

Abstract
Epstein–Barr virus encodes integral membrane proteins LMP1 and LMP2A in transformed lymphoblastoid cell lines. We now find that LMP1 associates with the cell cytoskeleton through a tumor necrosis factor receptor-associated factor-interacting domain, most likely mediated by tumor necrosis factor receptor-associated factor 3. LMP1 is palmitoylated, and the transmembrane domains associate with lipid rafts. Mutation of LMP1 cysteine-78 abrogates palmitoylation but does not affect raft association or NF-κB or c-Jun N-terminal kinase activation. LMP2A also associates with rafts and is palmitoylated but does not associate with the cell cytoskeleton. The associations of LMP1 and LMP2A with rafts and of LMP1 with the cell cytoskeleton are likely to effect interactions with cell proteins involved in shape, motility, signal transduction, growth, and survival.
Epstein–Barr virus (EBV) is causally associated with human malignancies including lymphoproliferative diseases in immune-compromised patients, Hodgkin's disease, and epithelial cancers (1). EBV can efficiently transform primary B lymphocytes into lymphoblastoid cell lines (LCLs) that proliferate indefinitely in vitro. Latent infection membrane proteins 1 (LMP1) and 2A (LMP2A) are viral-encoded, multiple membrane-spanning proteins expressed in EBV-transformed LCLs and also in Hodgkin's disease and nasopharyngeal carcinoma (2). LMP1 and 2A constitutively aggregate in a discrete patch in the plasma membrane (3, 4). LMP1 has transforming effects in rodent fibroblast assays (5); activates NF-κB, c-Jun N-terminal kinase (JNK), and p38 (6–10); up-regulates adhesion and activation marker expression in B lymphocytes (11, 12); and is essential for primary B lymphocyte transformation into LCLs (13). LMP2A constitutively engages src and syk family tyrosine kinases (14, 15) and has transforming or survival effects in some epithelial cell lines (16), but is not important in EBV-mediated LCL outgrowth (17).
LMP1 is composed of a 24-aa cytoplasmic amino terminus, 6 hydrophobic membrane-spanning domains separated by 5 reverse turns, and a 200-aa cytoplasmic carboxyl terminus (3). The six hydrophobic membrane-spanning domains are responsible for spontaneous aggregation in the plasma membrane and subsequent signal transduction from LMP1 (13, 18–20). The cytoplasmic carboxyl terminus has at least two domains that engage signal-transducing proteins, and both domains are important for B cell growth transformation. These two domains are named transformation effector sites 1 and 2 (TES1 and TES2). TES1 is critical for the recruitment of tumor necrosis factor receptor (TNFR)-associated factors (TRAFs) TRAF1, TRAF2, TRAF3, and TRAF5 (21–25). TES2 is critical for the recruitment of TNFR-associated death domain protein (TRADD) and TNFR-interacting protein (RIP) (26–28). LMP1 constitutively engages these proteins and mediates NF-κB, JNK, and p38 activation.
LMP2A is composed of a 119-aa cytoplasmic amino terminus, 12 hydrophobic transmembrane domains, and a 27-aa cytoplasmic carboxyl terminus (4). LMP2A also aggregates in the plasma membrane. Lyn and Syk tyrosine kinases constitutively associate with LMP2A, mediate constitutive phosphorylation of LMP2A, maintain a low level of forward signaling, and constitutively desensitize cells to further receptor-mediated src family tyrosine kinase activation (15, 29–32).
Plasma membranes have cholesterol- and sphingolipid-rich microdomains also known as lipid rafts or detergent-resistant membranes (DRMs). Lipid rafts are sites of receptor accumulation, signal transduction, endocytosis, and protein sorting (33, 34). Lipid rafts can be isolated based on their insolubility in Triton X-100 and their buoyant density in sucrose. Glycosylphosphatidylinositol (GPI)-anchored proteins, dually acylated src family kinases, heterotrimeric G proteins, and immune receptors, including the T cell receptor, the B cell receptor (BCR), and FcɛRI, associate with lipid rafts; the association contributes to receptor signaling (35, 36). Modification with saturated acyl chains targets GPI-anchored or cytosolic proteins to lipid rafts (37). For transmembrane proteins, palmitoylation, specific transmembrane sequences, or aggregation mediates raft association (37).
LMP1 and TRAF3, a TRAF that is tightly associated with LMP1, are enriched in rafts and associate with the cytoplasmic cytoskeleton (38–41). However, the mechanisms that target LMP1 to lipid rafts and to the cytoskeleton and the extent to which LMP2A shares these attributes have not been investigated. LMP1 has 3 cysteines within its membrane-spanning domain residues, and LMP2A has 10 cysteines that could be important for posttranslational acylation and membrane interactions.
Materials and Methods
Plasmids.
Flag-LMP1 expression vectors pSG5 Flag-LMP1(F-LMP1), pSG5 F-LMP1(P204A/Q206A) (F-LMP1 PQAA), pSG5 F-LMP1-ID, pSG5 F-LMP1(1–231), pSG5 F-LMP1(1–231)PQAA have been described previously (22, 23). Hemagglutinin (HA)-LMP2A expression vector pLMP2AHA was obtained from Richard Longnecker (Northwestern University, Chicago) (30). Flag-tagged LMP1 vectors with each single cysteine, cysteine-78, cysteine-84, or cysteine-116, mutated to alanine (C78A, C84A, C116A) or with all three cysteines mutated to alanines (C3A) were constructed by PCR. The JNK1 expression vector pcDNA3 Flag-JNK1 was provided by Roger Davis (Univ. of Massachusetts Medical School, Worcester).
Cell Lines and Transfections.
LCLs transformed by an EBV recombinant that expresses Flag-tagged LMP1 or wild-type LMP1 have been described previously (42). ES5 is an LCL infected with an EBV recombinant that expresses a null mutant LMP2A and was provided by Richard Longnecker (43). BJAB is an EBV-negative Burkitt's lymphoma (BL) cell line. 293 and 293T are human embryonic kidney cell lines. Cells were grown in RPMI 1640 (B cell lines) or DMEM (293 and 293T) supplemented with 10% FCS. Plasmid DNA was introduced by electroporation (BJAB) or by Superfect (293 and 293T) (Qiagen).
Antibodies.
The following antibodies were used: mouse anti-LMP1 mAb (S12) (44); rat anti-LMP2A mAb (14B7) (45); anti-Lyn (H-6), HA (F-7), TRAF1 (H-126 and 132), TRAF2 (C-20), TRAF3 (H-122), TRAF5 (H-257), and TRADD (C-20, N-19, and V-20) from Santa Cruz Biotechnology; anti-Flag M2, M5 mAb and M2 affinity gel from Sigma; anti-phospho-JNK and anti-JNK from New England Biolabs; and anti-CD71 from Zymed.
Detergent Extraction and Flotation Assay.
Cells (2 × 107) were lysed at 0°C in 1 ml of 1% or 0.2% Triton X-100 in MNE buffer (25 mM Mes, pH 6.5/150 mM NaCl/5 mM EDTA) containing 20 μg/ml aprotinin, 1 mM PMSF, and 1 mM sodium orthovanadate, subjected to 10 strokes of tight-fitting Dounce, and mixed with 1 ml of 80% sucrose in MNE buffer. The lysate was transferred to a centrifuge tube and overlaid with 2 ml of 30% sucrose and 1 ml of 5% sucrose. After centrifugation for 18 h at 200,000 × g, 0.4-ml fractions were collected from the top of the gradient. Fractions were mixed with 0.2 ml of 3× SDS sample buffer. The pellet fraction was suspended in 0.4 ml of 1% Triton X-100 in MNE buffer, mixed with SDS sample buffer, and sonicated. Twenty microliters of the individual fractions was subjected to SDS/PAGE, transferred to nitrocellulose membranes, and probed with indicated antibodies. For immunoprecipitation, fractions were mixed with 0.4 ml of 120 mM octyl β-glucoside in MNE buffer. The pellet fraction was suspended in 0.4 ml of 1% SDS, solubilized by sonication, and mixed with 0.4 ml of 10% Triton X-100 in MNE buffer.
Metabolic Labeling with [3H]Palmitate.
293T or BJAB cells were transiently transfected with pSG5 Flag-LMP1, pLMP2AHA, or empty vector and, after 24 h, were labeled for 3 h with 0.5 mCi [3H]palmitate in medium containing 5% FCS and 5 mM sodium pyruvate. Cells were lysed in ice-cold Nonidet P-40 lysis buffer (1% Nonidet P-40/50 mM Tris⋅HCl, pH 7.8/140 mM NaCl/1 mM EDTA/1 mM PMSF/20 μg/ml aprotinin). Cleared lysates were immunoprecipitated with anti-Flag M2 affinity gel or anti-HA antibody. Precipitated proteins then were separated by SDS/PAGE under nonreducing conditions, and the gels were fixed, treated with Amplify (Amersham Pharmacia), and exposed to film for 3 weeks at −80°C.
NF-κB and JNK Assays.
Briefly, 293 cells were transfected with wild-type or mutant Flag-LMP1 expression vectors, a luciferase reporter driven by three NF-κB-binding sites from the MHC class I promoter, and pGK-β-galactosidase to monitor transfection efficiency (23). For JNK assay, 293T cells were cotransfected with empty vector, wild-type or mutant Flag-LMP1 expression vectors, and with JNK1 expression vector. After 24 h, cells were lysed with SDS sample buffer, and the lysates were probed by Western blot with phospho-JNK or JNK antibody.
Results
LMP1 and LMP2A Associate with Lipid Rafts.
The association of LMP1 with lipid rafts was confirmed by using LCLs transformed by an EBV recombinant that expresses Flag-tagged LMP1. Cells were lysed in 0.2% Triton X-100, and the lysate in 40% sucrose was subjected to centrifugation and flotation in a 5%, 30%, 40% sucrose step gradient. Fractions were collected from the top and probed with anti-LMP1 antibody (Fig. 1A). Fractions 3 and 4, at the interface between 5% and 30% sucrose, are enriched for lipid rafts because of their buoyancy, whereas fractions 8–12 are the initial 40% sucrose fraction, and fraction 13 is the pellet. The pellet fraction contains the cell cytoskeleton and nucleus. About 10% of LMP1 was in the lipid raft fraction, 60–70% was in the soluble fractions, and 20–30% was in the pellet fraction (Fig. 1A). The presence of LMP1 in the pellet fraction is consistent with the known LMP1 association with vimentin intermediate filaments and the cell cytoskeleton (40). The same samples were probed with anti-Lyn and anti-CD71 (transferrin receptor) antibody. Lyn was mostly in the lipid raft and pellet fractions, whereas CD71 was completely excluded from the lipid raft and pellet fractions and only in the Triton-soluble fractions. This indicates that nonraft membranes were fully solubilized.
Figure 1.

Association of LMP1, LMP2A, and TRAF3 with lipid rafts. (A) LCLs expressing Flag-LMP1 were lysed in 0.2% Triton X-100 lysis buffer, the lysates were subjected to buoyant sucrose gradient centrifugation, and equal aliquots of each fraction from the top were probed with anti-LMP1, Lyn, CD71, and LMP2A antibodies. (B) The same samples were probed with anti-TRAF1, TRAF2, TRAF3, TRAF5, and TRADD antibodies. Fractions 3 and 4, 8–12, and 13 represent lipid rafts, soluble, and pellet fractions, respectively.
The association of LMP2A with lipid rafts then was investigated by probing the same samples with anti-LMP2A antibody. Similar to LMP1, about 30% of LMP2A is associated with lipid rafts and soluble fractions also contain LMP2A (Fig. 1A). LMP2A was absent from the pellet fraction, indicating that LMP2A is not associated with the insoluble cell cytoskeleton.
We also examined the lipid raft association of TRAFs and TRADD (Fig. 1B). TRAF1, TRAF2, TRAF5, and TRADD were mostly in the soluble fractions and not in lipid rafts. TRAF1 and 2 also were in the pellet fraction, whereas TRAF5 and TRADD were not. TRAF3 was associated partially with lipid rafts, consistent with a previous report (39). However, in contrast to the previous report, most of TRAF3 was in the pellet fraction, indicating a strong association with the cell cytoskeleton in LCLs. The partial localization of LMP1 and TRAF3 to noncytoskeletally associated or free lipid rafts and the extensive association of LMP1, TRAF3, and TRAF1 with the cell cytoskeleton in LCLs as opposed to non-EBV-infected BL cells (see below) likely are a result of the close association of these TRAFs with LMP1 and their activation by LMP1 (22–24).
LMP1 and LMP2A Are Palmitoylated.
To investigate whether LMP1 and LMP2A are palmitoylated as many other raft-associated proteins, 293T, a human embryonic kidney cell line, or BJAB, an EBV-negative BL cell line, were transiently transfected with Flag-LMP1 or HA-LMP2A expression vectors and metabolically labeled with [3H]palmitate. LMP1 or LMP2A then were immunoprecipitated with anti-Flag M2 or anti-HA antibody, respectively, and subjected to SDS/PAGE and fluorography. [3H]Palmitate was found to be incorporated into Flag-LMP1 and HA-LMP2A in both 293T and BJAB cells (Fig. 2A). To examine whether LMP1 is palmitoylated in LCLs, IB4 cells that express wild-type LMP1 and LCLs that express Flag-LMP1 were metabolically labeled with [3H]palmitate and immunoprecipitates with M2 affinity gel were analyzed as above. Again, [3H]palmitate was found to be incorporated into Flag-LMP1, and M2 affinity gel did not precipitate LMP1 without the Flag tag (Fig. 2B). These results demonstrate that LMP1 and LMP2A are palmitoylated similar to other lipid raft-associated proteins.
Figure 2.
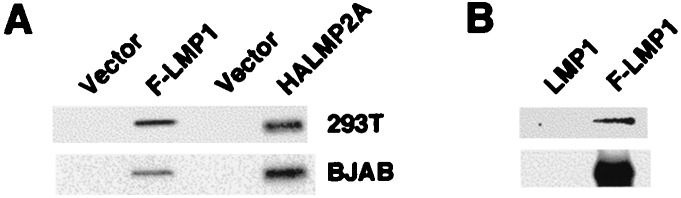
Palmitoylation of LMP1 and LMP2A. (A) 293T or BJAB cells were transiently transfected with pSG5 (Vector), pSG5 Flag-LMP1 (F-LMP1), or pLMP2AHA expression vectors and labeled with [3H]palmitate. The immunoprecipitates with anti-Flag M2 affinity gel or anti-HA antibody were analyzed by fluorography. (B) Wild-type LCLs (LMP1) or Flag-LMP1 expressing LCLs (F-LMP1) were labeled with [3H]palmitate, immunoprecipitated with M2 gel, and analyzed by fluorography (Upper). Ten percent of each immunoprecipitate was probed with LMP1 antibody (Lower).
Cysteine-78 Is the Principal Site for LMP1 Palmitoylation.
Palmitoylation of integral membrane proteins usually occurs on cysteines located near the cytoplasmic side of the plasma membrane (46). To test whether cysteine residues 78, 84, or 116, which are located within the membrane-spanning domains of LMP1, are palmitoylated, vectors for expression of Flag-tagged LMP1 with each single cysteine mutated to alanine (C78A, C84A, C116A) or with all three cysteines mutated to alanines (C3A) were constructed (Fig. 3A). BJAB cells were transiently transfected with wild-type or mutated Flag-LMP1 vectors and metabolically labeled with [3H]palmitate. Flag-LMP1 was immunoprecipitated with M2 affinity gel and subjected to SDS/PAGE and fluorography (Fig. 3B Upper). Whereas Flag-LMP1 incorporated [3H]palmitate, C3A Flag-LMP1 did not incorporate [3H]palmitate. This indicates that palmitoylation requires at least one of the three cysteines. Among the single cysteine-to-alanine mutants, only C78A mutant failed to incorporate [3H]palmitate whereas C84A and C116A incorporated [3H]palmitate (Fig. 3B Upper). Western blot analysis with Flag antibody M2 indicates that similar amounts of mutated Flag-LMP1 were expressed (Fig. 3B Lower). Thus, Flag-LMP1 is palmitoylated at cysteine-78, consistent with other reports that cysteines located near the cytoplasmic side of the plasma membrane are preferred sites for palmitoylation of integral membrane proteins.
Figure 3.

Effect of cysteine mutation on LMP1 palmitoylation. (A) Diagram of LMP1. LMP1 consists of a 24-aa N-terminal cytoplasmic domain, 6 hydrophobic membrane-spanning domains, and a 200-aa carboxyl-terminal cytoplasmic domain. The Flag epitope was introduced at the N terminus. The two signaling domains, amino acid 187–231 (TES1) and amino acid 352–386 (TES2), are represented by boxes. Residues directly implicated in TRAF or TRADD binding are indicated. Three cysteine residues in the membrane-spanning domains (cysteines 78, 84, and 116) are represented by open circles. (B) BJAB cells were transiently transfected with pSG5 Flag-LMP1 (WT) or cysteine-to-alanine-mutated C3A, C78A, C84A, or C116A Flag-LMP1 and labeled with [3H]palmitate. Cells were lysed and immunoprecipitated with M2 gel and analyzed by fluorography (Upper). Ten percent of the immunoprecipitates were subjected to Western blotting with Flag antibody (Lower).
LMP2A has 10 cysteine residues that are likely to be palmitoylated, and LMP2A may be multiply palmitoylated.
Palmitoylation Is Not Necessary for LMP1 Association with B Lymphoblast Lipid Rafts or for NF-κB or JNK Activation.
Palmitoylation is necessary for some membrane proteins such as influenza virus HA, HIV gp160, LAT, or CD8 to associate with lipid rafts (37, 47–49). To examine the role of palmitoylation in LMP1 association with rafts, BJAB cells that stably express wild-type Flag-LMP1 or nonpalmitoylated C78A Flag-LMP1 were generated. Wild-type Flag-LMP1 was associated with lipid rafts in BJAB cells as in LCLs (Fig. 4A). Lyn was more highly associated with lipid rafts and CD71 was excluded completely from them (data not shown). This result indicates that the association of LMP1 with lipid rafts does not require other latent EBV membrane proteins such as LMP2A and LMP2B. Nonpalmitoylated C78A Flag-LMP1 also was associated with lipid rafts, and the level of association was similar to wild-type Flag-LMP1 (Fig. 4A). Similarly, when using 1% Triton X-100, which is a more stringent condition than 0.2%, the raft association of wild-type and C78A Flag-LMP1 decreased slightly, but the level of association remained comparable (Fig. 4B). These results indicate that LMP1 is different from HA, LAT, or CD8 but similar to caveolin in not requiring palmitoylation for lipid raft association (50). C78A LMP1 aggregated in a patch in BJAB cells by immunofluorescence microscopy and was indistinguishable from wild-type LMP1 (data not shown).
Figure 4.
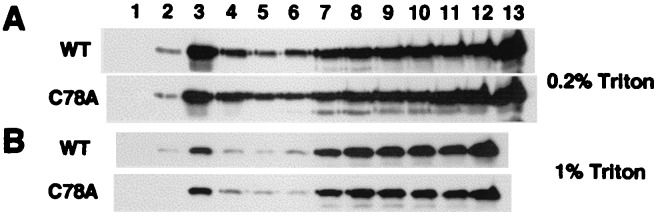
Lipid raft association of wild-type and nonpalmitoylated C78A Flag-LMP1. BJAB cells stably transfected with wild-type or C78A Flag-LMP1 expression vectors were lysed in 1% (A) or 0.2% (B) Triton X-100 lysis buffer, and the lysates were subjected to sucrose gradient centrifugation. Gradient fractions were probed with LMP1 antibody.
To investigate the relationship of LMP1 palmitoylation to signal transduction, 10, 30, 100, or 300 ng of wild-type or nonpalmitoylated C78A Flag-LMP1 was transfected into 293 cells with an NF-κB luciferase reporter (Fig. 5A Upper). As expected, larger amounts of DNA resulted in more LMP1 expression and more NF-κB activation (Fig. 5A Lower). Wild-type and C78A Flag-LMP1 were similar in NF-κB activation at all levels of expression. To examine the effect of palmitoylation on JNK activation, 293T cells were cotransfected with empty vector, wild-type Flag-LMP1, or C78A Flag-LMP1 expression vectors and with JNK1 expression vector, and the cell lysates were probed by Western blot with phospho-JNK and JNK antibody to assess JNK activation. Wild-type and nonpalmitoylated C78A Flag-LMP1 activated JNK similarly (Fig. 5B Upper) but did not alter JNK levels (Fig. 5B Lower). These results indicate that palmitoylation does not affect LMP1-mediated NF-κB or JNK activation. Consistent with these results, wild-type and C78A Flag-LMP1 did not differ in inducing expression of TRAF1 and cell surface markers, such as intercellular adhesion molecule-1, LFA-3, and Fas, in transiently transfected EBV-negative BL cell lines (data not shown). Thus, palmitoylation does not alter LMP1-signaling effects in lymphoblasts.
Figure 5.
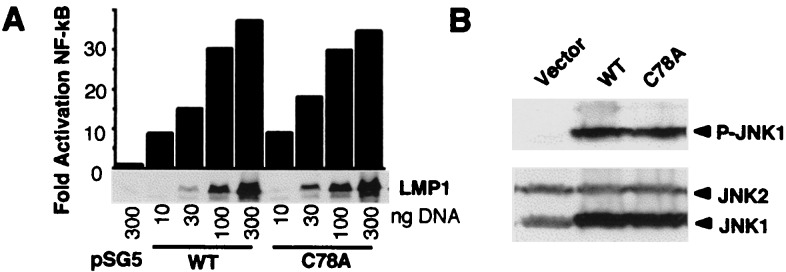
Activation of NF-κB and JNK by wild-type and nonpalmitoylated C78A Flag-LMP1. (A) 293 cells (7 × 105) were cotransfected with the indicated amount of wild-type (WT) LMP1 or C78A Flag-LMP1 expression vector or pSG5 vector control, 300 ng of an NF-κB-dependent luciferase reporter, and 300 ng of pGK-β-galactosidase vector as transfection control. Data are presented as luciferase activity normalized to β-galactosidase activity and adjusted to 1 for pSG5 control-transfected cells. Protein levels in the transfected cells were analyzed by Western blotting with LMP1 antibody. (B) 293T cells (7 × 105) were cotransfected with 500 ng of Flag-LMP1 or C78A Flag-LMP1 expression vector and 1 μg of pcDNA3 Flag-JNK1 and analyzed by Western blotting with phospho-JNK or JNK-specific antibody.
TRAF Binding Is Not Necessary for LMP1 Association with Lipid Rafts but Is Important for Its Cytoskeletal Association.
To investigate the involvement of the cytoplasmic carboxyl terminus of LMP1, especially TES1 and TES2, in LMP1 raft association, BJAB cell lines were made that stably express wild-type LMP1 or LMP1 PQAA (P204QQ206-to-AQA mutation) or ID (Y384YD386-to-ID mutation), which are mutated in the TRAF and TRADD association sites, respectively (22, 27) (Fig. 3A). The associations of mutant and wild-type LMP1 with lipid rafts were compared after solubilization of cells in 0.2% Triton X-100. Wild-type LMP1 was concentrated equally in the peak lipid raft (fraction 3), soluble (fraction 12), and pellet (fraction 13) fractions as assessed by Western blot analysis with LMP1 antibody S12 (Fig. 6A). The LMP1 distribution was similar to that in LCLs, although less LMP1 was associated with the cytoskeleteton in BJAB cells (data not shown). LMP1 PQAA, which is mutated in TES1, was barely detectable in the cytoskeletal fraction and was more concentrated in the soluble and free raft fraction. Because this mutation abrogates LMP1 association with TRAFs (23), the results indicate that TRAF binding is not critical for LMP1 presence in lipid rafts but is critical for LMP1 association with the cytoskeleton. In this regard, raft association is independent of cytoskeletal association, although, in general, cytoskeletal association may preclude flotation into the free raft fraction. The LMP1 ID mutant was found in all fractions, but the cytoskeletal fraction was greatly reduced, suggesting that TES2-mediated TRADD binding is not necessary for raft association but is important in wild-type LMP1 association with the cytoskeleton.
Figure 6.

Association of LMP1 mutants with lipid rafts and cytoskeleton. (A) Triton X-100 (0.2%) lysates from BJAB cells or cells stably expressing wild-type LMP1, PQAA, or ID mutant were subjected to sucrose gradient centrifugation, and aliquots of the peak lipid raft (r), soluble (s), and pellet (p) fractions were probed with anti-LMP1, TRAF3, TRAF2, or TRAF1 antibodies. (B) Gradient fractions from wild-type, 1–231, or 1–231 PQAA LMP1-expressing cells were immunoprecipitated with M2 gel and probed with Flag antibody. Arrows indicate wild-type or mutant LMP1. Ig heavy chain is indicated by asterisk. (C) Lysates from LMP2A null mutant, D1LMP1-expressing ES5 cells were subjected to sucrose gradient centrifugation, and the fractions were probed with LMP1 antibody.
The same samples were probed with anti-TRAF3, TRAF2, and TRAF1 antibodies (Fig. 6A). In parental BJAB cells, a small fraction of TRAF3 was in the raft fraction and a significant amount of TRAF3 was in the pellet fraction. In wild-type LMP1-expressing cells, the amount of TRAF3 in the raft fraction was increased and the amount in the cytoskeletal fraction was increased considerably, indicating that LMP1 causes the redistribution of TRAF3 to lipid rafts and the cytoskeleton. As expected, LMP1 PQAA, which is mutated in the TES1 TRAF-binding site, did not cause such effects and LMP1 ID showed increased raft association only. TRAF2 distribution was not affected by wild-type or mutant LMP1 expression. TRAF1 expression was up-regulated in wild-type and ID mutant-expressing BJAB cells, but TRAF1 remained predominantly in the soluble fraction and was only weakly distributed to the cytoskeletal fractions. Thus, TRAF3 was unique among the TRAFs in having a distribution that correlated with LMP1.
Membrane Spanning (and N-Terminal) Domains of LMP1 Mediate Raft Association and Require the C-Terminal Cytoplasmic-Signaling Domains for Cytoskeletal Association.
BJAB cell lines that stably express LMP1 mutants 1–231 (C terminus with TES1 only; Fig. 3A) or 1–231PQAA (C terminus with a nonfunctional TES1 only) were used to examine the role of the LMP1 transmembrane domains and of TES1 in lipid raft and cytoskeletal association. Cell lysates were subjected to sucrose gradient centrifugation, and LMP1 was immunoprecipitated with anti-Flag antibody from each fraction followed by Western blot analysis with anti-Flag antibody (Fig. 6B). The distribution of LMP1 1–231 was similar to WT LMP1. Surprisingly, LMP1 1–231PQAA was mostly in the raft fraction and not in the soluble or pellet fractions. This result indicates that the membrane-spanning and N-terminal cytoplasmic domains of LMP1 are sufficient for raft association, that these domains have strong affinity to lipid rafts, and that LMP1 association with TRAFs modifies the intrinsic raft-associating character of the LMP1 transmembrane domains, causing LMP1 to be mostly in the soluble, nonraft, and cytoskeletal fractions.
To investigate further the role of the LMP1 membrane-spanning and N-terminal domains in raft association, the distribution of D1LMP1, which is composed of the last two transmembrane domains and the entire LMP1 cytoplasmic domain, was examined. D1LMP1 does not aggregate in the plasma membrane and is nonfunctional (11, 19). For these experiments, we used an LCL that expresses both full-length LMP1 and D1LMP1. This LCL expresses D1LMP1 in latent infection because it is infected with an EBV recombinant that has an simian virus 40 promoter and enhancer that up-regulates the D1LMP1 promoter (51). Whereas full-length LMP1 was in the free lipid raft, soluble, and pellet fractions, D1LMP1 was found only in soluble fractions (Fig. 6C). This result confirms the importance of the multiple membrane-spanning domains and N terminus in LMP1 association with lipid rafts and the cytoskeleton. Previous genetic and biochemical analyses indicate that the N-terminal cytoplasmic domain primarily anchors the N terminus of the first transmembrane domain in the cytoplasm to enable the six hydrophobic transmembrane domains to mediate LMP1 aggregation and constitutive signaling through the C-terminal cytoplasmic domain (18, 22–24, 42). Thus, these data are most compatible with a model in which the LMP1 multiple hydrophobic transmembrane domains mediate raft association and homoaggregation, whereas homoaggregation causes TRAF aggregation on the LMP1 C-terminal cytoplasmic TRAF-binding site with signaling and cytoskeletal association mediated by TRAFs.
Discussion
The experiments described above demonstrate that both LMP1 and LMP2A are palmitoylated and associate with lipid rafts in B lymphoblasts. Further genetic and biochemical analyses of LMP1 identify cysteine-78 as the primary site for palmitoylation and indicate that palmitoylation is not critical for LMP1 association with lipid rafts or for NF-κB or JNK activation. The LMP1 six hydrophobic membrane-spanning domains expressed in the context of N- and short C-terminal cytoplasmic domains that do not appear to mediate significant protein–protein interactions are capable of high level and nearly exclusive association with lipid rafts. LMP2A is composed of 12 hydrophobic membrane-spanning domain, and these are likely candidates for mediating the higher level raft association that is characteristic of LMP2A. In part, this may be due to the absence of a cytoskeletal association for LMP2A, which eliminates the possibility of dragging rafts into the cytoskeleton fraction.
Lipid rafts are important for signal transduction from B cell surface receptors including CD40 and surface Ig (sIg). Stimulation of CD40 with CD40 ligand or crosslinking antibody results in translocation of CD40 to lipid rafts and the association of TRAF2 and 3 with CD40 within lipid rafts (52). Disruption of lipid rafts before stimulation abrogates this association, suggesting that lipid rafts play a critical role in initiation of CD40 signaling (52). Lipid raft association could be critical for LMP1 or LMP2A signaling, and LMP1 or LMP2A association with lipid rafts could effect signaling from other raft-interacting receptors, such as CD40 or sIg.
Immunoreceptors including FcɛRI, T cell receptor, and BCR complexes associate with lipid rafts after receptor clustering, and this association seems to be important for initial tyrosine phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) motifs and the recruitment and activation of cytosolic-signaling molecules (36). Like LMP1, LMP2A spontaneously aggregates in the plasma membrane, causing the N-terminal ITAM motif to be tyrosine-phosphorylated by Lyn and Syk. In analogy to immunoreceptors that associate with rafts, LMP2A association with lipid rafts may facilitate the interaction with Lyn, the subsequent tyrosine phosphorylation of the Lyn-binding and ITAM domains, Syk recruitment, and subsequent signaling and desensitizing events that are critical for LMP2A function. LMP2A partially activates Akt through phosphoinositide 3-kinase (PI3K), and this may have survival effects in some epithelial cell lines (16, 32). Lipid rafts are known to be enriched in a PI3K substrate, phosphatidylinositol-4,5-bisphosphate (56), and the LMP2A lipid raft association may be important for PI3K activation. In epithelial cells, LMP1 induces EGF receptor (EGFR) expression (57), and the presence of LMP1 and LMP2A in lipid rafts could effect EGFR activation, enhance EGFR signaling, and contribute to transforming phenotype of LMP1 and 2A expressing epithelial cells. During submission of this paper, LMP2A was reported to associate with rafts and block BCR raft association and signaling (58). Thus, LMP2A raft association may be important for constitutive BCR desensitization.
Because of the large number of cysteines in the LMP2A transmembrane domains, we do not know whether palmitoylation of LMP2A is required for LMP2A association with lipid rafts or tyrosine kinase-mediated effects. The LMP2A 12 transmembrane domains are rich in leucine and isoleucine residues and may not be required for raft association. Palmitoylation, however, may effect LMP2A interaction with raft-associated tyrosine kinases or downstream-signaling molecules.
Only a small fraction of wild-type LMP1 in LCLs is associated with noncytoskeleton or free lipid rafts. TRAF1, 2, and 5 and TRADD, which are important for NF-κB and JNK activation by LMP1, are not tightly associated with LMP1 in free lipid rafts. Only TRAF3, a TRAF that is tightly associated with LMP1 and does not directly activate NF-κB or JNK, was represented significantly in the raft fraction of LCLs. The association of LMP1 with other TRAFs and TRADD appears to be responsible for the extensive LMP1 localization to soluble membrane and cytoskeletal fractions. The LMP1 1–231PQAA mutant, which no longer binds TRAFs and TRADD, shows complete lipid rafts association, and LMP1 PQAA is distributed between the raft and soluble membrane fractions. LMP1 initially may aggregate in rafts. Recruitment of TRAFs and TRADD may cause LMP1 to move out of the raft fraction or cause the raft fraction to associate with the cytoskeleton. The activated BCR complex transiently associates with lipid rafts, indicating that raft association can be a dynamic process (53).
The unique association of TRAF3 with the raft fraction of LCLs may be due to its particularly strong binding to LMP1 or to a propensity for raft association because a small amount of TRAF3 is associated with rafts in BL cells in the absence of LMP1 expression. Given the intrinsic association of LMP1 transmembrane domains with rafts, binding to LMP1 is expected to enhance TRAF3 association with rafts. Other TRAFs may dissociate from LMP1 during raft purification or after activation by LMP1.
LMP1 directly or indirectly interacts with vimentin intermediate filaments and with the cytoplasmic cytoskeleton (40). The experiments described here show that point mutations in the TRAF-binding site of LMP1 abolish cytoskeletal association. This indicates that TRAFs have a critical role in bridging between LMP1 and the cytoskeleton. TRAF3 and TRAF2 associate with the BL cell cytoplasmic cytoskeleton in the absence of LMP1. LMP1 expression increases the association of TRAF3 with the cytoskeleton and causes TRAF1 to associate with the cytoskeleton. TRAF2 and TRAF3 can interact with actin and microtubule-binding proteins, respectively, and these interactions are likely to mediate TRAF1 and LMP1 cytoskeletal association (41, 55). Because the TRAF-binding site of LMP1 is critical for B cell transformation (42), cytoskeletal association may be important in LMP1 effects in B cell transformation.
Unlike influenza virus HA, HIV gp160, LAT, and CD8, palmitoylation of LMP1 is not necessary for LMP1 lipid rafts association. Rafts may be in a liquid-ordered phase with a high degree of acyl chain order, and lipids that have saturated acyl chains such as palmitate fit well into an ordered environment (34). However, dual palmitoylation is not sufficient for targeting of vesicular stomatitis virus glycoprotein G or transferrin receptor to lipid rafts (37), whereas the transmembrane domain sequences and palmitoylation are important for targeting HA to lipid rafts (37, 59). In addition, clustering of membrane proteins seems to increase their affinity for lipid rafts and is important for the formation of large rafts (34). The LMP1 membrane-spanning domains are particularly rich in leucine and isoleucine residues, and these short acyl R groups may suffice for LMP1 association with lipid rafts.
Despite our failure to demonstrate an effect of palmitoylation on LMP1 function, palmitoylation could affect an aspect of LMP1 signaling that is important for cell growth or survival. Palmitoylation may affect signals that originate from the LMP1 membrane-spanning domains or from raft-associated membrane proteins including CD40. An effect of the membrane-spanning domains has been suggested from differences in signaling between the nasopharyngeal carcinoma-derived Cao and prototype B95–8 LMP1 (60). Cysteine-78 is conserved in all published EBV LMP1 gene sequences, and the rhesus lymphocryptovirus LMP1 homologue has cysteine residues 76, 82, and 111 (61–64). The conservation of these residues suggests that they may be important for an aspect of EBV infections. The membrane-spanning domains of LMP1 are also important in Cdc42 activation and inhibiting of cell proliferation (65, 66). The phenotype of nonpalmitoylated LMP1 in the initiation and long-term maintenance of B lymphocyte transformation in vitro and the growth properties of such cells in vivo in severe combined immunodeficiency mice can be analyzed by introducing the C78A cysteine-mutated Flag-LMP1 into an EBV recombinant. Several primate lymphocryptovirus models have been described (67), and these offer additional possibilities for in vivo analysis of the effects of LMP1 palmitoylation in pathogenesis.
Acknowledgments
We thank Drs. Lian Wang, Ellen Cahir McFarland, George Mosialos, and Bakary Sylla for technical assistance and instructive discussions. We also thank Drs. Richard Longnecker and Roger Davis for the generous gifts of reagents. This work was supported by Public Health Service Grants CA47006 and CA85180 from the National Cancer Institute, National Institutes of Health, and by a grant from Uehara Memorial Foundation.
Abbreviations
- EBV
Epstein–Barr virus
- LMP
latent infection membrane protein
- LCL
lymphoblastoid cell line
- TNFR
tumor necrosis factor receptor
- TRAF
TNFR-associated factor
- TRADD
TNFR-associated death domain
- JNK
c-Jun N-terminal kinase
- BCR
B cell receptor
- TES1 and 2
transformation effector sites 1 and 2
- BL
Burkitt's lymphoma
- HA
hemagglutinin
References
- 1.Rickinson A B, Kieff E. In: Fields Virology. Fields B N, Knipe D M, Howley P M, editors. Philadelphia: Lippincott; 1996. pp. 2397–2446. [Google Scholar]
- 2.Kieff E. In: Fields Virology. Fields B N, Knipe D M, Howley P M, editors. Philadelphia: Lippincott; 1996. pp. 2343–2396. [Google Scholar]
- 3.Liebowitz D, Wang D, Kieff E. J Virol. 1986;58:233–237. doi: 10.1128/jvi.58.1.233-237.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Longnecker R, Kieff E. J Virol. 1990;64:2319–2326. doi: 10.1128/jvi.64.5.2319-2326.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Liebowitz D, Kieff E. Cell. 1985;43:831–840. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- 6.Eliopoulos A G, Gallagher N J, Blake S M, Dawson C W, Young L S. J Biol Chem. 1999;274:16085–16096. doi: 10.1074/jbc.274.23.16085. [DOI] [PubMed] [Google Scholar]
- 7.Eliopoulos A G, Young L S. Oncogene. 1998;16:1731–1742. doi: 10.1038/sj.onc.1201694. [DOI] [PubMed] [Google Scholar]
- 8.Hammarskjold M L, Simurda M C. J Virol. 1992;66:6496–6501. doi: 10.1128/jvi.66.11.6496-6501.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatzivassiliou E, Miller W E, Raab-Traub N, Kieff E, Mosialos G. J Immunol. 1998;160:1116–1121. [PubMed] [Google Scholar]
- 10.Laherty C D, Hu H M, Opipari A W, Wang F, Dixit V M. J Biol Chem. 1992;267:24157–24160. [PubMed] [Google Scholar]
- 11.Wang D, Liebowitz D, Wang F, Gregory C, Rickinson A, Larson R, Springer T, Kieff E. J Virol. 1988;62:4173–4184. doi: 10.1128/jvi.62.11.4173-4184.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang F, Gregory C, Sample C, Rowe M, Liebowitz D, Murray R, Rickinson A, Kieff E. J Virol. 1990;64:2309–2318. doi: 10.1128/jvi.64.5.2309-2318.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaye K M, Izumi K M, Kieff E. Proc Natl Acad Sci USA. 1993;90:9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burkhardt A L, Bolen J B, Kieff E, Longnecker R. J Virol. 1992;66:5161–5167. doi: 10.1128/jvi.66.8.5161-5167.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller C L, Burkhardt A L, Lee J H, Stealey B, Longnecker R, Bolen J B, Kieff E. Immunity. 1995;2:155–166. doi: 10.1016/s1074-7613(95)80040-9. [DOI] [PubMed] [Google Scholar]
- 16.Scholle F, Bendt K M, Raab-Traub N. J Virol. 2000;74:10681–10689. doi: 10.1128/jvi.74.22.10681-10689.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longnecker R, Miller C L, Miao X Q, Marchini A, Kieff E. J Virol. 1992;66:6461–6469. doi: 10.1128/jvi.66.11.6461-6469.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liebowitz D, Mannick J, Takada K, Kieff E. J Virol. 1992;66:4612–4616. doi: 10.1128/jvi.66.7.4612-4616.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D, Liebowitz D, Kieff E. J Virol. 1988;62:2337–2346. doi: 10.1128/jvi.62.7.2337-2346.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin J, Sugden B. J Virol. 1991;65:3246–3258. doi: 10.1128/jvi.65.6.3246-3258.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brodeur S R, Cheng G, Baltimore D, Thorley-Lawson D A. J Biol Chem. 1997;272:19777–19784. doi: 10.1074/jbc.272.32.19777. [DOI] [PubMed] [Google Scholar]
- 22.Devergne O, Hatzivassiliou E, Izumi K M, Kaye K M, Kleijnen M F, Kieff E, Mosialos G. Mol Cell Biol. 1996;16:7098–7108. doi: 10.1128/mcb.16.12.7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devergne O, McFarland E C, Mosialos G, Izumi K M, Ware C F, Kieff E. J Virol. 1998;72:7900–7908. doi: 10.1128/jvi.72.10.7900-7908.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 25.Sandberg M, Hammerschmidt W, Sugden B. J Virol. 1997;71:4649–4656. doi: 10.1128/jvi.71.6.4649-4656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eliopoulos A G, Blake S M, Floettmann J E, Rowe M, Young L S. J Virol. 1999;73:1023–1035. doi: 10.1128/jvi.73.2.1023-1035.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izumi K M, Kieff E D. Proc Natl Acad Sci USA. 1997;94:12592–12597. doi: 10.1073/pnas.94.23.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Izumi K M, McFarland E C, Ting A T, Riley E A, Seed B, Kieff E D. Mol Cell Biol. 1999;19:5759–5767. doi: 10.1128/mcb.19.8.5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fruehling S, Longnecker R. Virology. 1997;235:241–251. doi: 10.1006/viro.1997.8690. [DOI] [PubMed] [Google Scholar]
- 30.Fruehling S, Swart R, Dolwick K M, Kremmer E, Longnecker R. J Virol. 1998;72:7796–7806. doi: 10.1128/jvi.72.10.7796-7806.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Merchant M, Caldwell R G, Longnecker R. J Virol. 2000;74:9115–9124. doi: 10.1128/jvi.74.19.9115-9124.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swart R, Ruf I K, Sample J, Longnecker R. J Virol. 2000;74:10838–10845. doi: 10.1128/jvi.74.22.10838-10845.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown D A, London E. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 34.Brown D A, London E. J Biol Chem. 2000;275:17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 35.Simons K, Ikonen E. Nature (London) 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 36.Langlet C, Bernard A M, Drevot P, He H T. Curr Opin Immunol. 2000;12:250–255. doi: 10.1016/s0952-7915(00)00084-4. [DOI] [PubMed] [Google Scholar]
- 37.Melkonian K A, Ostermeyer A G, Chen J Z, Roth M G, Brown D A. J Biol Chem. 1999;274:3910–3917. doi: 10.1074/jbc.274.6.3910. [DOI] [PubMed] [Google Scholar]
- 38.Clausse B, Fizazi K, Walczak V, Tetaud C, Wiels J, Tursz T, Busson P. Virology. 1997;228:285–293. doi: 10.1006/viro.1996.8380. [DOI] [PubMed] [Google Scholar]
- 39.Ardila-Osorio H, Clausse B, Mishal Z, Wiels J, Tursz T, Busson P. Int J Cancer. 1999;81:645–649. doi: 10.1002/(sici)1097-0215(19990517)81:4<645::aid-ijc22>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 40.Liebowitz D, Kopan R, Fuchs E, Sample J, Kieff E. Mol Cell Biol. 1987;7:2299–2308. doi: 10.1128/mcb.7.7.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ling L, Goeddel D V. J Biol Chem. 2000;275:23852–23860. doi: 10.1074/jbc.M001095200. [DOI] [PubMed] [Google Scholar]
- 42.Izumi K M, Kaye K M, Kieff E D. Proc Natl Acad Sci USA. 1997;94:1447–1452. doi: 10.1073/pnas.94.4.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller C L, Lee J H, Kieff E, Longnecker R. Proc Natl Acad Sci USA. 1994;91:772–776. doi: 10.1073/pnas.91.2.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mann K P, Staunton D, Thorley-Lawson D A. J Virol. 1985;55:710–720. doi: 10.1128/jvi.55.3.710-720.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fruehling S, Lee S K, Herrold R, Frech B, Laux G, Kremmer E, Grasser F A, Longnecker R. J Virol. 1996;70:6216–6226. doi: 10.1128/jvi.70.9.6216-6226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunphy J T, Linder M E. Biochim Biophys Acta. 1998;1436:245–261. doi: 10.1016/s0005-2760(98)00130-1. [DOI] [PubMed] [Google Scholar]
- 47.Rousso I, Mixon M B, Chen B K, Kim P S. Proc Natl Acad Sci USA. 2000;97:13523–13525. doi: 10.1073/pnas.240459697. . (First Published November 28, 2000; 10.1073/pnas.240459697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang W, Trible R P, Samelson L E. Immunity. 1998;9:239–246. doi: 10.1016/s1074-7613(00)80606-8. [DOI] [PubMed] [Google Scholar]
- 49.Arcaro A, Gregoire C, Boucheron N, Stotz S, Palmer E, Malissen B, Luescher I F. J Immunol. 2000;165:2068–2076. doi: 10.4049/jimmunol.165.4.2068. [DOI] [PubMed] [Google Scholar]
- 50.Dietzen D J, Hastings W R, Lublin D M. J Biol Chem. 1995;270:6838–6842. doi: 10.1074/jbc.270.12.6838. [DOI] [PubMed] [Google Scholar]
- 51.Longnecker R, Miller C L, Tomkinson B, Miao X Q, Kieff E. J Virol. 1993;67:5068–5074. doi: 10.1128/jvi.67.8.5068-5074.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vidalain P O, Azocar O, Servet-Delprat C, Rabourdin-Combe C, Gerlier D, Manie S. EMBO J. 2000;19:3304–3313. doi: 10.1093/emboj/19.13.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrie R J, Schnetkamp P P, Patel K D, Awasthi-Kalia M, Deans J P. J Immunol. 2000;165:1220–1227. doi: 10.4049/jimmunol.165.3.1220. [DOI] [PubMed] [Google Scholar]
- 54.Liebowitz D, Kieff E. J Virol. 1989;63:4051–4054. doi: 10.1128/jvi.63.9.4051-4054.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leonardi A, Ellinger-Ziegelbauer H, Franzoso G, Brown K, Siebenlist U. J Biol Chem. 2000;275:271–278. doi: 10.1074/jbc.275.1.271. [DOI] [PubMed] [Google Scholar]
- 56.Hope H R, Pike L J. Mol Biol Cell. 1996;7:843–851. doi: 10.1091/mbc.7.6.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller W E, Mosialos G, Kieff E, Raab-Traub N. J Virol. 1997;71:586–594. doi: 10.1128/jvi.71.1.586-594.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dykstra M L, Longnecker R, Pierce S K. Immunity. 2001;14:57–67. doi: 10.1016/s1074-7613(01)00089-9. [DOI] [PubMed] [Google Scholar]
- 59.Scheiffele P, Roth M G, Simons K. EMBO J. 1997;16:5501–5508. doi: 10.1093/emboj/16.18.5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson R J, Stack M, Hazlewood S A, Jones M, Blackmore C G, Hu L F, Rowe M. J Virol. 1998;72:4038–4048. doi: 10.1128/jvi.72.5.4038-4048.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sample J, Kieff E F, Kieff E D. J Gen Virol. 1994;75:2741–2746. doi: 10.1099/0022-1317-75-10-2741. [DOI] [PubMed] [Google Scholar]
- 62.Miller W E, Edwards R H, Walling D M, Raab-Traub N. J Gen Virol. 1994;75:2729–2740. doi: 10.1099/0022-1317-75-10-2729. [DOI] [PubMed] [Google Scholar]
- 63.Fischer N, Kopper B, Graf N, Schlehofer J R, Grasser F A, Mueller-Lantzsch N. Virus Res. 1999;60:41–54. doi: 10.1016/s0168-1702(98)00147-6. [DOI] [PubMed] [Google Scholar]
- 64.Franken M, Devergne O, Rosenzweig M, Annis B, Kieff E, Wang F. J Virol. 1996;70:7819–7826. doi: 10.1128/jvi.70.11.7819-7826.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puls A, Eliopoulos A G, Nobes C D, Bridges T, Young L S, Hall A. J Cell Sci. 1999;112:2983–2992. doi: 10.1242/jcs.112.17.2983. [DOI] [PubMed] [Google Scholar]
- 66.Kaykas A, Sugden B. Oncogene. 2000;19:1400–1410. doi: 10.1038/sj.onc.1203365. [DOI] [PubMed] [Google Scholar]
- 67.Moghaddam A, Rosenzweig M, Lee-Parritz D, Annis B, Johnson R P, Wang F. Science. 1997;276:2030–2033. doi: 10.1126/science.276.5321.2030. [DOI] [PubMed] [Google Scholar]