

Abstract
Upregulated ERK1/2 activity is correlated with androgen receptor (AR) downregulation in certain prostate cancer (PCa) that exhibits androgen deprivation-induced neuroendocrine differentiation, but its functional relevance requires elucidation. We found that sustained ERK1/2 activation using active Raf or MEK1/2 mutants is sufficient to induce AR downregulation at mRNA and protein levels in LNCaP. Downregulation of AR protein, but not mRNA, was blocked by proteasome inhibitors, MG132 and bortezomib, indicating that the pathway regulation is mediated at multiple points. Ectopic expression of a constitutively active AR inhibited Raf/MEK/ERK-mediated regulation of the differentiation markers, neuron-specific enolase and neutral endopeptidase, and the cyclin-dependent kinase inhibitors, p16INK4A and p21CIP1, but not Rb phosphorylation and E2F1 expression, indicating that AR has a specific role in the pathway-mediated differentiation and growth inhibitory signaling. However, despite the sufficient role of Raf/MEK/ERK, its inhibition using U0126 or ERK1/2 knockdown could not block androgen deprivation-induced AR downregulation in an LNCaP neuroendocrine differentiation model, suggesting that additional signaling pathways are involved in the regulation. We additionally report that sustained Raf/MEK/ERK activity can downregulate full length as well as hormone binding domain-deficient AR isoforms in androgen-refractory C4-2 and CWR22Rv1, but not in LAPC4 and MDA-PCa-2b. Our study demonstrates a novel role of the Raf/MEK/ERK pathway in regulating AR expression in certain PCa types and provides an insight into PCa responses to its aberrant activation.
Keywords: Prostate cancer, androgen receptor, growth inhibition, differentiation, Raf, ERK1/2
Introduction
The Raf/MEK/extracellular signal-regulated kinase (ERK) pathway has pivotal roles in controlling cell survival, cell cycle progression and differentiation, and its dysregulated signaling is a central signature of many epithelial cancers [1, 2]. The Ser/Thr kinase Raf (c-Raf-1, Raf-B or Raf-A) activates the dual-specificity kinases MEK1 and MEK2 which, in turn, sequentially phosphorylate Tyr and Thr in the activation loop of the ubiquitously expressed Ser/Thr kinases ERK1 and its homologue ERK2. Activated ERK1/2 mediates diverse biological processes by activating/inactivating a wide variety of proteins, and different magnitudes of its activity can lead to distinct biological outputs [1]. Upregulated activity of the MEK/ERK pathway has been shown for its association with progression and poor prognosis of prostate cancer (PCa). For example, ERK1/2 phosphorylation is often detected in correlation with increased tumor grade of primary or metastatic PCa and tumor relapse after therapy [3–6]. In addition, although not frequent, mutations or chromosomal rearrangements of Raf genes that can confer neoplastic Raf kinase activity are detected in human PCa [7–11]. Together, these observations support pathogenetic relevance of the MEK/ERK pathway in prostate tumorigenesis and underscore the need for better understanding of the role of the pathway in PCa cells.
Androgen receptor (AR) is a member of the nuclear receptor superfamily that functions as a ligand-dependent nuclear transcription factor [12]. AR can control the growth regulatory and differentiation pathways in prostate epithelial cells, and its altered expression or signaling is a pivotal event in the carcinogenesis of prostate epithelium [13, 14]. While increased AR expression is a key feature of a subpopulation of castration-resistant PCa [15], loss of AR expression, accompanied by decreased mitotic activity, is also detected in certain PCa types that exhibit neuroendocrine (NE) differentiation [16, 17]. NE-differentiated cells in PCa are supposed to contribute to castration resistance of neighboring non-NE PCa cells by activating paracrine signaling mechanisms [18, 19]. Although the origin of NE differentiated cells in PCa is not fully understood, literatures support the possibility that prostate adenocarcinoma cells transdifferentiate into NE-like phenotype in response to androgen depletion. For example, NE differentiated cells in tumor lesions show identical genetic profiles with adjacent exocrine PCa cells [20], and certain human PCa cell lines, including LNCaP, CWR22 and PC310, exhibit expression of various makers for NE differentiation in vitro as well as in vivo in response to hormone withdrawal [21–23]. It has been shown that AR downregulation is required for NE differentiation [24], but the signaling mechanisms that underlie AR downregulation upon androgen withdrawal are as yet unclear.
Upregulated ERK1/2 activity is detected in NE cells in PCa as well as in NE-differentiating PCa cell lines while various stimulations that activate the Raf/MEK/ERK pathway can induce NE differentiation of PCa cells in in vitro and in vivo environments [19, 25]. Furthermore, ectopic expression of a constitutively active MEK1 mutant was indeed sufficient to mediate expression of NE markers in LNCaP cells [21]. We recently reported that sustained Raf/MEK/ERK activation can induce cell cycle arrest in G0/G1 phase in LNCaP [26], which may implicate the pathway in decreased mitotic activity of NE-transdifferentiating PCa cells. Together, these results indicate an involvement of Raf/MEK/ERK signaling in NE differentiation of PCa cells. Given the pivotal role of AR for PCa cell proliferation/survival and differentiation, it is conceivable that the Raf/MEK/ERK pathway may also have a role in mediating AR downregulation. Nevertheless, this possibility has never been addressed before.
In this study, we investigated whether the Raf/MEK/ERK pathway can mediate AR downregulation and, thereby, decrease cell proliferation during NE differentiation of LNCaP. We demonstrate that ERK1/2 activity and AR expression are inverse-correlated in an LNCaP model for hormone depletion-mediated NE differentiation and that sustained Raf/MEK/ERK activity is sufficient to mediate AR downregulation, although not necessary. We then examine how AR downregulation is mediated and whether it is required for the pathway-mediated growth arrest and differentiation signaling. Additionally, we show that the pathway can mediate AR downregulation in a subset of PCa lines regardless of androgen sensitivity. This study reveals previously unknown roles of the Raf/MEK/ERK pathway in PCa cells.
Materials and methods
Cell culture, generation of stable line
LNCaP (ATCC), PC3 (ATCC), Du145 (ATCC), and CWR22Rv1 (ATCC) were maintained in phenol red-deficient RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin and 100 μg of streptomycin per ml. LAPC-4 (ATCC) was grown in Iscove’s medium with 10% FBS. MDA-Pca-2b (ATCC) was grown in Hams F12 medium supplemented with 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 25 ng/ml cholera toxin, 10 ng/ml epidermal growth factor, 0.005 mM phosphoethanolamine, 100 pg/ml hydrocortisone, 45 nM selenious acid, 0.005 mg/ml insulin, and 20% FBS. C4-2, NE1.3 and NE1.8 cells were maintained in phenol red-deficient RPMI 1640 supplemented with 10% charcoal/dextran-stripped FBS (c.s.FBS). LNCaPRaf, stably transduced with lentivirus containing ΔRaf-1:ER, was previously described [26]. U0126, MG-132, Lactacystin, and 4-hydroxytamoxifen were obtained from Sigma (St. Louis, MO). Bortezomib was obtained from Selleck Chemicals (Houston, TX).
Viral infection
The lentiviral expression vector pHAGE and the lentiviral shRNA expression vector pLL3.7 (ATCC) were used as previously described [26]. Briefly, for viral production, pHAGE or pLL3.7 was co-transfected with packaging vectors into 293T cells and the resulting supernatant was collected after 48 h. Viral titers were determined by infecting the recipient cell lines with serially diluted viral supernatants mixed with polybrene (Sigma) at 8 μg/ml and scoring cells expressing GFP at 48 h post-infection. Cells were infected overnight and were switched into fresh culture media.
Recombinant lentiviral constructs and RNA interference
Generation of pHAGE-GFP-MEK1CA and pHAGE-GFP-MEK2CA containing constitutively active MEK1-R4F (ΔN3/S218E/S222D) and MEK2-KW71 (ΔN4/S222D/S226D), respectively, were previously described [26]. pHAGE-GFP-H-RasG12V and pHAGE-GFP-B-RafV600E contain the oncogenic human genes, H-RasG12V and B-RafV600E, respectively. pHAGE-GFP-AAø was constructed using the hormone binding domain-deficient AR mutant, AAø [27]. The siRNA oligomers targeting GACCTGAATTGTATCATC (ERK1 nucleotides 801–819), GAAACTACCTACAGTCTCT (ERK1 nucleotides 833–851), CCAAAGCTCTGGACTTATT (ERK2 nucleotides 851–869), GTACAGGGCTCCAGAAATT (ERK2 nucleotides 576–594), and the scrambled control were purchased from Dharmacon (Lafayette, CO). Cells were transfected using DharmaFECT 2 (Dharmacon) according to the manufacturer’s protocol. For stable expression of small hairpin RNA (shRNA), pLL3.7-shRNA systems targeting ERK1 (nucleotides 801–819) or ERK2 (nucleotides 851–869) were constructed and described previously [26].
Quantitative real time polymerase chain reaction (qPCR)
The qPCR analysis of AR was performed using the Mx3005P™ instrument (Stratagene, La Jolla, CA) by reverse transcription of 0.25 μg total RNA, and subsequent polymerase chain reaction using a Brilliant SYBR® Green QPCR Core Reagent Kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. Primers were CTACATCAAGGAACTCGATC and CTGGGTGTGGAAATAGATT (AR nucleotides 2,502–2,760) and the thermocycling conditions were 10 min at 95°C as first denaturation step, followed by 40 cycles at 95°C for 30 seconds, 55°C for 60 seconds and 72°C for 30 seconds. As the control for normalization, expression of phosphoglycerate kinase was measured using primers CAGTTTGGAGCTCCTGGAAG and TGCAAATCCAGGGTCCAGTG.
Calculation of the gene copy number was carried out using comparative threshold cycle, as previously described [28]. Briefly, the mean threshold cycle (mCT) was obtained from triplicate amplifications during the exponential phase of amplification. Then, mCT value of the reference gene was subtracted from mCT value of the target gene (AR) to obtain ΔCT. ΔΔCT values of each sample was calculated from corresponding CT values; where ΔΔCT = [mCT target (normal sample) − mCT reference (normal sample)] − [mCT target (test sample) − mCT reference (test sample)]. The calculated ΔΔCT was converted to ratio using the ratio formula (Ratio = 2−ΔΔCT).
Prostate-specific antigen (PSA)-luciferase reporter assay
Cells were transfected with the PSA-luciferase reporter construct harboring 6.0 kilo base pairs of PSA promoter DNA (obtained from Shuyuan Yeh, Univ. of Rochester) using Lipofectamine 2000 (Invitrogen). Transfected master cultures were then divided in triplicate into 24-well plates for further treatments, as described in the text. Cell lysates were analyzed using the Luciferase Assay System (Promega) according to the manufacturer’s instructions. Reporter activity data were normalized for protein concentration.
Immunoblot analysis
Cells harvested at various times were lysed in 62.5 mM Tris (pH 6.8)-2% SDS mixed with the protease inhibitor cocktail (Sigma) that contains 4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin, and aprotinin, and briefly sonicated before determining the protein concentration using the BCA reagent (Pierce, Rockford, IL). 50 μg of protein was resolved by SDS-PAGE, transferred to a polyvinylidene difluoride membrane filter (Bio-Rad, Hercules, CA), and stained with Fast Green reagent (Thermo Fisher Scientific, Waltham, MA). Membrane filters were then blocked in 0.1 M Tris (pH 7.5)-0.9% NaCl-0.05% Tween 20 with 5% nonfat dry milk, and incubated with appropriate antibodies. Antibodies were diluted as follows: ERK1/2, 1:2,500; phospho-ERK1/2 (Thr202/Tyr204), 1:2,500; phospho-p90RSK (Thr359/Ser363), 1:2,500; phospho-AKT/protein kinase B (Ser473), 1:5,000; AR, 1:2,500; phospho-Rb (Ser780), 1:2,500; Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 1:5,000 (Cell Signaling Technology, Danvers, MA); E2F1, 1:1,000; PSA, 1:1,000; neuron-specific enolase (NSE), 1:2,500; neutral endopeptidase (NEP), 1:1,000 (Thermo Fisher Scientific); p21CIP1, 1:2,500; p27KIP1, 1:1,000 (Santa Cruz Biotech, Santa Cruz, CA); p16INK4A, 1:2,500 (BD Bioscience, San Jose, CA). Nuclear and cytoplasmic fractions were extracted using the nuclear extraction kit (Pierce) according to the manufacturer’s instruction. The Supersignal West Pico and Femto chemiluminescence kits (Pierce) were used for visualization of the signal. For densitometry, immunoblots were scanned and analyzed using LabWorks™ (UVP BioImaging Systems, Upland, CA).
Results
ERK1/2 activation is correlated with AR downregulation during androgen deprivation-induced LNCaP differentiation
It was previously shown that LNCaP cells undergo NE differentiation in response to androgen depletion when cultured in c.s.FBS-containing medium [21]. We found that AR downregulation detected in LNCaP cells in c.s.FBS medium is also a specific response of the cells to androgen depletion because addition of R1881 nullified the c.s.FBS effect (Fig. 1A).
Figure 1. Correlation between AR expression and MEK/ERK activation changes during LNCaP response to androgen withdrawal.
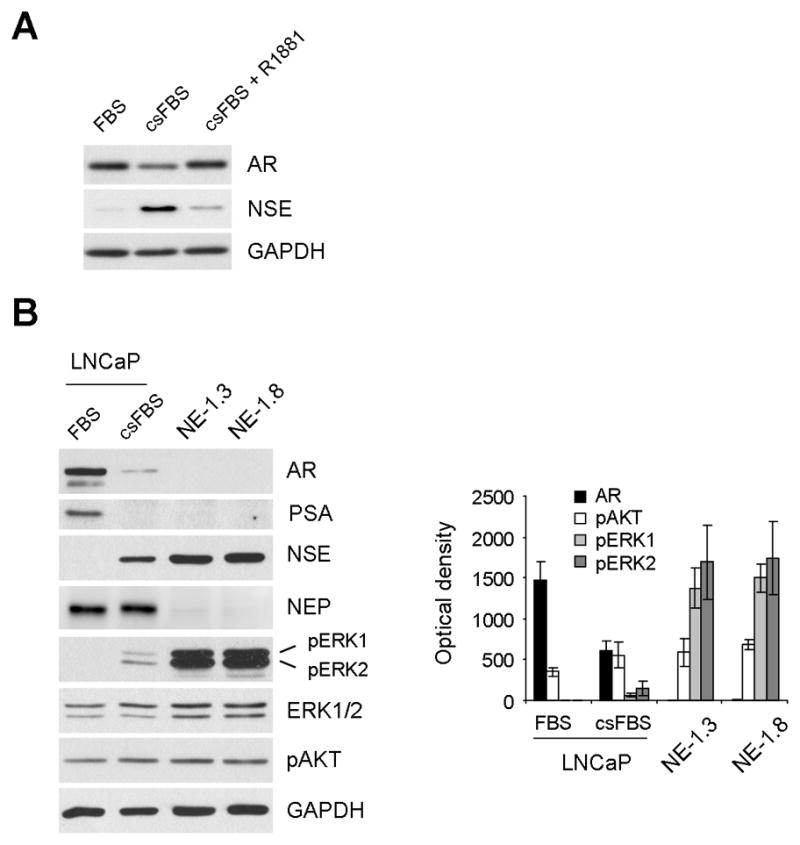
(A) AR downregulation in LNCaP cells cultured in c.s.FBS containing medium is a specific effect of androgen depletion. LNCaP cells maintained in the medium containing c.s.FBS for 6 days were moved to c.s.FBS medium with or without 1 nM R1881 and cultured for 4 days. Cell lysates were analyzed by Western blotting for expression of AR and NSE. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected to validate equal protein loading. (B) AR downregulation is correlated with ERK1/2 phosphorylation. LNCaP cells cultured in FBS or c.s.FBS medium for 10 days, and NE1.3 and NE1.8 cells maintained in c.s.FBS medium were analyzed by Western blotting for expression of AR, PSA, NSE, NEP, phosphorylated ERK1/2 (pERK1/2), ERK1/2, phosphorylated AKT (pAKT), and GAPDH. Western blotting results were analyzed by densitometry (right panel). Data (mean ± standard error) are from three independent experiments and show optical density normalized for GAPDH levels. P value is < 0.05 for AR, pERK1 and pERK2 in LNCaP-c.s.FBS compared to those in LNCaP-FBS or NE1.3 and NE1.8, respectively, and P value is < 0.05 for pAKT in LNCaP-c.s.FBS, NE1.3 and NE1.8, respectively, compared to that in LNCaP-FBS (Student’s t test).
To determine the role of the ERK1/2 pathway in regulating AR expression under a hormone withdrawal condition, we analyzed the correlation between ERK1/2 activation and AR levels in LNCaP cells, cultured in regular FBS- or c.s.FBS-containing medium for 10 days, and their NE derivatives, NE1.3 and NE1.8. NE1.3 and NE1.8 cells, derived under a prolonged (over 6 months) hormone-depleted culture condition, have served as a model to study NE differentiation of PCa because they exhibit the typical characteristics of NE differentiated PCa cells in both in vitro and in vivo microenvironments, i.e., hormone independency, absence of AR expression and increased expression of the NE differentiation markers, neuron-specific enolase (NSE), chromogranin B, neurotensin and parathyroid hormone related peptide [19, 21, 25]. In addition, these LNCaP derivatives exhibited low levels of neutral endopeptidase (NEP), which is known to antagonize NE differentiation and castration-resistance by degrading neuropeptides in prostate cancer [29] (Fig. 1B). We found that phosphorylation levels of the activation loop of ERK1 (T202/Y204) and ERK2 (T183/Y185) gradually increased during LNCaP responses to hormone depletion (Fig. 1B). Notably, about 20 and 10 fold increases in ERK1 and ERK2 phosphorylations, respectively, were detected in the NE derivatives when compared to the parental LNCaP cells cultured in c.s.FBS for 10 days. These increases in ERK1/2 phosphorylation were strongly correlated with decreases in AR, PSA and NEP protein levels, and increases in NSE expression (Fig. 1B). PSA is a bona fide indicator of the AR activity as a transcriptional regulator [13]. For a comparison, we also detected activation of AKT/protein kinase B, which is also implicated in NE differentiation of LNCaP [30]. AKT phosphorylation was also upregulated but the increase was mild and its correlation with AR downregulation was not as strong as ERK1/2 activation (Fig. 1B).
Sustained Raf activation is sufficient to induce AR downregulation in LNCaP cells
We determined the specific effect of MEK/ERK activation on AR expression using ΔRaf-1:ER that is the CR3 catalytic domain of Raf-1 fused to the hormone binding domain of the estrogen receptor [31]. ΔRaf-1:ER can be regulated by the estrogen analogue, 4-hydroxytamoxifen, and has been useful in many studies of Raf/MEK/ERK signaling [26, 32–36]. When LNCaP cells containing ΔRaf-1:ER (LNCaPRaf) was exposed to 1 μM 4-hydroxytamoxifen, cells exhibited highly increased ERK1/2 phosphorylation and significantly decreased AR protein levels within 48 hours similarly to the levels detected in NE1.8 cells (Fig. 2A). AR downregulation was detected not only in the cytosol but also in the nucleus (Fig. 2B) and, consistently, not only PSA protein expression but also its transcription was significantly decreased, as determined by the luciferase reporter containing DNA promoter region of PSA (Fig. 2A and 2C). Consistently with the previous result that the pathway activation is sufficient to mediate NE differentiation [21], NSE expression was elevated upon Raf activation while NEP levels were downregulated (Fig. 2A). However, the level of NSE induction at 48 hours after Raf activation was mild, indicating that Raf activation has more significant effects on AR and NEP regulation. All these effects were specific to Raf activation because 4-hydroxytamoxifen alone did not induce any similar effects.
Figure 2. Raf activation induces downregulation of AR in LNCaP and its NE derivatives.

LNCaP, NE1.3 and NE1.8 cells infected with lentiviral pHAGE (control) or pHAGE-Raf:ER were treated with 1 μM 4-hydroxytamoxifen (4-HT) in FBS medium for 2 days. (A) Total cell lysates of LNCaP were examined by Western blot analysis for expression of AR, PSA, NSE, NEP, pERK1/2, and ERK1/2. Cell lysates of NE1.3 or NE1.8 maintained in c.s.FBS medium were used for comparison. (B) Nuclear and cytoplasmic extracts of LNCaPRaf cells were compared for AR levels by Western blot analysis. GAPDH and PARP were used to validate the purity and equal protein loading of the cytoplasmic and nuclear fractions, respectively. (C) LNCaPRaf cells transfected with PSA-luciferase reporter were treated with 4-hydroxytamoxifen for indicated time periods before determining luciferase activity. The reporter assay data (mean ± standard error) are from three independent experiments and are expressed as percent changes induced by Raf activation relative to the untreated cells. P value is < 0.05 for Raf activation compared to the control at all time points (Student’s t test). (D) Total cell lysates of NE1.3 and NE1.8 were examined by Western blot analysis for expression of the indicated proteins. Uninfected or pHAGE-infected NE1.3 and NE1.8 cells showed identical responses to FBS medium (data not shown).
Interestingly, when NE1.3 and NE1.8 cells were removed from c.s.FBS medium to FBS medium, these cells exhibited restored expression of AR and strongly repressed ERK1/2 phosphorylation although NSE expression was not as strongly affected, especially in NE1.8 cells (Fig. 2D, first two lanes in each panel). When ΔRaf-1:ER was activated in these cells, AR expression was again downregulated to a similar level as repressed by hormone withdrawal (Fig. 2D), further supporting the specific effects of the pathway activation on AR levels.
Raf-induced AR downregulation is mediated by MEK/ERK but MEK/ERK inhibition cannot block long term androgen withdrawal-induced AR downregulation in LNCaP cells
Although the MEK/ERK pathway is known as the main effector of Raf, Raf can also mediate MEK/ERK-independent signaling, e.g., cell death responses [37]. Therefore, we determined whether MEK/ERK is required for Raf-induced AR downregulation. When Raf was activated in the presence of the MEK1/2-specific non-competitive inhibitor U0126, AR downregulation was significantly blocked in LNCaP cells (Fig. 3A). On the other hand, expression of the constitutively active mutants of MEK1 (ΔN3/S218E/S222D) or MEK2 (ΔN4/S222D/S226D) induced AR downregulation in LNCaP cells (Fig. 3B, first three lanes), indicating that MEK1/2 activity is necessary and sufficient to mediate Raf-induced AR downregulation.
Figure 3. MEK/ERK activation is necessary and sufficient to mediate Raf-induced AR downregulation, and ERK1 and ERK2 have overlapping roles.

(A) LNCaPRaf cells were treated for 1 or 2 days with 1 μM 4-hydroxytamoxifen for Raf activation in the presence or absence of 10 μM U0126 (MEK1/2 inhibitor), and examined for expression of AR, PSA, pERK1/2, and ERK1/2 by Western blot analysis. GAPDH was detected to validate equal protein loading. Western blotting results were then analyzed by densitometry (right panel). AR levels, normalized for GAPDH levels, are expressed as percent changes relative to the basal levels in the untreated cells at day 1. Data (mean ± standard error) are from three independent experiments. P value for blocking effects of U0126 is < 0.05 for day 1 and day 2 (Student’s t test). (B) LNCaP cells, co-infected with lentivirus containing shRNA targeting ERK1 (shERK1) or ERK2 (shERK2), were infected for 2 days with lentivirus containing constitutively active MEK1 (MEK1CA) or MEK2 (MEK2CA) before Western blot analysis of expression of the indicated proteins. Phosphorylated p90RSK (pRSK) indicates ERK1/2 activity. The empty pHAGE and pLL3.7 are the controls for MEK expression and ERK knockdown, respectively. Densitometry data of Western blotting results (right panel, mean ± standard error) are from two independent experiments. P value for blocking effects of shERK1/2 is < 0.05 for MEK1CA and MEK2CA (Student’s t test). (C) LNCaPRaf cells, transfected with two different sets of siRNA that target ERK1 or ERK2, were treated with 1 μM 4-hydroxytamoxifen for Raf activation for 2 days before Western blot analysis of expression of the indicated proteins. Densitometry data of Western blotting results (right panel, mean ± standard error) are from two independent experiments. AR levels are expressed as percent changes upon Raf activation, and data for ERK1 and ERK2 are GAPDH-normalized optical density (OD). P values for blocking effects of siERK1 and siERK2 are < 0.05 and <0.005, respectively (Student’s t test).
The requirement of MEK/ERK for Raf-mediated AR downregulation was further investigated at ERK1 and ERK2 levels using the RNA interference technique. Lentiviral shRNA-mediated knockdown of both ERK isoforms resulted in substantial depletion of ERK1/2 activity, as indicated by phosphorylation of ribosomal S6 kinase (p90RSK), and very effectively inhibited MEK1/2-mediated AR downregulation (Fig. 3B). p90RSK is an ERK1/2 substrate that serves as a bona fide readout of in vivo ERK1/2 kinase activity [26, 38]. Although ERK1 and ERK2 have overlapping roles in many different biological contexts, they are also known to have distinct roles for certain biological contexts, e.g., embryonic development or differentiation [39, 40]. Therefore, we investigated which ERK plays the key role in the pathway-mediated AR downregulation. When ERK1 and ERK2 were individually knocked down using two independent siRNA oligomers, ERK2 depletion generated stronger inhibitory effects on Raf-induced AR downregulation than ERK1 depletion (Fig. 3C), indicating that ERK2 is the stronger effector although ERK1 has redundant roles.
Nevertheless, despite the sufficient role of the Raf/MEK/ERK pathway, ERK1/2 depletion or U0126 treatment could not block c.s.FBS-induced AR downregulation in NE1.3 and NE1.8 cells (data not shown), indicating that the pathway activation is not necessary for the AR downregulation observed during LNCaP differentiation under a long term androgen withdrawal condition.
The Raf/MEK/ERK pathway mediates AR downregulation at mRNA and protein levels
We next investigated the mechanism by which the Raf/MEK/ERK pathway mediates AR downregulation. Our data showed that activation of the pathway, via ectopic expression of H-RasG12V, B-RafV600E, MEK1CA, or MEK2CA, can also downregulate AR at mRNA levels in LNCaP cells (Fig. 4A). When the rates of AR downregulation at mRNA and protein levels were compared at different time points after Raf activation, the rates of mRNA or protein downregulation were similar for the first 12 hours (Fig. 4B). However, thereafter, the rates of AR protein downregulation increased and exceeded the rates of mRNA downregulation, indicating that an additional mechanism(s) is present and mediates AR downregulation at protein levels as well (Fig. 4B). Indeed, the proteasome inhibitor MG132 could block Raf-mediated AR downregulation without significantly affecting the pathway activity, as indicated by phosphorylation of ERK1/2 and its substrate p90RSK (Fig. 4C, left panel). This inhibitory effect of MG132 was not due to delayed mRNA downregulation because MG132 treatment further decreased AR mRNA levels in Raf-activated cells (Fig. 4D); it was previously reported that MG132 suppresses AR transcription in LNCaP cells [41]. Raf-mediated AR downregulation was also inhibited by bortezomib, but not by lactacystin, indicating that the inhibitory effect is not common among different proteasome inhibitors (Fig. 4C, right panel). Furthermore, we could not detect increased AR ubiquitylation upon Raf activation even in the presence of MG132 (supplemental data 1), suggesting that proteasome activity may not be directly involved in AR downregulation.
Figure 4. Mechanisms of AR downregulation induced by Ras/Raf/MEK/ERK activation.

(A) LNCaP cells were infected with the lentivirus containing constitutively active H-RasG12V, B-RafV600E, MEK1CA, or MEK2CA for 2 days. Expression of AR mRNA was examined by the real time quantitative polymerase reaction (qPCR) assay. Data (mean ± standard error) are from a representative experiment performed in triplicate and are expressed as percent changes relative to the basal levels in the uninfected cells (No virus). P value is < 0.05 for each treatment compared to the pHAGE control (Student’s t test). (B) LNCaPRaf cells were treated with 4-hydroxytamoxifen for indicated periods. Changes in AR mRNA and protein levels were monitored by qPCR and Western blotting. Data (mean ± standard error) are from a representative experiment performed in triplicate and are expressed as percent changes relative to the basal levels in the untreated cells. P value at 48 hours is < 0.05, n = 3 (Student’s t test). (C) LNCaPRaf cells were treated with 4-hydroxytamoxifen for Raf activation for 24 hours in the presence of different doses of proteasome inhibitors, MG132, bortezomib, and lactacystin. Expression of AR, pERK1/2, pRSK, and GAPDH was examined by Western blot analysis. Equivalent volume of Dimethyl sulfoxide (DMSO) was used as the vehicle control. (D) LNCaPRaf cells were treated with 4-hydroxytamoxifen for Raf activation for 24 hours in the presence of different doses of MG132. Expression of AR mRNA was examined by qPCR. Data (mean ± standard error) are from a representative experiment performed in triplicate and are expressed as percent changes relative to the basal levels in DMSO-treated control.
AR downregulation is specifically required for Raf/MEK/ERK-mediated regulation of p16INK4A, p21CIP1, NSE, and NEP expression
Sustained Raf/MEK/ERK activation can mediate not only NE differentiation [21] but also cell cycle arrest in LNCaP cells [26], which supports the implication of the pathway signaling in decreased mitotic activity of NE transdifferentiating LNCaP cells. Because (i) AR controls G1/S cell cycle progression [14]; (ii) AR depletion is sufficient to induce growth arrest in LNCaP cells [42–45]; and (iii) AR downregulation was shown to induce NE differentiation of LNCaP cells [24], we questioned whether AR downregulation is a key regulatory mechanism for Raf/MEK/ERK-mediated growth inhibitory signaling and NE differentiation in LNCaP cells. To address this, we determined whether Raf-controlled expression of cell cycle regulators and NE differentiation could be abrogated by ectopic expression of AAø, a constitutively active AR that lacks the hormone binding domain [27].
AAø expression did not affect the MEK/ERK pathway activity, as indicated by phosphorylation of ERK1/2 and p90RSK (Fig. 5A), but it conferred sustained AR transcriptional activity in cells irrespective of the pathway activation, as determined by the PSA-luciferase reporter assay (Fig. 5B). Under this condition, Raf-induced NSE expression, NEP downregulation, and expression of the cyclin-dependent kinase inhibitors, p16INK4A and p21CIP1, were significantly blocked whereas downregulation of Rb phosphorylation and E2F1 expression was not affected (Fig. 5A), indicating that AR downregulation is required to mediate NE differentiation and to regulate a specific subset of the growth arrest signaling effectors while the pathway can also activate other mechanisms to negatively regulate Rb/E2F1 activity. Interestingly, despite the sustained AR transcriptional activity in these cells, as indicated by PSA-luciferase activity (Fig. 5B), PSA protein level was still downregulated upon Raf activation (Fig. 5A). This PSA downregulation required ERK1/2 because it was not observed in ERK1/2-depleted cells (Fig. 5C), indicating that the Raf/MEK/ERK pathway can regulate certain downstream effectors of AR via a separate mechanism independently of AR downregulation.
Figure 5. Ectopic expression of constitutively active AR mutant inhibits Raf-mediated regulation of NSE, NEP, p16INK4A, and p21CIP1 expression but not downregulation of Rb phosphorylation and E2F1 expression.
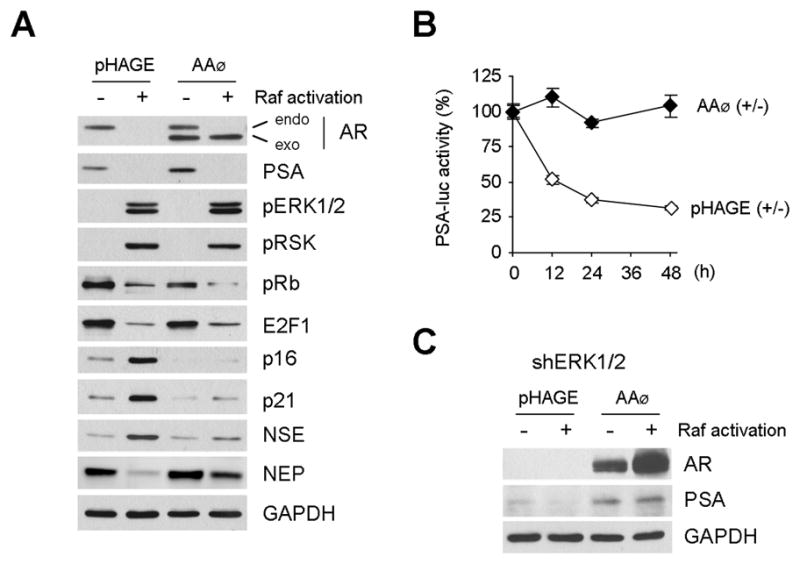
(A) LNCaPRaf cells, infected with the lentivirus containing the AR mutant lacking hormone binding domain (AAø), or the control empty pHAGE virus, were treated with 1 μM 4-hydroxytamoxifen for Raf activation for 2 days and were examined by Western blot analysis for expression of AR (endo: endogenous AR; exo: exogenously expressed AR mutant), PSA, pERK1/2, pRSK, phosphorylated Rb (pRb), E2F1, p16INK4A, p21CIP1, NSE and NEP. (B) LNCaPRaf cells, infected with the AAø virus or pHAGE, were transfected with PSA-luciferase reporter and treated with 4-hydroxytamoxifen for indicated time periods before determining luciferase activity. The reporter assay data (mean ± standard error) are from three independent experiments and are expressed as percent changes induced by Raf activation relative to the untreated cells. P value is < 0.05 for AR mutant effects compared to the pHAGE control at all time points (Student’s t test). (C) LNCaPRaf cells, co-infected with the shERK1 and shERK2 viruses, and then with the AR mutant lentivirus or pHAGE virus, were treated with 1 μM 4-hydroxytamoxifen for Raf activation for 2 days and were examined by Western blotting for expression of exogenous AR and PSA.
Raf/MEK/ERK activation can induce AR downregulation in different PCa cell lines irrespective of androgen sensitivity
Lastly, we determined whether the Raf/MEK/ERK pathway can also mediate AR downregulation in other PCa cell lines. AR is expressed in the androgen-sensitive LNCaP, LAPC4 and MDA-Pca-2b and the androgen-refractory CWR22Rv1 and C4-2 (an androgen-refractory LNCaP derivative [46]), but not in PC3 and Du145 (Fig. 6A). CWR22Rv1 expressed different sizes of AR proteins that migrate as two major bands on the SDS-PAGE gel; the faster migrating band was recently shown to contain three different AR isoforms that lack the ligand binding domain [47, 48]. The basal MEK/ERK activity in these PCa lines, except for CWR22Rv1 and Du145, was very low, as indicated by ERK1/2 phosphorylation levels (Fig. 6A).
Figure 6. Raf activation can induce AR downregulation regardless of hormone sensitivity.

(A) Hormone-dependent LNCaP, LAPC4, and MDA-Pca-2b; and hormone-independent C4-2, CWR22Rv1, PC3 and Du145 lines were examined for the basal expression levels of AR, AR variants lacking ligand binding domain (LBD), and ERK1/2 phosphorylation. (B and C) C4-2 and CWR22Rv1 cells were infected with pHAGE-Raf:ER and were treated with 1 μM 4-hydroxytamoxifen (4-HT) for 2 days. Cells were also infected with constitutively active B-RafV600E or MEK2CA viruses for 2 days. Expression of AR, NSE, NEP, p21CIP1, p27KIP1 and pERK1/2 was examined by Western blot analysis. pRb, E2F1, p16INK4A were also observed but were not changed (not shown). GAPDH is the loading control.
When the Raf/MEK/ERK pathway was activated using Raf:ER, B-RafV600E, or the constitutively active MEK2, AR levels were substantially downregulated in C4-2 and CWR22Rv1 cells (Fig. 6B and 6C), which was accompanied by strong expression of the cyclin-dependent kinase inhibitors, p21CIP1 or p27KIP1. These data indicate that Raf/MEK/ERK can also mediate AR downregulation and growth inhibitory signaling in these cell lines, and that the signaling can occur irrespective of androgen sensitivity. In C4-2 cells, Raf activation also induced NSE expression and mild NEP downregulation, which is consistent with the observation in LNCaP (Fig. 6B). Notably, in CWR22Rv1 cells, Raf activation downregulated not only the full length AR but also the ligand binding domain-truncated AR splicing variants (Fig. 6C), which are supposed to more potently stimulate androgen-insensitive growth of PCa cells [47, 48]. By contrast, Raf/MEK/ERK activation did not affect AR levels in LAPC4 and MDA-Pca-2b cells (data not shown), indicating that different types of PCa cells have different responsiveness to the pathway activation.
Discussion
Our study demonstrates that, in certain PCa cell types, (i) activation of the Raf/MEK/ERK pathway occurs in correlation with AR downregulation upon androgen deprivation and is sufficient, although not necessary, to mediate the AR regulation; (ii) AR downregulation is required for the pathway-mediated differentiation (i.e., regulation of NSE and NEP expression) and specific growth arrest signaling (i.e., p16INK4A and p21CIP1 expression but not Rb/E2F1 regulation); (iii) the pathway can regulate PSA via a mechanism independent of AR regulation; and (iv) the pathway activation can mediate AR downregulation irrespective of their hormone sensitivity (Fig. 7).
Figure 7. Raf/EMK/ERK-mediated AR regulation, growth inhibitory signaling, and differentiation.
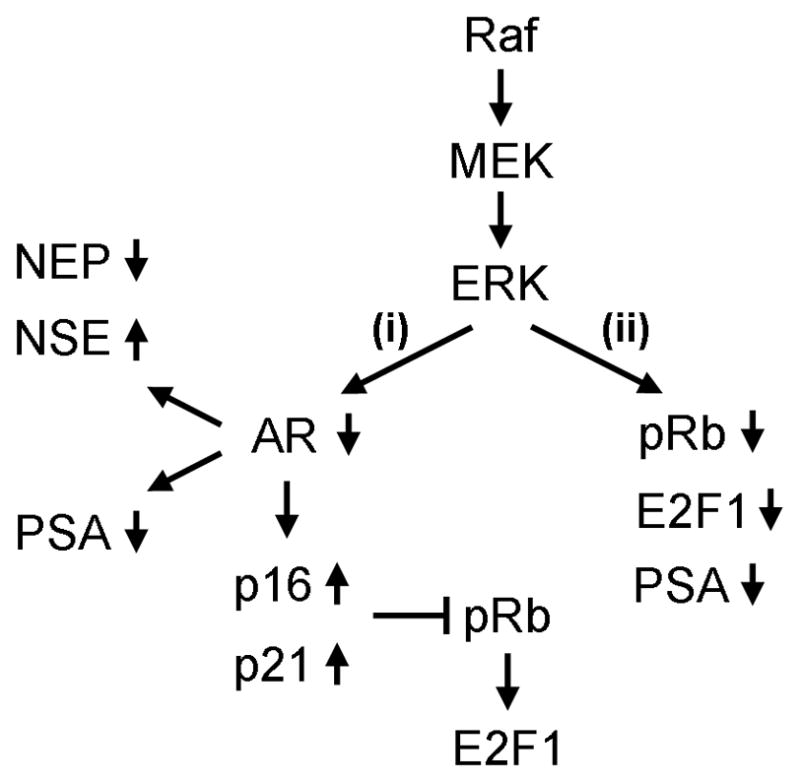
Constitutively high MEK/ERK activity can mediate AR downregulation, growth inhibitory signaling and differentiation. In this signaling, AR downregulation is required to control NSE, NEP, p16INK4A and p21WAF expression (i). However, Rb phosphorylation and expression of E2F1 can be downregulated independently of AR downregulation (ii). Decreased AR transcriptional activity leads to PSA downregulation (i) but PSA is also downregulated via a non-transcriptional mechanism independently of AR downregulation (ii). Raf/EMK/ERK-mediated AR downregulation is also detected in a subset of PCa cells regardless of hormone sensitivity.
Our results from an in vitro LNCaP model that mimics NE differentiation in PCa indicate that Raf/MEK/ERK signaling has a novel role in regulating AR expression. Contrary to most other castration-resistant PCa types, NE-like PCa cells do not express AR while exhibiting upregulated ERK1/2 activity. Consistently, these in vivo alterations were recapitulated in the NE derivatives of LNCaP that were derived from a long term hormone-deprived culture. Intriguingly, sustained activation of Raf/MEK/ERK was indeed sufficient for AR downregulation in the parental LNCaP, although inhibition of the pathway did not restore AR expression in the NE derivatives of LNCaP. This may indicate that not only Raf/MEK/ERK but also other parallel independent signaling pathways are involved in AR downregulation during NE differentiation. Indeed, it was previously reported that the PI3K/AKT/mTOR pathway is also involved in NE differentiation of LNCaP cells [30], and that ectopic expression of AKT can induce proteasomal degradation of AR via Mdm2 E3 ligase-mediated ubiquitylation [49]. However, in our study, AKT phosphorylation was only mildly upregulated in the NE derivatives of LNCaP and use of the PI3K specific inhibitor, LY294002 or overexpression of a dominant-negative AKT did not restore AR expression in the cells (data not shown), indicating the presence of additional pathways which remain to be identified.
It was previously shown that steady-state AR levels and its activity decreases upon prolonged inhibition of basal Raf/MEK/ERK activity in LNCaP cells [50]. Consistently, we observed similar effects using the MEK1/2 inhibitor U0126, but not with ERK1/2 knockdown, presumably because of its lower potency than the chemical inhibition. Intriguingly, our current study demonstrates that AR levels and its activity can also be downregulated by sustained high Raf/MEK/ERK activity. Taken together, these results suggest that the Raf/MEK/ERK pathway may have a capability to mediate an opposing context of AR regulation depending upon different signaling intensity, which is coherent with the biphasic pathway control on LNCaP cell proliferation (i.e., sustained Raf/MEK/ERK activation induces cell cycle arrest while depletion of its basal activity also delays cell proliferation [26, 50]). Indeed, it is known that different duration and strength of the kinase cascade signals can lead to distinct, and even opposing, cellular processes [1].
It appears that Raf/MEK/ERK-mediated AR downregulation is mediated via multiple mechanisms. AR was significantly downregulated at both protein and mRNA levels, while the rates of downregulation were higher at protein levels. Besides, decreases in AR protein, but not mRNA, were blocked by the proteasome inhibitors MG132 and bortezomib, indicating that the pathway also mediates AR downregulation via a mechanism independent of mRNA regulation and that the mechanisms may require proteasome activity. However, since increases in AR ubiquitylation were not detected upon Raf activation, proteasomal degradation does not appear to be a direct mechanism of AR downregulation but may be indirectly involved. Alternatively, since another proteasome inhibitor lactacystin could not exert similar inhibitory effects, AR downregulation may rather be due to non-proteasomal degradation. In support of this possibility, MG132 is known for its additional capability to inhibit certain non-proteasomal proteases such as calpains and cathepsins [51, 52]. Further exploration of the mechanism underlying AR downregulation is warranted in a future study.
Our study indicates that the Raf/MEK/ERK pathway-mediated growth inhibition and NE differentiation can be mediated via mechanisms dependent or independent of AR downregulation. Ectopic expression of the constitutively active AR could specifically inhibit Raf-mediated NSE, p16INK4A, and p21CIP1 expression, and NEP downregulation. This is coherent with the known AR function as a regulator of NE differentiation [24] and G1/S cell cycle progression in PCa cells [14], and indicates that AR can control the cyclin-dependent kinase/Rb/E2F1 axis via regulating those cyclin-dependent kinase inhibitors. Expression of p16INK4A or p21CIP1 can delay cell cycle progression by inhibiting G1/S cyclin-dependent kinases, which would promote Rb-mediated sequestration of the S-phase transcription factor E2F1 [14]. Nevertheless, despite the forced AR overexpression, the Raf/MEK/ERK pathway could still downregulate Rb phosphorylation and E2F1 expression, indicating that the pathway can mediate growth arrest signaling via additional mechanisms other than AR downregulation (Fig. 7). Consistently, E2F1 downregulation observed in our study indicates that the Raf/MEK/ERK pathway can inhibit E2F1 activity via a mechanism in addition to Rb-mediated E2F1 sequestration. Versatile signaling capability of Raf/MEK/ERK in PCa cells is further supported by its ability to mediate PSA downregulation independently of AR downregulation.
Raf/MEK/ERK-mediated AR downregulation in LNCaP and CWR22Rv1 extends the growth inhibitory context of the pathway signaling previously reported in LNCaP cells [26, 53]. Growth inhibitory MEK/ERK signaling in PCa cells has also been demonstrated in recent reports showing that ligand-mediated activation of the G-protein-coupled receptor 30 induces growth arrest of PC3 cells, for which MEK/ERK activation is necessary [54], and that expression of constitutively active MEK1 arrests PC3 cells in culture [55]. A question is thus raised: what would be the potential significance of such MEK/ERK signaling? The paradoxical growth inhibitory signaling of Ras or Raf in primary normal cells has been proposed as an anti-oncogenic cellular mechanism against aberrant pathway activation [32, 56, 57]. Similar growth arrest responses can also be elicited in different cell types, including malignant tumor cell lines in which aberrant Ras/Raf signaling is rarely detected [26, 33–36, 58, 59], suggesting that various cell types retain such mechanisms. Although not frequent, oncogenic mutations of Ras and Raf are detected in PCa, primarily from Japanese, Korean and Chinese men [7–10]. Furthermore, a recent study revealed that chromosomal rearrangements of Raf genes that can confer neoplastic Raf kinase activity occur in about 1–2% of human PCa [11]. Indeed, the oncogenic B-RafV600E mutant could drive PCa development in a transgenic mouse model [60]. Given these growing implications of Ras/Raf in prostate tumorigenesis, it is conceivable that certain AR-dependent prostate cell types may exhibit AR downregulation as a Ras/Raf-associated tumor suppressive mechanism when the pathway becomes deregulated.
Downregulation of AR has been recognized as a potential therapeutic strategy for PCa regardless of hormone sensitivity because recurrent PCa cells still depend on AR [15]. For examples, depletion or inhibition of AR using antisense oligonucleotides [42], neutralizing antibody [43], hammerhead ribozyme [43], RNA interference [44] and small molecules that mediate targeted proteolysis of AR [45], could induce growth inhibition and apoptosis in both androgen-dependent and androgen-independent PCa cells. Our results demonstrate that the Raf/MEK/ERK pathway is connected to a mechanism that can effectively induce AR downregulation in a subset of PCa cells regardless of hormone sensitivity and, importantly, the mechanism induces downregulation of not only full length AR but also the AR variants that lack the hormone binding domain and are known to more potently stimulate PCa cell growth [47, 48]. Further investigation of the mechanisms underlying Raf/MEK/ERK-mediated AR downregulation may lead to the development of a novel means to control AR levels in PCa cells.
In conclusion, our results demonstrate for the first time that the Raf/MEK/ERK pathway has a novel role in regulating AR levels in a subset of PCa cells and provide an insight into a PCa cell response in the face of aberrant pathway activation.
Supplementary Material
Acknowledgments
We thank Diane Robins, Julian Downward, Gavin Robertson, and Natalie Ahn for AR, H-Ras, B-Raf, and MEK cDNA; Amy Hudson, Stephen Duncan, and Richard Mulligan for lentiviral vectors; Shuyuan Yeh for PSA-luciferase reporter; Karen Knudson for C4-2; William See for R1881; and Selleck Chemicals for Bortezomib. This work was supported by the National Cancer Institute (1R01CA138441), American Cancer Society (RSGM-10-189-01-TBE), FAMRI Young Investigator Award (062438), DOD Prostate Cancer Research Program (W81XWH-07-01-0089), and Wisconsin Breast Cancer Showhouse to J.P. M.F.L. is supported in part by NCI CA88184 and DOD W81XWH-08-1-0459.
Abbreviation
- AR
androgen receptor
- c.s.FBS
charcoal/dextran-stripped fetal bovine serum
- ERK
extracellular signal-regulated kinase
- MEK
mitogen activated protein kinase kinase
- NEP
neutral endopeptidase
- NSE
neuron specific enolase
- PCa
prostate cancer
- PSA
prostate specific antigen
- RSK
ribosomal S6 kinase
Footnotes
Conflict of interest
The authors declare no conflict of interest for this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochimica et biophysica acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 2.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 3.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer research. 1999;59:279–284. [PubMed] [Google Scholar]
- 4.Royuela M, Arenas MI, Bethencourt FR, Sanchez-Chapado M, Fraile B, Paniagua R. Regulation of proliferation/apoptosis equilibrium by mitogen-activated protein kinases in normal, hyperplastic, and carcinomatous human prostate. Human pathology. 2002;33:299–306. doi: 10.1053/hupa.2002.32227. [DOI] [PubMed] [Google Scholar]
- 5.Uzgare AR, Kaplan PJ, Greenberg NM. Differential expression and/or activation of P38MAPK, erk1/2, and jnk during the initiation and progression of prostate cancer. The Prostate. 2003;55:128–139. doi: 10.1002/pros.10212. [DOI] [PubMed] [Google Scholar]
- 6.Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. The Journal of clinical investigation. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anwar K, Nakakuki K, Shiraishi T, Naiki H, Yatani R, Inuzuka M. Presence of ras oncogene mutations and human papillomavirus DNA in human prostate carcinomas. Cancer research. 1992;52:5991–5996. [PubMed] [Google Scholar]
- 8.Shiraishi T, Muneyuki T, Fukutome K, Ito H, Kotake T, Watanabe M, Yatani R. Mutations of ras genes are relatively frequent in Japanese prostate cancers: pointing to genetic differences between populations. Anticancer research. 1998;18:2789–2792. [PubMed] [Google Scholar]
- 9.Cho NY, Choi M, Kim BH, Cho YM, Moon KC, Kang GH. BRAF and KRAS mutations in prostatic adenocarcinoma. International journal of cancer. 2006;119:1858–1862. doi: 10.1002/ijc.22071. [DOI] [PubMed] [Google Scholar]
- 10.Shen Y, Lu Y, Yin X, Zhu G, Zhu J. KRAS and BRAF mutations in prostate carcinomas of Chinese patients. Cancer genetics and cytogenetics. 2010;198:35–39. doi: 10.1016/j.cancergencyto.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, Cao Q, Cao X, Suleman K, Kumar-Sinha C, Dhanasekaran SM, Chen YB, Esgueva R, Banerjee S, LaFargue CJ, Siddiqui J, Demichelis F, Moeller P, Bismar TA, Kuefer R, Fullen DR, Johnson TM, Greenson JK, Giordano TJ, Tan P, Tomlins SA, Varambally S, Rubin MA, Maher CA, Chinnaiyan AM. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nature medicine. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rochette-Egly C. Nuclear receptors: integration of multiple signalling pathways through phosphorylation. Cellular signalling. 2003;15:355–366. doi: 10.1016/s0898-6568(02)00115-8. [DOI] [PubMed] [Google Scholar]
- 13.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocrine reviews. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 14.Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nuclear receptor signaling. 2008;6:e001. doi: 10.1621/nrs.06001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eder IE, Haag P, Bartsch G, Klocker H. Targeting the androgen receptor in hormone-refractory prostate cancer--new concepts. Future oncology (London, England) 2005;1:93–101. doi: 10.1517/14796694.1.1.91. [DOI] [PubMed] [Google Scholar]
- 16.Bonkhoff H. Neuroendocrine differentiation in human prostate cancer. Morphogenesis, proliferation and androgen receptor status. Ann Oncol. 2001;12(Suppl 2):S141–144. doi: 10.1093/annonc/12.suppl_2.s141. [DOI] [PubMed] [Google Scholar]
- 17.Segawa N, Mori I, Utsunomiya H, Nakamura M, Nakamura Y, Shan L, Kakudo K, Katsuoka Y. Prognostic significance of neuroendocrine differentiation, proliferation activity and androgen receptor expression in prostate cancer. Pathology international. 2001;51:452–459. doi: 10.1046/j.1440-1827.2001.01226.x. [DOI] [PubMed] [Google Scholar]
- 18.Abrahamsson PA. Neuroendocrine cells in tumour growth of the prostate. Endocrine-related cancer. 1999;6:503–519. doi: 10.1677/erc.0.0060503. [DOI] [PubMed] [Google Scholar]
- 19.Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocrine-related cancer. 2007;14:531–547. doi: 10.1677/ERC-07-0061. [DOI] [PubMed] [Google Scholar]
- 20.Sauer CG, Roemer A, Grobholz R. Genetic analysis of neuroendocrine tumor cells in prostatic carcinoma. The Prostate. 2006;66:227–234. doi: 10.1002/pros.20338. [DOI] [PubMed] [Google Scholar]
- 21.Zhang XQ, Kondrikov D, Yuan TC, Lin FF, Hansen J, Lin MF. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene. 2003;22:6704–6716. doi: 10.1038/sj.onc.1206764. [DOI] [PubMed] [Google Scholar]
- 22.Huss WJ, Gregory CW, Smith GJ. Neuroendocrine cell differentiation in the CWR22 human prostate cancer xenograft: association with tumor cell proliferation prior to recurrence. The Prostate. 2004;60:91–97. doi: 10.1002/pros.20032. [DOI] [PubMed] [Google Scholar]
- 23.Jongsma J, Oomen MH, Noordzij MA, Van Weerden WM, Martens GJ, van der Kwast TH, Schroder FH, van Steenbrugge GJ. Androgen deprivation of the PC-310 [correction of prohormone convertase-310] human prostate cancer model system induces neuroendocrine differentiation. Cancer research. 2000;60:741–748. [PubMed] [Google Scholar]
- 24.Wright ME, Tsai MJ, Aebersold R. Androgen receptor represses the neuroendocrine transdifferentiation process in prostate cancer cells. Molecular endocrinology (Baltimore, Md) 2003;17:1726–1737. doi: 10.1210/me.2003-0031. [DOI] [PubMed] [Google Scholar]
- 25.Yuan TC, Veeramani S, Lin FF, Kondrikou D, Zelivianski S, Igawa T, Karan D, Batra SK, Lin MF. Androgen deprivation induces human prostate epithelial neuroendocrine differentiation of androgen-sensitive LNCaP cells. Endocrine-related cancer. 2006;13:151–167. doi: 10.1677/erc.1.01043. [DOI] [PubMed] [Google Scholar]
- 26.Hong SK, Yoon S, Moelling C, Arthan D, Park JI. Noncatalytic function of ERK1/2 can promote Raf/MEK/ERK-mediated growth arrest signaling. The Journal of biological chemistry. 2009;284:33006–33018. doi: 10.1074/jbc.M109.012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grad JM, Lyons LS, Robins DM, Burnstein KL. The androgen receptor (AR) amino-terminus imposes androgen-specific regulation of AR gene expression via an exonic enhancer. Endocrinology. 2001;142:1107–1116. doi: 10.1210/endo.142.3.8049. [DOI] [PubMed] [Google Scholar]
- 28.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 29.Patrikidou A, Vlachostergios PJ, Voutsadakis IA, Hatzidaki E, Valeri RM, Destouni C, Apostolou E, Daliani D, Papandreou CN. Inverse baseline expression pattern of the NEP/neuropeptides and NFkappaB/proteasome pathways in androgen-dependent and androgen-independent prostate cancer cells. Cancer cell international. 2011;11:13. doi: 10.1186/1475-2867-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu C, Huang J. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin pathway is essential for neuroendocrine differentiation of prostate cancer. The Journal of biological chemistry. 2007;282:3571–3583. doi: 10.1074/jbc.M608487200. [DOI] [PubMed] [Google Scholar]
- 31.Samuels ML, Weber MJ, Bishop JM, McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human raf-1 protein kinase. Molecular and cellular biology. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu J, Woods D, McMahon M, Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes & development. 1998;12:2997–3007. doi: 10.1101/gad.12.19.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fanton CP, McMahon M, Pieper RO. Dual growth arrest pathways in astrocytes and astrocytic tumors in response to Raf-1 activation. The Journal of biological chemistry. 2001;276:18871–18877. doi: 10.1074/jbc.M011514200. [DOI] [PubMed] [Google Scholar]
- 34.Ravi RK, Weber E, McMahon M, Williams JR, Baylin S, Mal A, Harter ML, Dillehay LE, Claudio PP, Giordano A, Nelkin BD, Mabry M. Activated Raf-1 causes growth arrest in human small cell lung cancer cells. The Journal of clinical investigation. 1998;101:153–159. doi: 10.1172/JCI831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JI, Strock CJ, Ball DW, Nelkin BD. The Ras/Raf/MEK/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/JAK/STAT pathway. Molecular and cellular biology. 2003;23:543–554. doi: 10.1128/MCB.23.2.543-554.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arthan D, Hong SK, Park JI. Leukemia inhibitory factor can mediate Ras/Raf/MEK/ERK-induced growth inhibitory signaling in medullary thyroid cancer cells. Cancer letters. 2010;297:31–41. doi: 10.1016/j.canlet.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7783–7788. doi: 10.1073/pnas.141224398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lazar DF, Wiese RJ, Brady MJ, Mastick CC, Waters SB, Yamauchi K, Pessin JE, Cuatrecasas P, Saltiel AR. Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. The Journal of biological chemistry. 1995;270:20801–20807. doi: 10.1074/jbc.270.35.20801. [DOI] [PubMed] [Google Scholar]
- 39.Fischer AM, Katayama CD, Pages G, Pouyssegur J, Hedrick SM. The role of erk1 and erk2 in multiple stages of T cell development. Immunity. 2005;23:431–443. doi: 10.1016/j.immuni.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 40.Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Molecular cell. 2010;38:114–127. doi: 10.1016/j.molcel.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin HK, Altuwaijri S, Lin WJ, Kan PY, Collins LL, Chang C. Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. The Journal of biological chemistry. 2002;277:36570–36576. doi: 10.1074/jbc.M204751200. [DOI] [PubMed] [Google Scholar]
- 42.Eder IE, Hoffmann J, Rogatsch H, Schafer G, Zopf D, Bartsch G, Klocker H. Inhibition of LNCaP prostate tumor growth in vivo by an antisense oligonucleotide directed against the human androgen receptor. Cancer gene therapy. 2002;9:117–125. doi: 10.1038/sj.cgt.7700416. [DOI] [PubMed] [Google Scholar]
- 43.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer research. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 44.Liao X, Tang S, Thrasher JB, Griebling TL, Li B. Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Molecular cancer therapeutics. 2005;4:505–515. doi: 10.1158/1535-7163.MCT-04-0313. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, Sakamoto KM. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. International journal of cancer. 1994;57:406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- 47.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer research. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, Pretlow TG, Kung HJ. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer research. 2002;62:6606–6614. [PubMed] [Google Scholar]
- 49.Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. The EMBO journal. 2002;21:4037–4048. doi: 10.1093/emboj/cdf406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agoulnik IU, Bingman WE, 3rd, Nakka M, Li W, Wang Q, Liu X, Brown M, Weigel NL. Target Gene Specific Regulation of Androgen Receptor Activity by p42/p44 MAPK. Molecular endocrinology (Baltimore, Md. 2008 doi: 10.1210/me.2007-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito H, Watanabe M, Kim YT, Takahashi K. Inhibition of rat liver cathepsins B and L by the peptide aldehyde benzyloxycarbonyl-leucyl-leucyl-leucinal and its analogues. Journal of enzyme inhibition and medicinal chemistry. 2009;24:279–286. doi: 10.1080/14756360802166921. [DOI] [PubMed] [Google Scholar]
- 52.Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. Journal of biochemistry. 1996;119:572–576. doi: 10.1093/oxfordjournals.jbchem.a021280. [DOI] [PubMed] [Google Scholar]
- 53.Ravi RK, McMahon M, Yangang Z, Williams JR, Dillehay LE, Nelkin BD, Mabry M. Raf-1-induced cell cycle arrest in LNCaP human prostate cancer cells. Journal of cellular biochemistry. 1999;72:458–469. doi: 10.1002/(sici)1097-4644(19990315)72:4<458::aid-jcb2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 54.Chan QK, Lam HM, Ng CF, Lee AY, Chan ES, Ng HK, Ho SM, Lau KM. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell death and differentiation. 2010;17:1511–1523. doi: 10.1038/cdd.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moro L, Arbini AA, Marra E, Greco M. Constitutive activation of MAPK/ERK inhibits prostate cancer cell proliferation through upregulation of BRCA2. International journal of oncology. 2007;30:217–224. doi: 10.3892/ijo.30.1.217. [DOI] [PubMed] [Google Scholar]
- 56.Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes & development. 1998;12:3008–3019. doi: 10.1101/gad.12.19.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 58.Park JI, Powers JF, Tischler AS, Strock CJ, Ball DW, Nelkin BD. GDNF-induced leukemia inhibitory factor can mediate differentiation via the MEK/ERK pathway in pheochromocytoma cells derived from nf1-heterozygous knockout mice. Experimental cell research. 2005;303:79–88. doi: 10.1016/j.yexcr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 59.Kim EJ, Park JI, Nelkin BD. IFI16 is an essential mediator of growth inhibition, but not differentiation, induced by the leukemia inhibitory factor/JAK/STAT pathway in medullary thyroid carcinoma cells. The Journal of biological chemistry. 2005;280:4913–4920. doi: 10.1074/jbc.M410542200. [DOI] [PubMed] [Google Scholar]
- 60.Jeong JH, Wang Z, Guimaraes AS, Ouyang X, Figueiredo JL, Ding Z, Jiang S, Guney I, Kang GH, Shin E, Hahn WC, Loda MF, Abate-Shen C, Weissleder R, Chin L. BRAF activation initiates but does not maintain invasive prostate adenocarcinoma. PloS one. 2008;3:e3949. doi: 10.1371/journal.pone.0003949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.