

Abstract
The 1,852,442-bp sequence of an M1 strain of Streptococcus pyogenes, a Gram-positive pathogen, has been determined and contains 1,752 predicted protein-encoding genes. Approximately one-third of these genes have no identifiable function, with the remainder falling into previously characterized categories of known microbial function. Consistent with the observation that S. pyogenes is responsible for a wider variety of human disease than any other bacterial species, more than 40 putative virulence-associated genes have been identified. Additional genes have been identified that encode proteins likely associated with microbial “molecular mimicry” of host characteristics and involved in rheumatic fever or acute glomerulonephritis. The complete or partial sequence of four different bacteriophage genomes is also present, with each containing genes for one or more previously undiscovered superantigen-like proteins. These prophage-associated genes encode at least six potential virulence factors, emphasizing the importance of bacteriophages in horizontal gene transfer and a possible mechanism for generating new strains with increased pathogenic potential.
Streptococcus pyogenes, also known as group A streptococci (GAS), is a strict human pathogen, and no other known reservoir or species is affected by diseases unique to this organism. As a member of the low G+C% family of Gram-positive bacteria, this pathogen is responsible for a wide variety of disease, including pharyngitis (streptococcal sore throat), scarlet fever, impetigo, erysipelas, cellulitis, septicemia, toxic shock syndrome, necrotizing fasciitis (flesh-eating disease) and the sequelae, rheumatic fever and acute glomerulonephritis. Genetic variability is known to occur, as evidenced by the appearance of strains associated with outbreaks of infection such as necrotizing fasciitis, toxic shock syndrome, and rheumatic fever (1–3). The GAS are remarkable for the number of extracellular proteins produced, many of which have been demonstrated to increase the virulence of the organism. These proteins often trigger a severe nonspecific immunological response in the human host. S. pyogenes strains are grouped into two classes on the basis of postinfectious sequelae associated with each strain, class I responsible for rheumatic fever and class II responsible for acute glomerulonephritis. Class I organisms, besides being associated with poststreptococcal rheumatic fever, possess an immunodeterminant contained in a surface-exposed conserved (C repeat domain) region of the M protein (class I M protein) that is lacking in class II proteins (4). In this report, we present the complete genomic sequence of a class I strain of S. pyogenes.
Methods
The S. pyogenes genome sequence was determined by using the whole-genome shotgun approach. Two genomic libraries were constructed from randomly sheared genomic DNA (1- to 2-kb insert and 3- to 5-kb insert), cloned into pUC18 (5) and end sequenced with fluorescent terminators by using an ABI377 (Applied Biosystems) automated DNA sequencer. A third library was constructed from Sau3aI partially digested genomic DNA and cloned into the λ replacement vector, λBlueSTAR (Novagen). End sequences from λ clones were used in determining contig linkage for gap closure and final genome linkage verification. All sequences were assembled by using the phred/phrap/consed software package (http://bozeman.mbt.washington.edu) (6, 7). Gap closure was accomplished through a primer-walking plasmid template and direct sequencing of combinatorial PCR products.
Initial ORF prediction was accomplished with glimmer 2.0 by using the default parameters (http://www.tigr.org) (8, 9). ORFs showing significant overlap were visually examined and removed as needed. Initial identification of ORFs was made on the basis of blastp analysis against the nonredundant protein database. Frame-shift and point mutations were corrected when appropriate, with ORFs containing sequence-verified frameshift or point-verified mutations designated as putatively inactive genes. Further identification of ORFs was performed through analysis with the pfam (Rel. 4.4) (10), cogs (11), and blocks (Blocks Database Ver. 11.0) (12) databases. A sequence E value of <10–4 was used as the cutoff for all database searches. toppred 2 (13) was used to identify transmembrane domains, and signalp (http://www.cbs.dtu.dk/services/SignalP-2.0) (14) was used for prediction of signal peptide regions. Functional assignment to cogs categories was determined on the basis of the results of the cogs database and agreement with results obtained from the other database searches. Annotation was accomplished by using the Genome Annotation Tool Kit from the Los Alamos National Laboratory (Los Alamos, NM).
Detailed sequencing protocols and methodology are provided on our web site (http://microgen.ouhsc.edu/ and at the University of Oklahoma Advanced Center for Genome Technology web site (http://www.genome.ou.edu/proto.html). The complete S. pyogenes genome sequence has been deposited in the Genome Sequence Database with accession no. AE004092. Strain SF370 is available through the American Type Culture Collection (ATCC 700294).
Sequence Analysis
S. pyogenes strain SF370 was originally isolated from a patient with a wound infection and its M1 serotype confirmed serologically and by sequence analysis of the emm1 gene. The M1 serotype is among the most prevalent in terms of involvement in severe invasive infections and as a class I organism may be associated with rheumatic fever. This strain is also known to contain an inducible bacteriophage containing streptococcal erythrogenic toxin C (speC), also known as pyrogenic exotoxin C, but no other previously identified mobile genetic elements.
The completed genome sequence was derived from over 42,000 sequence reads generated by the mass sequencing of a whole genome shotgun library cloned into pUC vectors (5) followed by end-sequencing of a large insert λ library and direct sequencing of PCR products to facilitate gap closure. The average read length was 477 base pairs, and the final genome coverage was 9.5-fold. The final contiguous proofread sequence had a CONSED calculated accuracy of greater than 99.98%, and the deduced physical map is consistent with the previously established physical and genetic map of SF370 by Suvorov and Ferretti (15). After initial ORF prediction by using glimmer 2.0 (8, 9) under default settings, annotation was performed by using the Genome Annotation Tool Kit from the Los Alamos National Laboratory.
The S. pyogenes 370 genome is a circular chromosome with a size of 1,852,442 base pairs and an average G+C content of 38.5%. The average G+C content of the protein-coding sequences is 39.1%. Fig. 1 presents a circular map of the chromosome with the direction of transcription emanating in both directions from oriC. The starting point of base numbering is located at the origin of bidirectional replication adjacent to the dnaA gene in Box region C, similar to that described for Bacillus subtilis (16). A linear map of the SF370 chromosome is presented in Fig. 4 (which is published as supplemental data on the PNAS web site, www.pnas.org), along with the putative functional designation of each gene in Table 2, which is published as supplemental data on the PNAS web site. The genes are predominantly transcribed in the direction of DNA replication; i.e., genes transcribed in the clockwise direction from oriC to the replication terminus represent 83% of the genes, whereas 76% of the genes are transcribed in the counterclockwise direction from oriC. The location of the replication terminus appears to be somewhat skewed from the expected position at 180° from oriC, possibly because of the presence of two complete bacteriophage genomes present on one side. A replication termination protein and ter site have not been identified at this time. However, a putative dif-like termination sequence, identical to that found in many bacteria, including Escherichia coli, is found starting at base pair 929,320, roughly at the point opposite oriC (17). This sequence, along with recombinases XerC and XerD (SPy1196 and SPy1092, respectively), most likely plays a role in the resolution of newly replicated daughter chromosomes.
Figure 1.
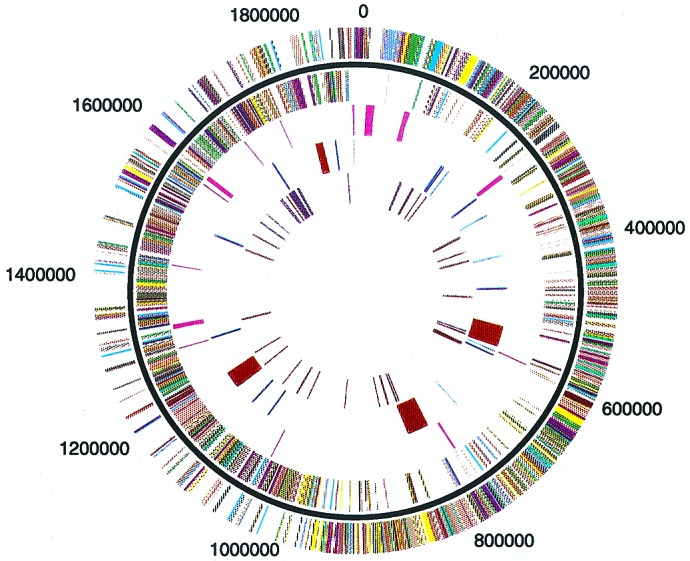
Circular representation of the S. pyogenes strain SF370 genome. Outer circle, predicted coding regions transcribed on the forward (clockwise) DNA strand. Second circle, predicted coding regions transcribed on the reverse (counterclockwise) DNA strand. Third circle, stable RNA molecules. Fourth circle, mobile genetic elements: burgundy, bacteriophage; blue, transposons/IS elements; light cyan, transposons/IS elements (pseudogenes). Fifth circle, known and putative virulence factors: purple, previously identified ORFs; brown, ORFs identified as a result of genome sequence. The lines in each concentric circle indicate the position of the represented feature. Colors: dark gray, amino acid transport and metabolism; light gray, carbohydrate transport and metabolism; green, cell division and chromosome portioning; olive green, cell envelope biogenesis, outer membrane; salmon, cell motility and secretion; tan, coenzyme metabolism; violet, DNA replication, recombination and repair; yellow, energy production and conversion; light pink, function unknown; rose, general function prediction only; light brown, inorganic ion transport and metabolism; light purple, lipid metabolism; light blue, nucleotide transport and metabolism; orange, posttranslational modification, protein turnover, chaperones; red, signal transduction mechanisms; cyan, transcription; green, translation, ribosomal structure and biogenesis; purple, virulence factors; magenta, stable RNA; burgundy, bacteriophage; medium blue, pseudogenes; brown, newly identified virulence factors; blue, transposons/IS elements.
Classification of Gene Products
Of the total of 1,752 ORFs predicted in the genome, 1,282 (83%) could be assigned a putative function or had an identifiable homologue from another bacterial species. There are 79 stable RNA genes, including 6 rRNA operons. Fully 10% of the ORFs (176) are associated with prophage genomes harbored in the SF370 chromosome. The greatest extent of similarity to proteins from other species in the currently available databases was found with B. subtilis, Lactococcus lactis, and various streptococci.
The overall distribution of protein-coding sequences according to functional groups is presented in Table 1. Metabolic pathways present include a complete glycolytic pathway, fatty acid synthesis, nucleotide synthesis and transport, and carbohydrate transport and metabolism. Notable in its absence is a complete tricarboxylic acid cycle pathway and its accompanying electron transport system, consistent with its homofermentative metabolism and the facultative anaerobic environment in which this organism resides. Additionally, only a few amino acids are synthesized, in accord with the fastidious growth requirements of the organism. This synthetic deficiency is offset by scavenging resources from the environment; S. pyogenes SF370 has six ABC transporters putatively identified as amino acid uptake systems, as well as two additional transporter systems that appear to mediate the uptake of dipeptides and oligopeptides.
Table 1.
Distribution of proteins among functional categories
| Functional category | ORFs |
|---|---|
| Amino acid transport and metabolism | 101 |
| Carbohydrate transport and metabolism | 109 |
| Cell division and chromosome partitioning | 21 |
| Cell envelope biogenesis, outer membrane | 57 |
| Cell motility and secretion | 21 |
| Coenzyme metabolism | 32 |
| DNA replication, recombination and repair | 90 |
| Energy production and conversion | 58 |
| Function unknown | 615 |
| General function prediction only | 172 |
| Inorganic ion transport and metabolism | 48 |
| Lipid metabolism | 40 |
| Nucleotide transport and metabolism | 65 |
| Posttranslational modification, protein turnover, chaperones | 38 |
| Signal transduction mechanisms | 41 |
| Transcription | 66 |
| Translation, ribosomal structure and biogenesis | 132 |
| Virulence factors | 46 |
| TOTAL | 1,752 |
| Stable RNA | 79 |
| Putatively inactive genes | 15 |
Regulation and Signaling
The number of σ factors present in bacterial species varies considerably; for example, 18 σ factors are present in B. subtilis, whereas only 3 are present in the genome of Haemophilus influenzae RD (18, 19). S. pyogenes contains a major σ factor [σ70 (rpoD)] as well as an identifiable minor σ factor (homolog of σE). The σE (also known as σ24) is one of the major factors necessary for transcription of heat-induced proteins in E. coli (20), and the homolog found in S. pyogenes may play a similar role when the organism encounters elevated temperatures in the host. Another putative σ factor is a homolog of the Streptococcus pneumoniae proposed σ factor com X that is a transcriptional regulator of competence-specific genes (21). A protein with sequence similarity to the σ-54 modulator protein (SPy1613) is present in the genome; however, a σ-54 homolog could not be conclusively identified. It may be that this potential regulator interacts with one of the identified σ factors or may play some other undiscovered regulatory role. Overall, the number of σ factors present in S. pyogenes (4 probable) is consistent with that found in other bacterial pathogens with small genomes, which can range from 1 to 4 (22).
As with other organisms, the presence of alternate transcription signals allows the streptococcus to respond to environmental changes (23, 24). S. pyogenes encodes the genes for a number of stress-related proteins, which includes several proteases involved in the stress response and most of the highly conserved SOS regulon genes. Highly conserved genes responsible for osmoregulation and genes involved in uptake and synthesis of the osmoprotectants glycine–betaine, proline, and trehalose also are present. In addition to stress pathways common to eubacteria, lactic acid-producing bacteria must deal with acidification of their local environment. The principal means of protection against acid stress in S. pyogenes is most likely the action of the proton translocating F0F1 ATPase, a mechanism that has been shown to efficiently protect Streptococcus mutans against an acidified environment (25, 26). Additionally, the arginine deiminase pathway is used by some species of lactococci, streptococci, and lactobacilli to survive such a decrease in pH. The genes responsible for this system have recently been examined in Lactobacillus sakei (27), and an operon resembling this one is found in S. pyogenes. The relA/spoT proteins are key components of the bacterial stringent response. The genomic sequence revealed the presence of a gene (SPy1981, relA) encoding a bifunctional enzyme involved in the synthesis (Rel-like function) and hydrolysis (SpoT-like function) of (p)ppGpp during amino acid starvation (28). Thus rel fulfills functions that reside separately in the proteins encoded by the relA and spoT genes of E. coli.
Among 13 identified 2-component regulators, 6 can be assigned to a specific function. Three are sensor–responder pairs that appear to be associated with small peptide signaling systems; one pair is associated with the salivaricin lantibiotic operon and a second with the competence factor response system, ComD and ComE (29). The third is the recently described two-component regulator (csrS/csrR; covR/covS) that affects the expression of streptolysin S as well as hyaluronic acid capsule synthesis and pyrogenic exotoxin B expression (30–33). Another two-component system (SPy2026 and SPy2027) may be involved in bacterial virulence, being positioned near the major virulence regulon controlled by Mga and located immediately upstream from the immunogenic secreted protein gene, isp (34). An additional two-component system (SPy0528 and SPy0529) is homologous to YycF-AucG and to hk02-rr02, two-component systems that are essential for growth in B. subtilis and S. pneumoniae, respectively (35).
Thirty-six ABC transporters are found in strain SF370, and the roles of many are associated with conserved systems controlling the transport of iron and ferrichrome, phosphate, inorganic ions, sugars, dipeptides/oligopeptides, and amino acids. One transport system appears to be dedicated to the uptake of polyamines, offsetting the lack of de novo polyamine synthesis. Several of the transporters are related to multidrug-resistance/efflux systems and may play important roles in environmental stress responses. Additionally, the ABC transporter for choline uptake (OpuA and OpuB) is present. This system provides the substrate for the synthesis of glycine–betaine, an important osmoprotectant. Of these 36 transport systems, 8 apparently have alternate ATP-binding proteins, and 15 have no readily identifiable substrate specificity.
Two nine-gene operons encoding information for the synthesis of bacteriocin-like peptide toxins have been identified. The first is salivaricin A, a bacteriocin originally described in S. salivarius and that is present in 90% of S. pyogenes strains (36). The second is streptolysin S, a pore-forming hemolysin that has escaped identification for over 40 years (37). The genetic organizations of both of these operons resemble that of the lantibiotic nisin produced by L. lactis (38) and the cytolysin of Enterococcus faecalis (39).
Horizontal Gene Transfer, Bacteriophages, and Mobile Genetic Elements
Horizontal gene transfer between bacterial species can occur by several mechanisms, including competence-mediated transformation and bacteriophage infection. Although many of the related streptococci are naturally competent, transformation via a competence pathway has never been described for S. pyogenes. A number of genes present in strain SF370 specify proteins with varying degrees of sequence similarity to competence-related genes from the oral streptococci, S. pneumoniae, and B. subtilis. In pneumococci, the genes for recA, cinA, and dinF are transcribed as a single 5.7-kb transcript, where their coordinated expression appears to be necessary for efficient incorporation of donor DNA during transformation (40). The recA and cinA genes of SF370 are also positioned together; however, the only possible gene similar to dinF is found in a distant part of the genome. Because the products of these genes also mediate functions unrelated to transformation, such as SOS repair, any role they play in the incorporation of foreign DNA is contingent on the presence of other competence genes. Nevertheless, the intriguing observation has been made that a binding site for the ComX transcription factor for late-competence genes (“cin box”) is found in the promoter region of cinA (21, 41). As in S. pneumoniae, two copies of comX are present in SF370, each positioned next to duplicated ribosomal operons. Additional copies of the cin box sequence (TACGAATA) have been identified in the genome, some positioned in front of ORFs for competence gene homologs such as the B. subtilis comGA. A number of other genes similar to competence-related genes from several Gram-positive bacteria have been identified including, comG ORFs ABCD, comE ORFs CA, and comF ORFs CA (29). The crucial late genes for expression of competence, comABC, cannot be identified in the genome. Thus, whereas a significant portion of the transformation mechanism appears to be present in S. pyogenes, whether these genes have ever mediated such an event in GAS cannot be determined.
Bacteriophage and transposon genes account for ≈10% of the total genome, including the complete or partial sequence of four bacteriophage genomes. One of the bacteriophages, identified as phage 370.1, may be induced into the lytic cycle by mitomycin C treatment (not shown). This phage contains the speC gene and an adjacent gene (SPy0712; mf2) that has sequence similarity to the previously described streptococcal mitogenic factor (SPy2043), as well as to the nucleases EndA (competence-specific nuclease) from S. pneumoniae and streptodornase from S. pyogenes (42). A second phage genome (phage 370.2) appears to be complete, but attempts to induce the lytic cycle produce no phage particles. Analysis of the bacteriophage-related genes revealed a point mutation within the putative portal protein that results in a stop codon within the coding region that would eliminate the ability of this phage to package its chromosome into a prohead. Phage 370.2 carries two superantigen-like genes identified as speH/I. The third complete phage genome (370.3) also appears to be defective because it is also not inducible, although no obvious genetic defects have been identified within the predicted coding regions. Phage 370.3 also carries two genes with implications of horizontal transfer and virulence. These genes are located, as is the case for all known phage-associated toxin genes, at the end of the phage genome and are transcribed in an opposite direction from the bacteriophage genes. The first gene is paralogous to the MF2 gene of phage 370.2, with a similar, although reversed, order of sequence similarity with MF, EndA, and streptodornase. The second gene, although possessing several predicted transmembrane domains, appears to be completely unique to S. pyogenes, unlike anything found in the current databases. The fourth phage genome, phage 370.4, is incomplete and has an extensive deletion that includes all identifiable structural and lysis genes. No virulence-associated gene can be identified in this phage genome. Of particular interest is the location of the virulence-associated genes near the integration site of each complete phage. Because the GC content of these genes is in the range of 26–30%, whereas the other adjacent phage genes are at or above the 38.5% average GC content of the overall chromosome (Fig. 2), it is likely that at some point in its evolution, these genes were acquired from an unrelated organism and transferred to S. pyogenes. The ubiquitous presence of phages in the GAS (43) assures the possibility of horizontal gene transfer of these virulence determinants, playing an important role in increasing the pathogenic potential of the organism as well as in its overall evolution.
Figure 2.

%G+C profiles of phage genomes. A plot of the average %G+C (100-base window) along the length of each phage complete genome is shown with the residue numbers in the horizontal axis. The regions encoding the known or putative virulence factors associated with each phage are enclosed within the boxed regions; these regions all show a marked decrease in average %G+C compared with the remainder of the genomes. Analysis was done by using the Genetics Computer Group software package.
Ten predicted transposons or insertion sequence (IS) elements are dispersed evenly over the SF370 genome as well as seven additional transposons that contain sequence mutations or deletions resulting in gene inactivation. The only IS element that appears to be directly associated with virulence factors is IS1562 (SPy2013), which is associated with the scpA and SIC genes (44). Interestingly, one of the inactive IS elements (SPy0858; IS861-like element) is located adjacent to a putative gene fragment (SPy0860), encoding a peptide with sequence similarity to the C-terminal region of SpeC.
Virulence Factors
Putative virulence associated genes are abundant in the genome, with many of the encoded proteins predicted to be localized to the cell surface or secreted as extracellular products. Virtually all previously identified and sequenced genes of a class I organisms were identified in the genome. These genes are located randomly throughout the chromosome and are not grouped together as a pathogenicity island with the exception of the cluster of virulence-associated factors in the region of emm. Extending from mitogenic exotoxin Z (smeZ; SPy1998) through the mitogenic factor (mf1; SPy2043), this region includes many of the best-studied virulence factors and their associated regulatory elements such as the Mga regulon. Although it has been proposed that this region may be a pathogenicity island (45), it does not appear to have the organization of the well-studied virulence regions of other bacteria (46).
Thirteen predicted surface proteins contain an LPXTG motif, such as the M protein, protein F, and C5a peptidase (see Table 3, which is published as supplemental data on the PNAS web site). Proteins containing this motif are known to anchor their C-terminal ends to the cell surface (47). Although not identified as a virulence factor, the T protein is a protease-resistant cell-surface protein found in all GAS that is important in serological typing. The gene for the serotype 1 T protein in SF370 may vary significantly from the previously identified gene such that it cannot be identified by gapped blast search. All genes essential for capsule synthesis, another important GAS virulence factor, are present.
At least six genes encoding new superantigen-like proteins also are found, many of which are associated with mobile genetic elements, making a total of 14 superantigen-like molecules identified to date in the GAS. These known or putative streptococcal proteins all have at least one related protein identified from another Gram-positive bacterial species, suggesting that these genes may have been disseminated by horizontal transfer (Fig. 3). The products of several of these genes have now been characterized and indeed shown to be among the class of superantigens (48). Genes encoding previously proposed virulence activities that had not been identified or cloned before the start of this sequencing project have now been located such as NADase, hyaluronidase, streptolysin S, amylase, phosphatase, and proteinase. Several known virulence genes have not yet been definitively identified, including those that encode the four DNase activities. The sequence similarity between MF3, MF2, and MF with EndA and streptodornase, both known nucleases, suggests that all might possess DNase activity and represent the genes for DNases. It was surprising to find the gene for cAMP factor in the GAS genome, as this gene was thought to be found only in the GBS or in Streptococcus uberis (49). Three putative novel hemolysins are present in the genome, having similarity to theoretical proteins from either S. mutans or B. subtilis, as well as numerous other known and theoretical hemolysins. A list of identified putative virulence factors is presented in Table 4, which is published as supplemental data on the PNAS web site.
Figure 3.
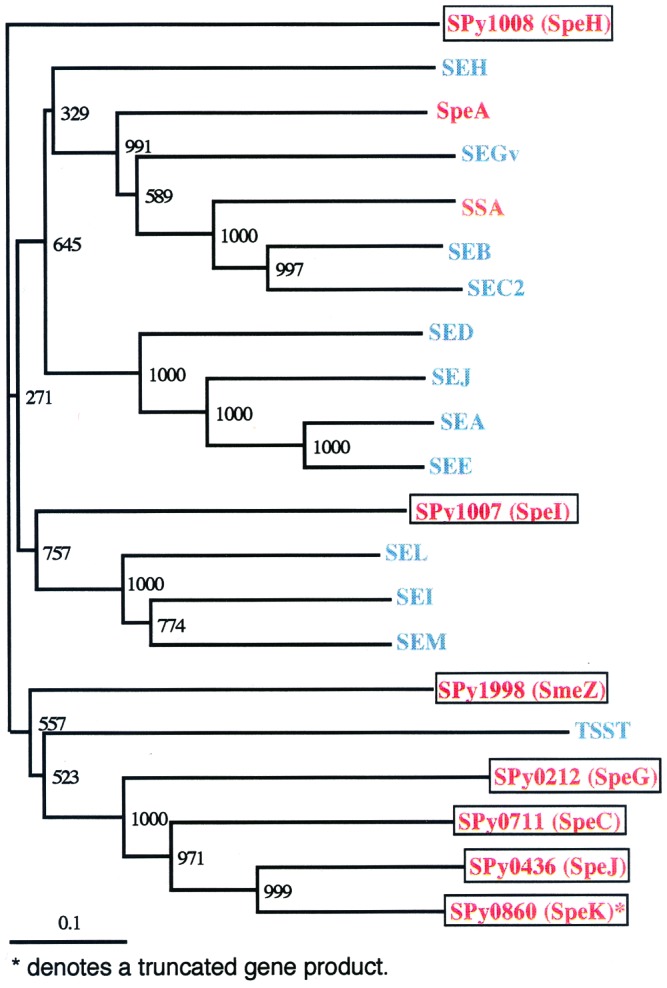
Phylogram of superantigen-like proteins identified in S. pyogenes SF370. The protein alignment was generated by using clustalx (by using the blossum matrix and a bootstrap trial of 1,000). The graphical representation of the tree was generated by using treeview. Gene products encoded by SF370: red; encoded by S. pyogenes but not present in SF370: green and encoded by S. aureus. Scale bar represents the length of the branches. Bootstrap values are displayed at each internal node. Note: SpeK is present in SF370 as a partial product only; an intact copy of speK has not yet been identified in S. pyogenes. Gene products encoded by S. pyogenes are in red with those proteins specifically encoded by strain SF370 also enclosed in a box. The products encoded by S. aureus are in blue. GenBank accession nos.: S. pyogenes proteins: SSA (gb: AAA65928.1); SpeA (gb: AAC48868.1). S. aureus proteins: SEA (prf:1704203A); SEB (gb: AAA88550.1); SEC2 (gb: AAA26624.1); SED (gb: AAB06195.1); SEE (gb: AAA26617.1); SEGv (dbj: BAA36693.1); SEH (gb: AAA19777.1); SEI (gb: AAC26661.1); SEJ (gb: AAC78590.1); SEL (gb: AAG29598.1); SEM (gb: AAG36952.1); TSST (gb: AAA26682.1). Supplementary information is available on the world wide web sites for our laboratories at the University of Oklahoma (http://www.genome.ou.edu/) and the University of Oklahoma Health Sciences Center (http://www.microgen.ouhsc.edu/).
In S. pneumoniae and several other streptococci, the spread of resistance to β-lactam antibiotics in natural populations has occurred when segments of penicillin-binding proteins (PBPs) from sensitive strains were replaced by homologous blocks originating from resistant strains, resulting in gene mosaics. These transfers have most likely been mediated by natural transformation with exogenous DNA and can cross species boundaries (50). The two most important PBPs associated with penicillin resistance in S. pneumoniae are encoded by pbp1A and pbp2X. Phylogenetic comparisons of the S. pyogenes homologs of these PBPs to the proteins from S. pneumoniae and the oral streptococci show the relatedness of these proteins (see Fig. 5, which is published as supplemental data on the PNAS web site); however, blocks analysis (50) showed that pbp1A and pbp2X from S. pyogenes contain no lengthy regions of homology with the genes from the other streptococci. Thus, the acquisition of penicillin resistance by homologous recombination with genetic material from a related species is unlikely. Additionally, because there is no evidence that GAS are competent for transformation, it is probable that penicillin resistance in GAS would have to arise de novo.
Several putative genes encoding proteins with internal repeats of the motif sequence Gly-X-Y were identified in the genome. These amino acid triplet repeats resemble the characteristic repeating sequences found in collagen. Genes SPy1983 and SPy1054 encode proteins that contain 50- and 38-aa triplet repeats, respectively, and 2 bacteriophage hyaluronidase proteins each contain 10 of the amino acid triplets. The GC content of genes SPy1983 and SPy1054 are 50.3 and 47.1%, respectively, both considerably higher than the average of 38.5% for the genome. The origin and function of these sequences are unknown; however, availability of these proteins to the human immune system during infection could possibly lead to antibodies directed against collagen in connective tissue. The formation of such autoantibodies could result in the polyarthritis generally associated with rheumatic fever, one of the postinfection sequelae of a GAS infection, similar to the onset of rheumatic heart disease resulting from the crossreactivity of cardiac myosin and the M protein (52). Further, at least one of these proteins (SclA; SPy1983) has been shown to be expressed on the cell surface and under the control of the Mga regulator (53), suggesting a link to virulence.
Conclusions
The complete sequence of the S. pyogenes genome and the resulting initial analysis that reveals the numerous encoded virulence factors reflect how this organism has adapted to be an obligate and versatile human pathogen. The continued analysis of this genome should provide new insights not only into how adaptations have shaped the overall genetic organization of the GAS chromosome but also into the role regulatory elements play in physiologic responses to environmental stress and in the expression of virulence factors. Additionally, several approaches to developing a GAS vaccine are currently under way (54) and further genome analysis coupled with functional genomic studies and gene distribution surveys should suggest new or alternate candidates, especially from the gene products unique to GAS and highly conserved among all strains. The eventual sequencing of additional GAS strains, including a class II strain, should provide answers to the classical question concerning the difference between throat and skin strains (55, 56). The discovery of additional new putative virulence factors should allow future research to be directed toward answering important questions relating to the physiology and pathogenesis of streptococcal diseases, which will ultimately lead to improved prevention and treatment of these diseases.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institute of Allergy and Infectious Diseases, National Institutes of Health. We are appreciative of the assistance in annotation by Gerry Myers and Thomas Brettin of the Los Alamos National Laboratory. We thank Carolyn Thompson, Min Zhan, RunYing Tian, C. A. Reece, and Linda Ray for technical assistance.
Abbreviations
- GAS
group A streptococci
- PBP
penicillin-binding protein
- IS
insertion sequence
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AE004092).
References
- 1.Stevens D L. Emerg Infect Dis. 1995;1:69–78. doi: 10.3201/eid0103.950301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens D L, Tanner M H, Winship J, Swarts R, Ries K M, Schlievert P M, Kaplan E. N Engl J Med. 1989;321:1–7. doi: 10.1056/NEJM198907063210101. [DOI] [PubMed] [Google Scholar]
- 3.Veasy L G, Wiedmeier S E, Orsmond G S. N Engl J Med. 1987;316:421–427. doi: 10.1056/NEJM198702193160801. [DOI] [PubMed] [Google Scholar]
- 4.Bessen D K, Fischetti V A. J Exp Med. 1990;172:1757–1764. doi: 10.1084/jem.172.6.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodenteich A, Chissoe S, Wang Y F, Roe B A. In: Automated DNA Sequencing and Analysis Techniques. Ventor C, editor. London: Academic; 1993. [Google Scholar]
- 6.Ewing B, Hillier L, Wendl M, Green P. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 7.Gordon D, Abajian C, Green P. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- 8.Delcher A L, Harmon D, Kasif S, White O, Salzberg S L. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salzberg S, Delcher A, Kasif S, White O. Nucleic Acids Res. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bateman A, Birney E, Durbin R, Eddy S R, Howe K L, Sonnhammer E L L. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tatusov R L, Galperin M Y, Natale D A, Koonin E V. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henikoff S, Henikoff J G. Genomics. 1994;19:97–107. doi: 10.1006/geno.1994.1018. [DOI] [PubMed] [Google Scholar]
- 13.Heijne G v. J Mol Biol. 1992;225:487–494. doi: 10.1016/0022-2836(92)90934-c. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen H, Engelbrecht J, Brunak S, Heijne G V. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Suvorov A N, Ferretti J J. J Bacteriol. 1996;178:5546–5549. doi: 10.1128/jb.178.18.5546-5549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moriya S, Ogasawara N. Gene. 1996;176:81–84. doi: 10.1016/0378-1119(96)00223-5. [DOI] [PubMed] [Google Scholar]
- 17.Hill T M. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 2. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1602–1614. [Google Scholar]
- 18.Kunst F, Ogasawara N, Moszer I, Albertini A, Alloni G, Azevedo V, Bertero M, Bessieres P, Bolotin A, Borchet S, et al. Nature (London) 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 19.Fleischmann R D, Adams M D, White O, Clayton R A, Kirkness E F, Kerlavage A R, Bult C J, Tomb J-F, Dougherty B A, Merrick J M, et al. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 20.Gross C A. In: Escherichia coli and Salmonella: Cellular and Molecular Biology. Neidhardt F C, editor. Vol. 1. Washington, DC: Am. Soc. Microbiol.; 1996. pp. 1382–1399. [Google Scholar]
- 21.Lee M S, Morrison D A. J Bacteriol. 1999;181:5004–5016. doi: 10.1128/jb.181.16.5004-5016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koonin E V, Aravind L, Galperin M Y. In: Bacterial Stress Responses. Storz G, Hengge-Aronis R, editors. Washington, DC: Am. Soc. Microbiol.; 2000. pp. 417–444. [Google Scholar]
- 23.Granok A, Parsonage D, Ross R, Caparon M. J Bacteriol. 2000;182:1529–1540. doi: 10.1128/jb.182.6.1529-1540.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heyningen T V, Fogg G, Yates D, Hanski E, Caparon M G. Mol Microbiol. 1993;9:1213–1222. doi: 10.1111/j.1365-2958.1993.tb01250.x. [DOI] [PubMed] [Google Scholar]
- 25.Bender G, Sutton S, Marquis R. Infect Immun. 1986;53:331–338. doi: 10.1128/iai.53.2.331-338.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki T, Tagami J, Hanada N. J Appl Microbiol. 2000;88:555–562. doi: 10.1046/j.1365-2672.2000.00840.x. [DOI] [PubMed] [Google Scholar]
- 27.Foster J W. In: Bacterial Stress Responses. Storz G, Hengge-Aronis R, editors. Washington, DC: Am. Soc. Microbiol.; 2000. pp. 99–116. [Google Scholar]
- 28.Mechold U, Malke H. J Bacteriol. 1997;179:2658–2667. doi: 10.1128/jb.179.8.2658-2667.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dubnau D. In: Bacillus subtilis and Other Gram-Positive Bacteria. Sonenshein A L, Hoch J A, Losick R, editors. Washington, DC: Am. Soc. Microbiol.; 1993. pp. 555–584. [Google Scholar]
- 30.Bernish B, van de Rijn I. J Biol Chem. 1999;274:4786–4793. doi: 10.1074/jbc.274.8.4786. [DOI] [PubMed] [Google Scholar]
- 31.Heath A, Di R V, Barg N, Engleberg N. Infect Immun. 1999;67:5298–5305. doi: 10.1128/iai.67.10.5298-5305.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levin J, Wessels M. Mol Microbiol. 1998;30:209–219. doi: 10.1046/j.1365-2958.1998.01057.x. [DOI] [PubMed] [Google Scholar]
- 33.Federle M, McIver K, Scott J R. J Bacteriol. 1999;181:3649–3657. doi: 10.1128/jb.181.12.3649-3657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McIver K S, Subbarao S, Kellner E M, Heath A S, Scott J R. Infect Immun. 1996;64:2548–2555. doi: 10.1128/iai.64.7.2548-2555.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lange R, Wagner C, de, S. A, Flint N, Molnos J, Stieger M, Caspers P, Kamber M, Keck W, Amrein K. Gene. 1999;237:223–234. doi: 10.1016/s0378-1119(99)00266-8. [DOI] [PubMed] [Google Scholar]
- 36.Simpson W, Ragland N, Ronson C, Tagg J. Dev Biol Stand. 1995;85:639–643. [PubMed] [Google Scholar]
- 37.Nizet V, Beall B, Bast D, Datta V, Kilburn L, Low D, De Azevado J. Infect Immun. 2000;68:4245–4254. doi: 10.1128/iai.68.7.4245-4254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ra S, Qiao M, Immonen T, Pujana I, Saris E. Microbiology. 1996;142:1281–1288. doi: 10.1099/13500872-142-5-1281. [DOI] [PubMed] [Google Scholar]
- 39.Booth M, Bogie C, Sahl H, Siezen R, Hatter K, Gilmore M. Mol Microbiol. 1996;21:1175–1184. doi: 10.1046/j.1365-2958.1996.831449.x. [DOI] [PubMed] [Google Scholar]
- 40.Mortier-Barriere I, de Saizieu A, Claverys J P, Martin B, Mercenier A. Mol Microbiol. 1998;27:159–170. doi: 10.1046/j.1365-2958.1998.00668.x. [DOI] [PubMed] [Google Scholar]
- 41.Claverys J-P, Martin B. Mol Microbiol. 1998;29:1126–1127. doi: 10.1046/j.1365-2958.1998.01005.x. [DOI] [PubMed] [Google Scholar]
- 42.Podbielski A, Zarges I, Flosdorff A, Weber-Heynemann J. Infect Immun. 1996;64:5349–5356. doi: 10.1128/iai.64.12.5349-5356.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hynes W L, Hancock L, Ferretti J J. Infect Immun. 1995;63:3015–3020. doi: 10.1128/iai.63.8.3015-3020.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berge A, Rasmussen M, Bjorck L. Infect Immun. 1998;66:3449–3453. doi: 10.1128/iai.66.7.3449-3453.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hacker J, Blum-Oehler G, Muhldorfer I, Tschape H. Mol Microbiol. 1997;23:1089–1097. doi: 10.1046/j.1365-2958.1997.3101672.x. [DOI] [PubMed] [Google Scholar]
- 46.Blum G, Ott M, Lischewski A, Ritter A, Imrich H, Tschape H, Hacker J. Infect Immun. 1994;62:606–614. doi: 10.1128/iai.62.2.606-614.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fischetti V A. In: Gram-Positive Pathogens. Fischetti V A, Novick R P, Ferretti J J, Portnoy D A, Rood J I, editors. Washington, DC: Am. Soc. Microbiol.; 2000. pp. 11–24. [Google Scholar]
- 48.Proft T, Moffatt S L, Berkahn C J, Fraser J D. J Exp Med. 1999;189:89–102. doi: 10.1084/jem.189.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gase K, Ferretti J J, Primeaux C, McShan W M. Infect Immun. 1999;67:4725–4731. doi: 10.1128/iai.67.9.4725-4731.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dowson C, Hutchison A, Woodford N, Johnson A, George R, Spratt B. Proc Natl Acad Sci USA. 1990;87:5858–5862. doi: 10.1073/pnas.87.15.5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schuler G D, Altschul S F, Lipman D J. Proteins. 1991;9:180–190. doi: 10.1002/prot.340090304. [DOI] [PubMed] [Google Scholar]
- 52.Cunningham M W. In: Gram-Positive Pathogens. Fischetti V A, Novick R P, Ferretti J J, Rood J I, editors. Washington, DC: Am. Soc. Microbiol.; 2000. pp. 66–77. [Google Scholar]
- 53.Rasmussen M, Eden A, Bjorck L. Infect Immun. 2000;68:6370–6377. doi: 10.1128/iai.68.11.6370-6377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fischetti V A. In: Gram-Positive Pathogens. Fischetti V A, Novick R P, Ferretti J J, Portnoy D A, Rood J I, editors. Washington, DC: Am. Soc. Microbiol.; 2000. pp. 96–104. [Google Scholar]
- 55.Wannamaker L. N Engl J Med. 1970;282:23–31. doi: 10.1056/NEJM197001012820106. [DOI] [PubMed] [Google Scholar]
- 56.Wannamaker L. Circulation. 1973;48:9–18. doi: 10.1161/01.cir.48.1.9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}