

Abstract
Methods for synthetically manipulating protein structure enable greater flexibility in the study of protein function. Previous characterization of the E. coli aminoacyl tRNA transferase (AaT) has shown that it can modify the N-terminus of a protein with an amino acid from a tRNA or a synthetic oligonucleotide donor. Here, we demonstrate that AaT can efficiently use a minimal adenosine substrate, which can be synthesized in one to two steps from readily available starting materials. We have characterized the enzymatic activity of AaT with aminoacyl adenosyl donors and found that reaction products do not inhibit AaT. The use of adenosyl donors removes the substrate limitations imposed by the use of synthetases for tRNA charging and avoids the complex synthesis of an oligonucleotide donor. Thus, our AaT donors increase the potential substrate scope and reaction scale for Nterminal protein modification under conditions that maintain folding.
INTRODUCTION
The conjugation of synthetic molecules to proteins contributes to biomedical research by enabling the immobilization of proteins on surfaces, modifications with chromophores to make in vitro sensors, or tagging with in vivo imaging agents.1–5 In most cases, the formation of well-defined conjugates is valuable, if not essential. The protein termini are attractive targets for conjugation, because in many cases appending synthetic molecules at the termini will have minimal undesired effects on protein folding and function.6–26 This is particularly applicable in cases where the terminus is relatively unstructured. As part of our research on minimalist labeling strategies for biophysical studies, we sought a method for N-terminal modification that could be carried out easily under conditions that maintain folding without substantial prior protein manipulation.27
Selective N-terminal modification has been achieved by a variety of chemical and enzymatic methods, each with benefits and drawbacks. Small molecule strategies permit the attachment of a variety of molecules, but they are subject to side reactions, incomplete specificity for the N-terminus, and may need to be carried out in organic solvent mixtures.16,28–37 Reverse proteolysis methods can be used to modify the N-terminus under conditions that do not require protein unfolding, but the reaction can be difficult to drive to completion without high protein concentrations.38–51 Several other chemoenzymatic methods are valuable in functioning under benign conditions, but have moderate-sized target sequences which must be appended to the protein.52–59 Here, we describe a minimal system for N-terminal protein labeling that utilizes only adenonsine esters of natural or unnatural aminoacids and a single, readily available enzyme. This process gives high yields of modified proteins under non-denaturing conditions and requires only a single basic amino acid for specific recognition.
Aminoacyl tRNA transferases (AaTs) are members of a growing class of enzymes that use aminoacyl tRNAs in secondary metabolism.60 The E. coli AaT catalyzes the transfer of Leu, Phe, or Met from an aminoacyl tRNA to a protein bearing an N-terminal Arg or Lys.61–63 The addition of Leu or Phe targets that protein for degradation by ClpA as part of the N-end rule pathway.64–67 Kaji et al. first observed AaT aminoacylation activity in crude E. coli preparations.61,68,69 Soffer and Leibowitz subsequently reconstituted the purified enzyme and characterized its specificity for both the RNA and amino acid componenets of the donor molecule, demonstrating its first use in transferring an unnatural amino acid, p-fluorophenylalanine. 70,71 (Fig. 1, Top)
Figure 1.
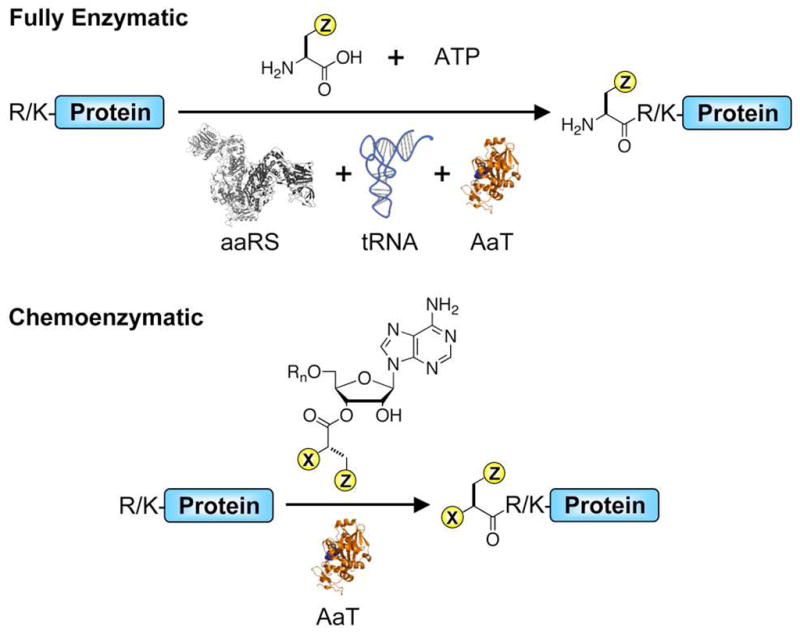
Transferase-mediated N-terminal protein modification. Top: Fully enzymatic methods use aminoacyl tRNA synthetase (aaRS), tRNA, aminoacyl transferase (AaT), amino acid, and ATP. Bottom: Chemoenzymatic methods use only a synthetic nucleic acid donor and transferase. In this work, an aminoacyl mononucleoside (Rn = H) was used.
Recently, purified AaT has been used to modify proteins in vitro with a variety of unnatural amino acids charged onto tRNAs, either by chemical semi-synthesis or the use of a mutant aminoacyl tRNA synthetase (aaRS).19,72–75 Abramochkin and Shrader discovered that AaT was tolerant of variation in the acceptor stem (aminoacylation site) of the tRNA, and Sisido and coworkers have shown that much shorter oligonucleotides (Fig. 1 Bottom, Rn = 2 – 22 nucleotides) can act as donors. 74,76,77 As part of their structural characterization of AaT, Tomita and coworkers found that AaT could bind phenylalanyl adenosine and transfer Phe to peptides in trace amounts.63 However, the use of adenosine mononucleoside as a donor substrate for AaT has not, to our knowledge, been further explored.
We have found that a variety of hydrophobic amino acids can be transferred from adenosine donors with high yields at relatively low protein concentrations. (Fig. 1 Bottom, Rn = H) These donors can be synthesized from commercially-available active esters of the amino acids. Reduction of the two step synthetase/AaT reaction sequence to the one step AaT-only sequence allows us to explore the substrate specificity of AaT without limitation by the specificity of the synthetase in the first step. While this is also possible with Sisido’s pdCpA dinucleotide donors, the shorter synthesis of the simple adenosine donors (1–2 steps vs. 7 steps) makes this exploration easier. We have characterized the substrate scope of the wild type AaT enzyme using a reporter peptide, determined the kinetic parameters for the adenosyl substrates, and demonstrated the efficient modification of a full-sized protein, 23 kilodalton α-casein.
RESULTS AND DISCUSSION
The synthesis of adenosyl donor compounds, shown in Scheme 1, can be carried out on any amino acid with appropriate acid-labile protecting groups (e.g. N-Boc). This synthesis is not limited to α-amino acids, and our donor synthesis has also enabled us to test the transfer of analogs such as α-azidophenylalanine (N3f, 4g). Conversion to the cyanomethyl ester (1a–h) is typically high-yielding, although yields for the subsequent acylation of 5′-O-dimethoxytrityl-adenosine ((DMT)-A) varied between 60 and 90%. Acylation yields generally decreased with sidechain bulk; derivatives such as N-1,8-naphthyldiaminopropionic acid coupled poorly (data not shown). Uncatalyzed reactions took one to five days, but in all cases, addition of tetrabutylammonium acetate increased reaction rates.
Scheme 1. Synthesis of Adenosyl Donors and Use in AaT-Catalyzed Protein Modification.

Donor synthetic intermediates: 1a, 2a, 3a X = NHBoc, Z = Ph; 1b, 3b X = NHBoc, Z = i-Pr; 1c, 3c X = NHBoc, Z = p-Azidophenyl; 1d, 3d X = NHBoc, Z = 2-naphthyl; 1e, 3e X = NHMeBoc, Z = Ph; 1f, 3f X = NHAc, Z = Ph; 1g, 3g X = N3, Z = Ph; 1h, 3h X = NHBoc, Z = 7-Methoxycoumarinyl; 1i, 3i X = NHMeBoc, Z = p-Benzoylphenyl
The Phe donor 4a was synthesized from both the cyanomethyl ester (1a) and N-hydroxysuccinimidyl (OSu) ester (2a). All other adenosyl donors were synthesized from the cyanomethyl ester (1b–h). After trifluoroacetic acid (TFA) deprotection and CH2Cl2 washing (or Et2O precipitation) to remove DMT byproducts, the compounds could be taken on directly to AaT enzymatic reactions (contaminants were limited to incompletely-deprotected compounds which are not transferred by AaT, see Supporting Information). However, in most cases the deprotected adenosyl donor was purified by HPLC, and higher transfer yields were generally observed with these purified donors.
Transfer extent was evaluated by HPLC analysis of reactions with adenosyl donor, AaT, and an aminocoumarinlabeled reporter peptide used previously by Tirrell and coworkers (LysAlaAcm, Scheme 1).78 Monitoring LysAlaAcm acylation in crude reaction mixtures using absorbance at 325 nm conveniently eliminates background due to other proteins and nucleic acids, although some unnatural amino acids such as napthylalanine (Nap) and methoxycoumarinylalanine (Mcm) also absorb in this range. Example HPLC chromatograms from Phe ligations are shown in Figure 2. LysAlaAcm starting material eluted at 11.8 minutes (Fig. 2A); hydrophobic products eluted at 12 to 15 minutes (PheLysAlaAcm 5a elutes at 13.0 minutes in Fig. 2B, NapLysAlaAcm 5d elutes at 14.1 minutes in Fig. 2D).
Figure 2.

Reversed-phase HPLC Analysis of Transferase Reactions. Top: AaT-mediated transfer of Phe from adenosyl donor 4a to LysAlaAcm reporter peptide. HPLC chromatograms obtained after (A) 0 h or (B) 4 h show conversion of LysAlaAcm (11.8 min retention time) to PheLysAlaAcm (13.0 min retention time). Conversion after one addition of donor 4a is 92 % (B). A second addition of 1 mM 4a drives the reaction to completion with a small amount of double Phe addition (C). Bottom: HPLC analysis of transfer of Nap from adenosyl donor 4d to LysAlaAcm after (D) 4 h shows conversion to NapLysAlaAcm (14.1 min retention time). Product identities confirmed by observation of indicated masses by MALDI MS.
To evaluate substrate scope, reactions were analyzed after 4 hours, when transfer should be complete for the fully enzymatic reaction with Phe tRNA synthetase.78 Phe (4a) and Leu (4b) were transferred efficiently. (Table 1) Our PheLysA-laAcm yields from donor 4a were comparable to yields from Phe-pdCpA, the dinculeotide used by Sisido. Transfer yields for other substrates varied with sidechain size, where Mcm (4h) is too large and benzoylphenylalanine (Bzf, 4i) is a poor substrate. (See Table 1) Sisido has shown that AaT mutants can use substrates with larger sidechains; we are currently exploring some of these mutations.74 While transfer yields after 4 hours varied, most reactions could be driven to completion by a bolus of donor molecule. (Fig. 2C)
Table 1.
N-terminal Modification of Reporter Peptide After Single Addition of Adenosyl Donor
| Donor | Additivesa | Yield (%)b |
|---|---|---|
| 4a (Phe) | 92.7 ± 5.2 | |
| Phe-pdCpA | 95.2 ± 4.3 | |
| 4a (Phe) | 1 mM ATP | 86.1 ± 0.8 |
| 4a (Phe) | 5 mM ATP | 79.1 ± 0.4 |
| 4a (Phe) | 5 mM AMP | 88.2 ± 0.8 |
| 4a (Phe) | 5 mM A | 93.1 ± 1.1 |
| 4a (Phe) | 1 mM pdCpA | 90.4 ± 0.9 |
| 4b (Leu) | 80.4 ± 0.9 | |
| 4c (Azf) | 78.5 ± 5.2 | |
| 4d (Nap) | 95.4 ± 0.4 | |
| 4d (Nap) | 100 μM PheLysAlaAcm | 93.8 ± 0.3 |
| 4e (Mef) | 0.6 ± 0.1 | |
| 4f (Acf) | 5f not observed | |
| 4g (N3f) | 5g not observedc | |
| 4h (Mcm) | 5h not observed | |
| 4i (Bzf) | 9.2 ± 0.3 |
Standard AaT-catalyzed transfer conditions with or without potential inhibitor added. See description under Experimental Procedures.
Yield determined by integration of HPLC chromatogram peak areas at 325 nm, described in Experimental Procedures. Standard deviations are reported for an average of at least 6 experiments with at least 2 different AaT preparations.
HPLC and MALDI MS analysis indicate that the observed product is 5a not 5g.
One of the advantages of adenosyl donors over in situ tRNA aminoacylation with a tRNA synthetase is that we are not limited by the substrate specificity of the synthetase. In particular, non-amino acid substrates can be attached to adenosyl donors to assess their transferability by AaT. N-methyl phenylalanine (Mef, 4e) can be transferred, albeit with low yield. We have also tested N-acetyl phenylalanine (Acf, 4f) and α-azidophenylalanine (4g) and found them to be poor substrates. In some experiments, substantial conversion of LysAlaAcm was found when incubated with 4g, but MALDI MS analysis has shown the product to be 5a, presumably obtained by transfer of Phe after prior reduction (i.e. Phe donor 4a is formed from 4g under the reaction conditions). Steps taken to alter the concentration of β-mercaptoethanol reductant did not improve the yield of 5g (data not shown). We believe that disproportionation of the azide is incomplete, but that AaT is selective for the α-amine substrate (Phe-A, 4a), so far more 5a than 5g is formed. This may be due to an important interaction of the α-amine with Q188, which also acts as a catalytic base to deprotonate the peptide substrate for amide bond formation. The Q188/α-amine interaction was defined by the structural and enzymological work of Watanabe and coworkers.63 We note that the preference of AaT for unmodified N-termini was previously documented with full-length tRNA by Soffer and Leibowitz.71
To better understand limitations on the yield from AaT reactions, we assessed the possibilities of inhibition by hydrolyzed substrate or aminoacylated product. Since the acyl adenosine donor substrate is not regenerated during the reaction (unlike the aaRS/tRNA/AaT reaction), we hypothesized that the adenosine byproduct might inhibit further acylation by binding to AaT. To investigate this possibility, we added adenosine to the reaction at varying concentrations and monitored the reaction timecourse by fluorescence spectroscopy and by HPLC. Addition from the Phe donor 4a was not inhibited by the addition of 5 mM concentrations of adenosine. (Table 1) A comparison of adenosine compounds, including pdCpA, the dinucleotide donor used by Sisido, showed that only adenosine triphosphate (ATP) inhibited the reaction.
We were also able to monitor product formation in real time based on partial quenching of coumarin fluorescence upon addition of Nap to the LysAlaAcm peptide to form 5d. The fluorescence intensity at 390 nm of NapLysAlaAcm is 28% of the fluorescence of LysAlaAcm, and the overall fluorescence of mixtures of the two peptides can be used to determine the proportions of each peptide if the total concentration is known. (See Supporting Information) Thus, the change in fluorescence intensity was used to monitor NapLysAlaAcm formation in real time, and HPLC injection of the reaction endpoints were used to confirm the final product distribution. We found that exogenous adenosine did not significantly inhibit the reaction, even at concentrations up to 1 mM. (Fig. 3A)
Figure 3.

Kinetic Analysis of AaT Reactions. (A) Real time monitoring of Nap transfer to LysAlaAcm from 4d by quenching of Acm fluorescence. No significant inhibition is observed in the presence of 1 mM adenosine. (B) Saturation curve used to determine Michaelis-Menten kinetic parameters for LysAlaAcm modification by Phe donor 4a.
We also assessed the possibility of product inhibition by the aminoacylated LysAlaAcm product. In our standard 4 hour transfer assay, addition of 100 μM (1 equiv) PheLysAlaAcm did not significantly inhibit production of NapLysAlaAcm (5d) when Nap-A (4d) was used as a donor. (Table 1) MALDI MS analysis and differences in HPLC retention time allowed us to easily distinguish LysAlaAcm, PheLysAlaAcm, from NapLysAlaAcm. (See Fig. 2 and Supporting Information). Finally, we investigated the question of whether donor hydrolysis slowed reaction rates after 30 - 60 minutes. Analysis of mock reactions lacking transferase or LysAlaAcm indicates that donor hydrolysis may contribute significantly to slowing reactions, which is why a donor bolus can be used to drive the reaction to completion. (See Supporting Information)
In order to characterize the enzymatic activity of AaT toward our acyl adenosyl substrates, we used our HPLC assay to determine initial reaction rates to be fit to a standard Michaelis- Menten kinetic model. Equations describing this kinetic analysis are included in Supporting Information. Since adenosine does not inhibit the enzyme, by keeping LysAlaAcm concentrations high (100 μM), we measured the rate of transfer as a function of the concentration of Phe donor 4a and fit this to a single-substrate kinetic model to determine a kcat of 1.68 ± 0.09 × 10−1 s−1 and a KM of 1.24 ± 0.23 × 10−4 M. The enzyme efficiency (kcat/KM = 1.35 × 103 M−1 s−1) is relatively low.79 While KM is higher for Phe-A than what Shrader reported for aminoacyl tRNALeu-4 (0.3 μM), the kcat is comparable (0.13 × 10−1 s−1).77 Since it is trivial to run our reactions at donor concentrations well above KM, the lower apparent affinity for our substrates should not affect the utility of the reaction. We note that the binding affinity, which gives a KM of 124 μM for 4a must arise primarily from Phe binding interactions since the KI for adenosine inhibition is greater than 5 mM.
It is possible that larger protein substrates are modified with different efficiencies than peptide substrates.71,74,77 To address this question and to demonstrate the utility of our method in tagging full-sized proteins with useful fluorophores or affinity purification tags, we modified the milk protein α-casein (bearing a native N-terminal Arg after proteolysis of a leader sequence). 80 First, modification using Phe donor 4a was assessed by Edman degradation, where successful, quantitative addition of Phe was observed. (See Supporting Information)
In a more complex set of experiments, we first modified α-casein with Azf using donor 4c, and then used a Cu-catalyzed Huisgen cycloaddition (“click” reaction) to append a fluorescein label.81–84 (See Fig. 4A) Prior to fluorescent-labeling, the initial transfer of Azf was analyzed by trypsin digest and MALDI MS of α-casein. (Fig. 4C) The N-terminal peptide ArgProLys disappeared from the mass spectrum following modification and a peptide corresponding to AzfArgProLys was observed (also a mass corresponding to AzfArgProLys after loss of N2 during ionization; N2 loss was also observed for AzfLysAlaAcm). Complete conversion of α-casein was observed in MALDI MS due to double addition of 4c to drive reaction.
Figure 4.

AaT-Catalyzed Modification and “Click” Reaction of α-Casein N-terminus. (A) α-Casein modification scheme. (B) PAGE gel analysis of α-casein modification. Lanes (left to right): 1) molecular weight (MW) markers (Masses in kDa: 17, 25, 30, 46, 58, 80, 175); 2) α-casein; 3) α-casein mixed with fluorescein alkyne (FlAlk); 4) α-casein mixed with FlAlk, CuSO4, THPTA, and sodium ascorbate; 5) α-casein mixed with Azf-A (4c) and AaT; 6) α-casein mixed with 4c and AaT, then FlAlk; 7) α-casein mixed with 4c and AaT, then propargylamine (Alk), CuSO4, Tris- (3-hydroxypropyltriazolylmethyl) amine (THPTA), and sodium ascorbate; 8) α-casein mixed with 4c and AaT, then FlAlk, CuSO4, THPTA, and sodium ascorbate; 9) MW markers; 10) Cell Lysate Labeling: α-casein reaction carried out using conditions of lane 8 with unpurifed AaT in cleared E. coli lyaste. (C) MALDI MS analysis of trypsinized N-terminal fragment of α-casein with or without modification by Azf using AaT and 4c (double addition).
After AaT-catalyzed modification of α-casein with Azf, residual 4c was removed by dialysis, AaT (His10-tagged) was removed by treatment with Ni beads, and the azide-bearing protein was reacted with fluoescein alkyne (FlAlk) in the presence of Cu (I). The results of this labeling experiment, as well as appropriate control reactions are shown in Figure 4B. One can see that fluorescent α-casein is only observed under the proper conditions (Lane 8), where both the chemoenzymatic labeling by AaT with 4c and the subsequent click reaction are successful. Finally, we carried out a similar labeling experiment in crude E. coli lysate with unpurified AaT. Azf transfer was successful, as shown by the observation of fluorescence after labeling with FlAlk. Control reactions shown in Supporting Information) show no observable labeling of any protein, implying that endogenous levels of proteins terminating in Arg or Lys are too low to be observed here.
CONCLUSION
Substantial recent effort has been devoted to developing methods for labeling proteins under conditions that maintain protein folding and activity.85–87 E. coli aminoacyl tRNA transferase can work under these conditions, and specifically modifies the N-terminus, a useful point for conjugation. However, certain aspects of its native mechanism are non-ideal for its use as a preparative tool (i.e. a multi-step, multienzyme reaction sequence). The work of Leibowitz, Shrader, and Sisido showed that E. coli aminoacyl tRNA transferase can modify the N-terminus of a protein ending in Arg or Lys with amino acids from an aminoacyl oligonucleotide donor.71,74,77 Watanabe et al. crystallized AaT with Phe-A bound and showed that Phe could be transferred from Phe-A to the N-terminus of the peptide RYLGYL in trace amounts. These two results provide crucial precedent for our own work, in which we show that AaT can efficiently use a variety of easily-synthesized acyl adenosine substrates. Enzymological analysis demonstrates that AaT is able to use the Xaa-A substrates with turnover numbers comparable to full-length aminoacyl tRNAs. Furthermore, we have shown that neither the adenosine reaction byproduct nor aminoacylated protein product (at least in the case of our reporter peptide) appreciably inhibit the reaction. In fact, the major obstacle to efficient transfer is donor hydrolysis, but this can be overcome by a second addition of donor molecule since the KI for inhibition by adenosine is greater than 5 mM.
Our AaT-only reaction sequence allows us to explore the compatibility of Phe analogs lacking a primary α-amine with transfer by AaT. The wild type transferase seems to have a strong preference for the α-amine, as only N-methylphenylalanine was transferred. In contrast, the sidechain pocket is quite permissive: we have observed the transfer of bicyclic amino acid sidechains and Sisido has reported mutants capable of transferring tricyclic amino acids.74 We are currently investigating the transfer of larger sidechains such as benzophenone, biotin, and fluorescein derivatives.
Our reaction conditions and yields compare favorably to two well-established methods in the field. Subtiligase (reverse proteolysis) reactions can be carried out in a few hours, but rarely go to completion.44,45,51 The pryidoxyl-5-phosphate (PLP) transamination reactions of Francis and coworkers can require elevated temperatures to achieve high yields and are subject to a number of side reactions.36,37 AaT transfer reactions are highly specific (no Lys sidechain modifications were observed in our work or the work of others) and can be driven to completion by subsequent repeated additions of donor. Our discovery of efficient transfer from simple adenosyl donors removes the limitations imposed by the selectivity of synthetases on the range of molecules that can be AaT substrates without restricting reaction scale by necessitating the synthesis of oligonucleotide donors. Our yields are comparable to those obtained with pdCpA donors, but pdCpA synthesis requires 6 steps, and overall yields are typically 25%. We see our methods as complementary to the fully enzymatic synthetase-based methods (Fig. 1, Top). Fully enzymatic methods do not require prior synthesis and aminoacyl tRNAs are efficient substrates for AaT. On the other hand, our methods can be easily scaled up to modify larger quantities of protein since they do not require the use of purified tRNA.
We have shown that our method can be used to transfer reactive handles to the N-terminus under “protein friendly” (pH ~ 7, high salt, 37 °C) conditions and that the labeled protein product can be easily purified afterward. Given the previous success of Sisido and coworkers, we expect that further mutation of AaT will allow us to transfer larger sidechains that may permit direct transfer of fluorescent probes or affinity tags. We also hope to use the available cystal structures to redesign AaT to act on other N-terminal sequences through a combination of rational design and selection. Finally, our demonstration that AaT can modify α-casein in crude lysates, coupled with the cell-permeability of our moderately polar donors raises the exciting possibility of using AaT as part of an in vivo labeling strategy.
EXPERIMENTAL PROCEDURES
General Information
Chloroacetonitrile, N,N-diisopropylethylamine (DIPEA), 5′-O-(4,4′-Dimethoxytrityl) adenosine ((DMT)-A), tetrabutylammonium acetate (TBAAc), trifluoroacetic acid (TFA), and triisopropyl silane (TIPSH) were purchased from Sigma-Aldrich (St. Louis, MO). Note: (DMT)-A production was discontinued by Sigma-Aldrich, after which it was purchased as a custom order from Chem- Genes Corporation (Wilmington, MA) and was additionally synthesized in-house following the protocol outlined by Ogilvie et al.88 N-Boc-L-phenylalanine-O-succinimide (Boc-Phe- OSu) and Lysylalanylaminomethylcoumarin (LysAlaAcm) were purchased from Bachem (Torrence, CA). N-Boc-L-phenylalanine (Boc-Phe-OH) and all solvents were purchased from Fisher Scientific (Pittsburgh, PA). All deuterated solvents were purchased from Cambridge Isotopes Laboratories, Inc. (Andover, MA). E. coli BL21(DE3) cells were purchased from Stratagene (La Jolla, CA). The pEG6 plasmid, containing His10-tagged E. coli AaT, was a gift from Alexander Varshavsky (California Institute of Technology). Sequencing-grade trypsin was purchased from Promega (Madison, WI). All other reagents were purchased from Fisher Scientific (Pittsburgh, PA).
Milli-Q filtered (18 MΩ) water was used for all aqueous solutions (Millipore; Billerica, MA). Matrix-assisted laser desorption ionization (MALDI) mass spectra were collected using a Bruker Ultraflex III MALDI-TOF-TOF mass spectrometer (Billerica, MA). UV absorbance spectra were obtained with a Hewlett-Packard 8452A diode array spectrophotometer (currently Agilent Technologies; Santa Clara, CA). Donor molecule purification was conducted on a BioCad Sprint FPLC (GMI Inc.; Ramsey, MN; originally from Perseptive Biosystems) with a Waters Sunfire Prep C18-prep OBD™ column, 5 μm, 17 × 150 mm (Milford, MA). Analytical HPLC assays were performed on an Agilent 1100 HPLC using a Waters Symmetry Shield C18 column. NMR spectra, 1H and 13C, were collected with a Bruker DRX 500 MHz instrument. “Low resolution” electrospray ionization (ESI) mass spectra (LRMS) were obtained on a Waters Acquity Ultra Performance LC connected to a single quadrupole detector (SQD) mass spectrometer. “High resolution” ESI mass spectra (HRMS) were obtained on a Waters LCT Premier XE LC/MS. DNA sequencing was performed at the University of Pennsylvania DNA sequencing facility. Fluorescence spectra were collected with a Varian Cary Eclipse fluorescence spectrophotometer fitted with a Peltier multicell holder (currently Agilent Technologies).
Donor (4a–h) Synthesis
The synthesis of Phe donor 4a is given as a general procedure, starting from commercially available Boc-Phe-OH or Boc-Phe-OSu. The carboxyterminus of the amino acid was activated as a cyanomethyl ester using chloroacetonitrile to give 1a. Next, the protected amino acid was attached to one of the available hydroxyls (2′ OH or 3′ OH) of (DMT)-A without preference. The resulting protected adenylate (3a) was deprotected using a 50/50 mixture of TFA and THF with TIPSH present as a scavenger. The final product, deprotected adenylate (4a), was purified on C18 reverse-phase HPLC column. The synthesis and characterization of donor 4a is provided below, characterization of other donor compounds (4b–h) and precursors is provided in Supporting Information.
(S)-cyanomethyl 2-((tert-butoxycarbonyl)amino)-3- phenylpropanoate (Boc-Phe-OCH2CN, 1a)
Boc-Phe-OH (2.01 g, 7.57 mmol) was dissolved in 10 mL tetrahydrofuran under ambient conditions. 10 equivalents of chloroacetonitrile (4.78 mL, 75.4 mmol) and 1.1 equivalents of DIPEA (8.30 mmol, 1.45 mL) were added to the reaction mixture and allowed to stir overnight. 1a was purified on silica gel (20% EtOAc in hexanes) to give 2.474 g (100%) of a pale yellow oil after evaporation. Rf 0.4 in 20% EtOAc in hexanes; 1H NMR (500 MHz, CDCl3) δ 7.26 (t, J = 7.0 Hz, 2H), 7.20 (t, J = 7.1 Hz, 1H), 7.11 (d, J = 7.3 Hz, 2H), 5.11 (d, J = 7.8 Hz, 1H), 4.69 - 4.57 (m, 3H), 3.09 - 2.98 (m, 2H), 1.36 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.8, 155.1, 135.4, 129.2, 128.7, 127.3, 114.0, 80.9, 80.2, 54.4, 48.8, 37.8, 28.2; HRMS (ESI) calcd m/z for C16H20N2O4Na [M + Na]+ 327.132, found 327.133.
(S)-(2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-((bis(4- methoxyphenyl)(phenyl)methoxy) methyl)-4- hydroxytetrahydrofuran-3-yl 2-((tert-butoxycarbonyl) amino)-3-phenylpropanoate (Boc-Phe- (DMT)-A, 3a)
(DMT)-A (25 mg, 44 μmol) was dissolved in 5 mL tetrahydrofuran (dried with 4 Å molecular sieves) in an over-dried 4 dram reaction vessel. 20 equivalents of 1a (272 mg, 0.895 mmol) were added to the reaction mixture. TBAAc (4.9 mg, 16 μmol), the reaction catalyst, was added last to the reaction mixture. In reproductions of this reaction, TBAAc additions varied according to reaction progress. The reaction stirred under argon overnight at room temperature for 1 - 4 days. After evaporation of solvent, the product was purified on silica gel (gradient solvent system: 20 - 0% petroleum ether in EtOAc, 5% methanol in EtOAc) to give 27 mg (75%) of a pale yellow foam after evaporation. Rf 0.5 in 5% methanol in EtOAc. 1H NMR (500 MHz, THF-d8) δ 8.08 (s, 1H), 8.00 (s, 1H), 7.44 (d, J = 3.68 Hz, 2H), 7.32 (d, J = 8.85 Hz, 4H), 7.20 (dd, J = 8.33 Hz, J = 6.66, 2H), 7.13 (t, J = 7.25 Hz, 1H), 7.21-7.10 (m, 4H), 6.77 (d, J = 4.30 Hz, 2H), 6.75 (d, J = 4.29 Hz, 2H), 5.92 (d, J = 4.87 Hz, 1H), 5.49 (t, J = 4.34 Hz, 1H), 5.13 (t, J = 5.54 Hz, 1H), 4.51 - 4.47 (m, 1H), 4.14 (t, J = 4.07 Hz, 1H), 3.71 (s, 6H) (S), 3.42 - 3.32 (m, 2H), 3.12 - 2.96 (m, 3H), 1.37 (s, 9H); 13C NMR (125 MHz, THF-d8) δ 172.2, 159.9, 157.6, 156.7, 153.8, 146.3, 138.3, 146.3, 138.3, 1367.0, 136.9, 131.2, 131.1, 130.3, 129.3, 129.2, 129.1, 128.6, 128.5, 127.5, 114.0, 113.9, 89.7, 87.5, 82.5, 79.7, 75.1, 73.5, 64.7, 56.4, 55.5, 38.9, 30.6, 28.8; LRMS (ESI) calcd m/z for C45H48N6O9H (M + H)+ 816.9, C45H48N6O9Na (M + Na)+ 839.3, found 817.3, 839.3.
(S)-(2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-5-((bis(4- methoxyphenyl)(phenyl)methoxy) methyl)-4- hydroxytetrahydrofuran-3-yl 2-((tert-butoxycarbonyl) amino)-3-phenylpropanoate (3a) from 2a
(DMT)-A (26 mg, 45 μmol) was dissolved in 5 mL tetrahydrofuran (dried with 4 Å molecular sieves) in an over-dried 4- dram reaction vessel. 20 equivalents of 2a (321 mg, 0.885 mmol) were added to the reaction mixture. TBAAc (19 mg, 64 μmol), the reaction catalyst, was added last to the reaction mixture in two separate additions, 6 mg, at the beginning of the reaction, and 13 mg after the first 24 h of reaction. The reaction stirred under argon overnight at room temperature for 2 days. After evaporation of solvent, the product was purified on silica gel (gradient solvent system: 20-0% petroleum ether in EtOAc, 5% methanol in EtOAc) to give 19.1 mg (52.1%) of a pale yellow foam after evaporation. LRMS (ESI) calcd m/z for C45H48N6O9H (M + H)+ 816.9, C45H48N6O9Na (M + Na)+ 839.3, found 817.4, 839.4.
(S)-(2R,3R,4R,5R)-2-(6-amino-9H-purin-9-yl)-4- hydroxy-5-(hydroxymethyl)tetrahydrofuran-3-yl 2-amino- 3-phenylpropanoate (Phe-A, 4a)
3a (50 mg) was dissolved in 1 mL THF and 1 mL trifluoroacetic acid (TFA) and reacted overnight with four equiv of TIPSH. Upon addition of TFA, the reaction turned a bright orange color and at the completion of the reaction, the color had become a dark brown. The reaction mixture was reduced to dryness by rotary evaporation and extracted using 1 mL dichloromethane and 1 mL water twice with a 1 mL water final back-extraction against the dichloromethane layer. The water-soluble layer containing 4a was then HPLC-purified on a C18-prep column using an increasing acetonitrile gradient (Gradient 2: 1 – 100% acetonitrile with water and 0.1% TFA, 1% per min) to remove any protected material. Prep. HPLC purified to give 13.6 mg (53.5%) of a white solid after evaporation. MALDI MS calcd m/z for C19H22N6O5H (M + H)+ 415.4, C19H22N6O5Na (M + Na)+ 437.4, found 415.0, 437.0.
LysAlaAcm Ligation Assay
Each ligation reaction, 125 μL total volume, contained the following reagents: aminoacyl adenosine donor (1 mM), recombinant His10-tagged E. coli aminoacyl transferase (2.26 μM), and LysAlaAcm (100 μM) in the AaT Ligation Buffer (50 mM HEPES pH 8.0, 150 mM KCl, 10 mM MgCl2). The reaction mixtures were incubated at 37 °C for four hours and quenched with 1% acetic acid. The proteins were extracted from the reactions via acetone precipitation. The reactions were precipitated using 4X reaction volume of acetone and cooled at − 20 °C for 1 hour. Next, the reactions were centrifuged at 13, 200 rpm at 4 °C for 20 min to separate the reaction from precipitated protein. The supernatant was transferred to fresh 1.5 mL centrifuge tubes and allowed for acetone evaporation overnight at room temperature. After acetone evaporation, the supernatant was dried in a Speedvac (Savant, Thermo Scientific, Fisher Inc.) for 30 min to remove residual acetone. The resulting reaction volume was dissolved up to 1.2 mL using MilliQ water and analyzed by HPLC (gradient below) to determine ligation yield by integration of separated peak intensities monitored at 325 nm. Collected HPLC fractions were characterized through MALDI MS analysis. Reactions were performed with 6 trials on at least two protein preparations for all successful reactions, 3 trials for failed reactions. Reactions using Phe-pdCpA as donor were carried out in an identical fashion.89
A/AMP/ATP/pdCpA Inhibition Studies
Each ligation reaction was performed as described above. The addition of four types of adenosyl compounds (adenosine, AMP, ATP, or pdCpA) were monitored individually in ligation reactions with Phe donor 4a. Adenosyl compound concentrations tested were 1 mM, 2.5 mM, and 5 mM. HPLC analysis was used to determine inhibition of ligation by integration of HPLC reagent and product peaks. The LysAlaAcm ligation assay was used to analyze the kinetics of AaT with substrate 4a. The ligation assay was modified for ease of analysis as follows: The total reaction volume was scaled up to 590 μL and all reagents were maintained at the same concentrations as mentioned above. Five concentrations of 4a were monitored for a total reaction time of 30 min, with concentrations ranging from 0.05 mM to 1 mM.
Each 30 min kinetic experiment was done in triplicate with a total of fourteen 40 μL aliquots for all substrate concentrations. The synthesis of pdCpA was carried out essentially as previously described.89
AaT Ligation Reaction Analysis by HPLC
All HPLC analyses of LysAlaAcm ligations were monitored on an Agilent HPLC using a Waters C18 column (Milford, MA). The solvents used for peptide purification were the following: 0.1% trifluoroacetic acid in water (Solvent A) and 0.1% trifluoroacetic acid in acetonitrile (Solvent B). The HPLC method had the following solvent gradient (Gradient 1): 0 min 1% B, 5 min 1% B, 10 min 30% B, 15 min 40% B, 20 min 100% B, 25 min 100% B, 27 min 1% B, 30 min 1% B. Peptides were monitored at two absorption wavelengths during HPLC analysis, 215 nm for peptide absorption, and 325 nm for Acm absorption. MALDI MS analyses confirmed identity of peptide, Xaa-A donor, and ligated products.
Fluorometric Analysis of Transferase Experiments
Real time fluorescent monitoring of Nap addition to LysA-laAcm was carried out using reaction solutions prepared on the 500 μL scale in stirred quartz cuvettes with 1.00 cm path lengths. Reagent concentrations were as described above, except that donor 4d was withheld until the cuvette was placed in the fluorometer and temperature equilibrated. Reactions were then initiated by addition of 20 μL of 25 mM Nap- A (4d) stock or water (for negative controls). Fluorescence emission at 390 nm was monitored after excitation at 325 nm. Prior calibration had shown that NapLysAlaAcm (5d) concentrations could be estimated from fluorescence intensity readings (F5d) using the equation [5d] = (1− (F5d/FControl))•1.39•[LysAlaAcm], where FControl is the fluorescence reading of the reaction with water added instead of 4d. Calibration details are given in Supporting Information. Measurements were taken on the Cary Eclipse fluorometer in the “Kinetics” mode at 37 °C, acquiring data every 15 s. The excitation and emission slit widths were 5 nm and the averaging time 1 s.
α-Casein N-Terminal Modification
α-Casein (4.8 mg) was modified with Azf-A (4c) in a reaction volume of 1 mL in modified AaT buffer (50 mM HEPES pH 8.0, 150 mM KCl, 10 mM MgCl2) and AaT (0.05 mg). The reaction mixture was incubated at 37 °C for 12 h, AaT was removed using Ni2+ resin (Ni-NTA Superflow, Qiagen), and then buffer exchanged four times into phosphate buffered saline (PBS, 12 mM NaH2PO4, 50 mM NaCl, 4.7 mM KCl, pH 8.0) by Spectra/Por 1 dialysis tubing (Spectrum Labs; Rancho Dominguez, CA). Azf-modified α-casein was used directly after buffer exchange into PBS. Aliquots (1 nmol) of α-casein were diluted into PBS (22 μL) containing either fluorescein-alkyne (FlAlk, 91 μM) or propargylamine (Alk, 227 μM) for copper-catalyzed azide-alkyne cycloaddition (CuAAC). The CuAAC reaction was initiated by addition of CuSO4 (100 μM), tris-(3- hydroxypropyltriazolylmethyl) amine (THPTA, 500 μM), and sodium ascorbate (5 mM). For control reactions, equivalent volumes were replaced with either PBS or water. The reaction mixtures were incubated at 4 °C for 2h. Reactions were boiled with gel loading dye LDS (Pierce; Rockford, IL) for 10 min at 95 °C and analyzed by SDS-PAGE. Fluorescence images were obtained with a Geldoc (BioRAD; Hercules, CA) using an excitation wavelength of 302 nm for detection of fluorescein. α-Casein was also directly visualized by Coomassie Brilliant Blue staining.
Tryptic Digest Analysis of α-Casein Modification
α-Casein-Azf (33 μg), or α-casein (33 μg), was digested with sequencing grade modified Trypsin (0.6 μg) in 27 μL of 25 mM ammonium bicarbonate pH 7.5 (freshly prepared). Digestions were carried out at 37 °C for 14 hours. Trypsin digest aliquots (1 μL) were combined with α-cyano-4- hydroxycinnamic acid (1 μL of a saturated solution in 1:1 H2O/CH3CN with 1% TFA) and analyzed by MALDI MS.
α-Casein Labeling in Cleared Cell Lysate
AaT was expressed in E. coli BL21-Gold (DE3) cells as previously described. A cleared cell lysate was obtained by centrifugation following cell lysis using sonication. Protein were modified in a final reaction volume of 110 μL with Azf-A (4c, 3 mM) using AaT (25 μL) from cleared lysate in AaT buffer (50 mM HEPES pH 8.0, 150 mM KCl, 10 mM MgCl2). α-Casein (0.012 mg or 0.12 mg) was added to the reaction and incubated at 37 °C for 2 h, after 1 h, an additional 0.3 μmole of 4c was added. For control reactions, equivalent volumes were replaced with water. Excess 4c was removed by buffer exchange four times against phosphate buffered saline (PBS, 12 mM NaH2PO4, 50 mM NaCl, 4.7 mM KCl, pH 8.0) by Spectra/Por 1 dialysis tubing. Aliquots (18 μL) of 4c modified lysates were diluted to a final volume of 22 μL using PBS containing FlAlk (182 μM) for copper-catalyzed “click” reactions. The “click” reaction was initiated by addition of CuSO4 (100 μM), THPTA (500 μM), and sodium ascorbate (5 mM). The reaction mixtures analyzed by PAGE gel using Coomassie staining and fluorescence as above. The full gel is shown in Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by funding from the University of Pennsylvania. The authors thank Alexander Varshavsky for the pEG6 plasmid, containing a gene for the His10-tagged E. coli AaT. We also thank David Tirrell and Frank Truong for the gift of the His6- tagged yeast Phe synthetase plasmid pQE32_yPheRS (used in control experiments not described) and for helpful advice. We are grateful to Eileen Moison for expression and purification of AaT and Lee Speight for pdCpA and Phe-pdCpA. We thank Jeff Saven for use of the fluorometer, and Rakesh Kohli for assistance with HRMS (NIH RR-023444) and MALDI-MS (supported by NSF MRI-0820996).
ABBREVIATIONS
- AaT
E. coli Aminoacyl tRNA Transferase
- aaRS
Aminoacyl tRNA Synthetase
- DMT-A
5′-O-dimethoxytrityl-adenosine
- OSu
N-Hydroxysuccinimidyl
- Azf
p-Azidophenylalanine
- Nap
2- Naphthylalanine
- Mef
N-Methylphenylalanine
- Acf
NAcetylphenylalanine
- N3f
α-Azidophenylalanine
- Mcm
7- Methoxycoumarinylalanine
- Bzf
p-Benzoylphenylalanine
- Acm
7-Aminomethylcoumarin
- FlAlk
Fluorescein 5- and 6- propargylamide
Footnotes
Supporting Information. Synthesis and characterization of 1b–h, 3b–h, and 4b–9h; synthesis of reagents for “click” chemistry; AaT expression and purification; analysis of transferase reactions with some representative HPLC chromatograms; kinetics derivations for enzymology; analysis of α-casein N-terminal modification by Edman degradation; full PAGE gel analysis of Azf labeling in cell lysates; and descriptions of AaT mutation experiments. Complete author lists for references 13 and 15. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wu PG, Brand L. Fluorescence Spectroscopy. 1997;278:321–330. doi: 10.1016/s0076-6879(97)78017-0. [DOI] [PubMed] [Google Scholar]
- 2.Harris JM, Chess RB. Nat Rev Drug Disc. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Schultz PG. Angew Chem, Int Ed. 2005;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 4.Basle E, Joubert N, Pucheault M. Chem Biol. 2010;17:213–227. doi: 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Canalle LA, Lowik D, van Hest JCM. Chem Soc Rev. 2010;39:329–353. doi: 10.1039/b807871h. [DOI] [PubMed] [Google Scholar]
- 6.Alouani S, Gaertner HF, Mermod JJ, Power CA, Bacon KB, Wells TNC, Proudfoot AEI. Eur J Biochem. 1995;227:328–334. doi: 10.1111/j.1432-1033.1995.tb20393.x. [DOI] [PubMed] [Google Scholar]
- 7.Kinstler OB, Brems DN, Lauren SL, Paige AG, Hamburger JB, Treuheit MJ. Pharm Res. 1996;13:996–1002. doi: 10.1023/a:1016042220817. [DOI] [PubMed] [Google Scholar]
- 8.Gale TF, Gorlitzer J, O’Brien SW, Williams DH. J Chem Soc Perkin 1. 1999:2267–2270. [Google Scholar]
- 9.Ramachandiran V, Willms C, Kramer G, Hardesty B. J Biol Chem. 2000;275:1781–1786. doi: 10.1074/jbc.275.3.1781. [DOI] [PubMed] [Google Scholar]
- 10.Jones DS, Cockerill KA, Gamino CA, Hammaker JR, Hayag MS, Iverson GM, Linnik MD, McNeeley PA, Tedder ME, Ton-Nu HT, Victoria EJ. Bioconj Chem. 2001;12:1012–1020. doi: 10.1021/bc015512x. [DOI] [PubMed] [Google Scholar]
- 11.Kinstler O, Molineux G, Treuheit M, Ladd D, Gegg C. Adv Drug Delivery Rev. 2002;54:477–485. doi: 10.1016/s0169-409x(02)00023-6. [DOI] [PubMed] [Google Scholar]
- 12.Chelius D, Shaler TA. Bioconj Chem. 2003;14:205–211. doi: 10.1021/bc025605u. [DOI] [PubMed] [Google Scholar]
- 13.Arduini RM, et al. Protein Exp Purification. 2004;34:229–242. doi: 10.1016/j.pep.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Mamaev S, Olejnik J, Olejnik EK, Rothschild KJ. Anal Biochem. 2004;326:25–32. doi: 10.1016/j.ab.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 15.Baker DP, et al. Bioconj Chem. 2006;17:179–188. doi: 10.1021/bc050237q. [DOI] [PubMed] [Google Scholar]
- 16.Scheck RA, Francis MB. ACS Chem Biol. 2007;2:247–251. doi: 10.1021/cb6003959. [DOI] [PubMed] [Google Scholar]
- 17.Merkel L, Beckmann HSG, Wittmann V, Budisa N. ChemBioChem. 2008;9:1220–1224. doi: 10.1002/cbic.200800050. [DOI] [PubMed] [Google Scholar]
- 18.Sharon JL, Puleo DA. Biomaterials. 2008;29:3137–3142. doi: 10.1016/j.biomaterials.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebhardt HA, Xu ZZ, Fung AW, Fahlman RP. Anal Chem. 2009;81:1937–1943. doi: 10.1021/ac802423d. [DOI] [PubMed] [Google Scholar]
- 20.Gao WP, Liu WG, Mackay JA, Zalutsky MR, Toone EJ, Chilkoti A. Proc Natl Acad Sci USA. 2009;106:15231–15236. doi: 10.1073/pnas.0904378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sayers CT, Mantovani G, Ryan SM, Randev RK, Keiper O, Leszczyszyn OI, Blindauer C, Brayden DJ, Haddleton DM. Soft Matter. 2009;5:3038–3046. [Google Scholar]
- 22.Xiao JP, Tolbert TJ. Org Lett. 2009;11:4144–4147. doi: 10.1021/ol9016468. [DOI] [PubMed] [Google Scholar]
- 23.Jia J, Chen W, Ma HM, Wang K, Zhao CA. Mol Biosys. 2010;6:1829–1833. doi: 10.1039/c005223j. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Hu T, Liu YD, Zhang GF, Ma GH, Su ZG. Anal Biochem. 2010;412:114–116. doi: 10.1016/j.ab.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 25.Wildes D, Wells JA. Proc Natl Acad Sci USA. 2010;107:4561–4566. doi: 10.1073/pnas.0914495107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu JJ, Peng HT, Shek PN. J App Poly Sci. 2010;118:3269–3273. [Google Scholar]
- 27.Goldberg JM, Batjargal S, Petersson EJ. J Am Chem Soc. 2010;132:14718–14720. doi: 10.1021/ja1044924. [DOI] [PubMed] [Google Scholar]
- 28.Dixon HBF, Weitkamp LR. Biochem J. 1962;84:462–468. doi: 10.1042/bj0840462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dixon HBF. Biochem J. 1964;92:661–666. doi: 10.1042/bj0920661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geoghegan KF, Ybarra DM, Feeney RE. Biochemistry. 1979;18:5392–5399. doi: 10.1021/bi00591a021. [DOI] [PubMed] [Google Scholar]
- 31.Dixon HBF. J Protein Chem. 1984;3:99–108. [Google Scholar]
- 32.Acharya AS, Manjula BN. Biochemistry. 1987;26:3524–3530. doi: 10.1021/bi00386a041. [DOI] [PubMed] [Google Scholar]
- 33.Qasmi D, Rene L, Badet B. Tetrahedron Lett. 1994;35:4343–4344. [Google Scholar]
- 34.Li XF, Zhang LS, Hall SE, Tam JP. Tetrahedron Lett. 2000;41:4069–4073. [Google Scholar]
- 35.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew Chem, Int Ed. 2006;45:5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 36.Scheck RA, Dedeo MT, Lavarone AT, Francis MB. J Am Chem Soc. 2008;130:11762–11770. doi: 10.1021/ja802495w. [DOI] [PubMed] [Google Scholar]
- 37.Witus LS, Moore T, Thuronyi BW, Esser-Kahn AP, Scheck RA, Iayarone AT, Francis MB. J Am Chem Soc. 2010;132:16812–16817. doi: 10.1021/ja105429n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuhl P, Konnecke A, Doring G, Daumer H, Jakubke HD. Tetrahedron Lett. 1980;21:893–896. [Google Scholar]
- 39.Jakubke HD, Kuhl P, Konnecke A. Angew Chem, Int Ed. 1985;24:85–93. [Google Scholar]
- 40.Schellenberger V, Jakubke HD. Angew Chem, Int Ed. 1991;30:1437–1449. [Google Scholar]
- 41.Chang TK, Jackson DY, Burnier JP, Wells JA. Proc Natl Acad Sci USA. 1994;91:12544–12548. doi: 10.1073/pnas.91.26.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson DY, Burnier J, Quan C, Stanley M, Tom J, Wells JA. Science. 1994;266:243–247. doi: 10.1126/science.7939659. [DOI] [PubMed] [Google Scholar]
- 43.Bordusa F, Ullmann D, Elsner C, Jakubke HD. Angew Chem, Int Ed. 1997;36:2473–2475. [Google Scholar]
- 44.Braisted AC, Judice JK, Wells JA. Solid-Phase Peptide Synthesis. 1997;289:298–313. doi: 10.1016/s0076-6879(97)89053-2. [DOI] [PubMed] [Google Scholar]
- 45.Atwell S, Wells JA. Proc Natl Acad Sci USA. 1999;96:9497–9502. doi: 10.1073/pnas.96.17.9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bordusa F. Chem Rev. 2002;102:4817–4867. doi: 10.1021/cr010164d. [DOI] [PubMed] [Google Scholar]
- 47.Tolbert TJ, Wong CH. Angew Chem, Int Ed. 2002;41:2171–2174. doi: 10.1002/1521-3773(20020617)41:12<2171::aid-anie2171>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 48.Muir TW. Ann Rev Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 49.Wehofsky N, Koglin N, Thust S, Bordusa F. J Am Chem Soc. 2003;125:6126–6133. doi: 10.1021/ja0344213. [DOI] [PubMed] [Google Scholar]
- 50.Gentle IE, De Souza DP, Baca M. Bioconj Chem. 2004;15:658–663. doi: 10.1021/bc049965o. [DOI] [PubMed] [Google Scholar]
- 51.Yoshihara HAI, Mahrus S, Wells JA. Bioorg Med Chem Lett. 2008;18:6000–6003. doi: 10.1016/j.bmcl.2008.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mazmanian SK, Liu G, Hung TT, Schneewind O. Science. 1999;285:760–763. doi: 10.1126/science.285.5428.760. [DOI] [PubMed] [Google Scholar]
- 53.Mao HY, Hart SA, Schink A, Pollok BA. J Am Chem Soc. 2004;126:2670–2671. doi: 10.1021/ja039915e. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka T, Kamiya N, Nagamune T. FEBS Lett. 2005;579:2092–2096. doi: 10.1016/j.febslet.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 55.Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Nat Chem Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- 56.Fontana A, Spolaore B, Mero A, Veronese FM. Adv Drug Delivery Rev. 2008;60:13–28. doi: 10.1016/j.addr.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 57.Heal WP, Wickramasinghe SR, Leatherbarrow RJ, Tate EW. Org Biomol Chem. 2008;6:2308–2315. doi: 10.1039/b803258k. [DOI] [PubMed] [Google Scholar]
- 58.Tsukiji S, Nagamune T. ChemBioChem. 2009;10:787–798. doi: 10.1002/cbic.200800724. [DOI] [PubMed] [Google Scholar]
- 59.Nelson JW, Chamessian AG, McEnaney PJ, Murelli RP, Kazmiercak BI, Spiegel DA. ACS Chem Biol. 2010;5:1147–1155. doi: 10.1021/cb100195d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lahoud G, Hou YM. Nat Chem Biol. 2010;6:795–796. doi: 10.1038/nchembio.459. [DOI] [PubMed] [Google Scholar]
- 61.Kaji A, Novelli GD, Kaji H. Biochem Biophys Res Commun. 1963;10:406–409. doi: 10.1016/0006-291x(63)90546-1. [DOI] [PubMed] [Google Scholar]
- 62.Suto K, Shimizu Y, Watanabe K, Ueda T, Fukai S, Nureki O, Tomita K. EMBO J. 2006;25:5942–5950. doi: 10.1038/sj.emboj.7601433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watanabe K, Toh Y, Suto K, Shimizu Y, Oka N, Wada T, Tomita K. Nature. 2007;449:867–871. doi: 10.1038/nature06167. [DOI] [PubMed] [Google Scholar]
- 64.Mogk A, Schmidt R, Bukau B. Trends Cell Biol. 2007;17:165–172. doi: 10.1016/j.tcb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 65.Tobias JW, Shrader TE, Rocap G, Varshavsky A. Science. 1991;254:1374–1377. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]
- 66.Varshavsky A. Nat Struct Mol Biol. 2008;15:1238–1240. doi: 10.1038/nsmb1208-1238. [DOI] [PubMed] [Google Scholar]
- 67.Schuenemann VJ, Kralik SM, Albrecht R, Spall SK, Truscott KN, Dougan DA, Zeth K. EMBO Reports. 2009;10:508–514. doi: 10.1038/embor.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaji A, Kaji H, Novelli GD. J Biol Chem. 1965;240:1185–1191. [PubMed] [Google Scholar]
- 69.Kaji A, Kaji H, Novelli GD. J Biol Chem. 1965;240:1192–1197. [PubMed] [Google Scholar]
- 70.Leibowitz MJ, Soffer RL. Biochem Biophys Res Commun. 1969;36:47–53. doi: 10.1016/0006-291x(69)90647-0. [DOI] [PubMed] [Google Scholar]
- 71.Leibowitz MJ. J Biol Chem. 1971;246:5207–5212. [PubMed] [Google Scholar]
- 72.Taki M, Kuno A, Matoba S, Kobayashi Y, Futami J, Murakami H, Suga H, Taira K, Hasegawa T, Sisido M. ChemBioChem. 2006;7:1676–1679. doi: 10.1002/cbic.200600181. [DOI] [PubMed] [Google Scholar]
- 73.Taki M, Sisido M. Biopolymers. 2007;88:263–271. doi: 10.1002/bip.20678. [DOI] [PubMed] [Google Scholar]
- 74.Taki M, Kuroiwa H, Sisido M. ChemBioChem. 2008;9:719–722. doi: 10.1002/cbic.200700721. [DOI] [PubMed] [Google Scholar]
- 75.Ebisu K, Tateno H, Kuroiwa H, Kawakami K, Ikeuchi M, Hirabayashi J, Sisido M, Taki M. ChemBioChem. 2009;10:2460–2464. doi: 10.1002/cbic.200900430. [DOI] [PubMed] [Google Scholar]
- 76.Abramochkin G, Shrader TE. J Biol Chem. 1995;270:20621–20628. doi: 10.1074/jbc.270.35.20621. [DOI] [PubMed] [Google Scholar]
- 77.Abramochkin G, Shrader TE. J Biol Chem. 1996;271:22901–22907. doi: 10.1074/jbc.271.37.22901. [DOI] [PubMed] [Google Scholar]
- 78.Connor RE, Piatkov K, Varshavsky A, Tirrell DA. ChemBioChem. 2008;9:366–369. doi: 10.1002/cbic.200700605. [DOI] [PubMed] [Google Scholar]
- 79.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. 3. W. H. Freeman; 1998. [Google Scholar]
- 80.Mercier JC, Grosclau F, Ribadeau B. Eur J Biochem. 1971;23:41–51. doi: 10.1111/j.1432-1033.1971.tb01590.x. [DOI] [PubMed] [Google Scholar]
- 81.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 82.Wang Q, Chan TR, Hilgraf R, Fokin VV, Sharpless KB, Finn MG. J Am Chem Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 83.Huisgen R. Pure App Chem. 1989;61:613–628. [Google Scholar]
- 84.Meldal M, Tornoe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 85.Hackenberger CPR, Schwarzer D. Angew Chem, Int Ed. 2008;47:10030–10074. doi: 10.1002/anie.200801313. [DOI] [PubMed] [Google Scholar]
- 86.Sletten EM, Bertozzi CR. Angew Chem, Int Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carrico IS. Chem Soc Rev. 2008;37:1423–1431. doi: 10.1039/b703364h. [DOI] [PubMed] [Google Scholar]
- 88.Ogilvie KK, Letsinge RI. J Org Chem. 1967;32:2365–2366. doi: 10.1021/jo01282a066. [DOI] [PubMed] [Google Scholar]
- 89.Ellman J, Mendel D, Anthonycahill S, Noren CJ, Schultz PG. Methods Enzymol. 1991;202:301–336. doi: 10.1016/0076-6879(91)02017-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.