediated mRNA decay rescues the phenotype in Ullrich’s disease. Ann Neurol. 2004;55(5):740–4. doi: 10.1002/ana.20107. [DOI] [PubMed] [Google Scholar]
- 84.Usuki F, Yamashita A, Kashima I, Higuchi I, Osame M, Ohno S. Specific inhibition of nonsense-mediated mRNA decay components, SMG-1 or Upf1, rescues the phenotype of ullrich disease fibroblasts. Mol Ther. 2006;14(3):351–60. doi: 10.1016/j.ymthe.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 85.Abraham RT. The ATM-related kinase, hSMG-1, bridges genome and RNA surveillance pathways. DNA Repair (Amst) 2004;3(8-9):919–25. doi: 10.1016/j.dnarep.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 86.Durand S, Cougot N, Mahuteau-Betzer F, Nguyen CH, Grierson DS, Bertrand E, Tazi J, Lejeune F. Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J Cell Biol. 2007;178(7):1145–60. doi: 10.1083/jcb.200611086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gong Q, Stump MR, Zhou Z. Inhibition of nonsense-mediated mRNA decay by antisense morpholino oligonucleotides restores functional expression of hERG nonsense and frameshift mutations in long-QT syndrome. J Mol Cell Cardiol. 2011;50(1):223–9. doi: 10.1016/j.yjmcc.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci U S A. 2010;107(27):12186–91. doi: 10.1073/pnas.1007336107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC. Rent1, a transeffector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet. 2001;10(2):99–105. doi: 10.1093/hmg/10.2.99. [DOI] [PubMed] [Google Scholar]
- 90.Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, Porse BT. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008;22(10):1381–96. doi: 10.1101/gad.468808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sharifi NA, Dietz HC. Physiologic substrates and functions for mammalian NMD. In: Maquat LE, editor. Nonsense-Mediated mRNA Decay. Landes Bioscience; Georgetown, Texas: 2006. pp. 97–109. [Google Scholar]
- 92.Seoighe C, Gehring C. Heritability in the efficiency of nonsense-mediated mRNA decay in humans. PLoS One. 2010;5(7):e11657. doi: 10.1371/journal.pone.0011657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ashton LJ, Brooks DA, McCourt PA, Muller VJ, Clements PR, Hopwood JJ. Immunoquantification and enzyme kinetics of alpha-L-iduronidase in cultured fibroblasts from normal controls and mucopolysaccharidosis type I patients. Am J Hum Genet. 1992;50(4):787–94. [PMC free article] [PubMed] [Google Scholar]
- 94.Wraith JE. The first 5 years of clinical experience with laronidase enzyme replacement therapy for mucopolysaccharidosis I. Expert Opin Pharmacother. 2005;6(3):489–506. doi: 10.1517/14656566.6.3.489. [DOI] [PubMed] [Google Scholar]
- 95.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106(44):18825–30. doi: 10.1073/pnas.0904709106. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rowe SM, Varga K, Rab A, Bebok Z, Byram K, Li Y, Sorscher EJ, Clancy JP. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol. 2007;37(3):347–56. doi: 10.1165/rcmb.2006-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29(8):1037–47. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]