

Abstract
Focal adhesion kinase is a key component of cell-substratum adhesions, known as focal adhesion complexes. Growing evidence indicates that FAK is important in maintenance of normal cell survival and that disruption of FAK signaling results in loss of substrate adhesion and anoikis (apoptosis) of anchorage-dependent cells, such as endothelial cells. Basal FAK activity in non-stimulated endothelial cells is important in maintaining cell adhesion to integrins via PI3 kinase/Akt signaling. FAK activity is dependent upon small GTPase signaling. FAK also appears to be important in cardiomyocyte hypertrophy and hypoxia/reoxygenation-induced cell death. This review summaries the signaling pathways of FAK in prevention of apoptosis and the role of FAK in mediating adenosine and homocysteine-induced endothelial cell apoptosis and in cardiovascular diseases.
Introduction
Normally, endothelial cells are linked to underlying basement membrane by focal adhesion complexes. Formation of focal adhesion complexes is initiated with creation of links between extracellular matrix protein ligands in the basement membrane with specific integrin receptors on the endothelial cells (Hynes, 1992). Integrins are heterodimers of α and β subunits with ectodomains linked to arginine-glycine-aspartate (RGD) amino acid motifs in extracellular matrix proteins (Pierschbacher and Ruoslahti, 1984). There are 19 α and 8 β integrin subunits; endothelial cells possess α1β1, which bind to collagen; α2β1, α3β1, α6β1, and α6β4, which bind to laminin; α4β1 and α5β1, which are fibronectin receptors; and αvβ3 and αvβ5, which selectively bind vitronectin (Albelda et al., 1989; Cheng and Kramer, 1989; Dejana et al., 1988; Mechtersheimer et al., 1994). Upon integrin binding to extracellular matrix RGD domains, the smaller cytoplasmic integrin domains are associated with actin binding proteins, including vinculin, α-actinin, paxillin, talin, zyxin, tensin, and filamin (Geiger et al., 2001; Zamir and Geiger, 2001). The actin binding proteins in turn associate with specific target molecules and signaling networks. Focal adhesion kinase (FAK) is activated by integrins and integrates integrin signals through directly binding to paxillin, p130Cas, and other signaling molecules (Parsons, 2003).
Anchorage-dependent cells, such as endothelial cells, undergo programmed cell death (apoptosis) when detached from underlying extracellular matrix (Ruoslahti and Reed, 1994). This was first described in endothelial cells suspended in serum-free medium or seeded on plastic coated with anti-integrin β1 antibody (Meredith et al., 1993). The term “anoikis”, or “homelessness”, was first used by Frisch and Francis to describe this process in MDCK epithelial cells (Frisch and Francis, 1994). Malignant cells that have undergone oncogenic transformation can grow in an anchorage-independent fashion and not undergo anoikis. FAK phosphorylation is important in maintaining normal cell adhesion (Guan and Shalloway, 1992; Guan et al., 1991).
Endothelial cell detachment from underlying basement membrane in the form of “blebs” has been described in microvessels in human atherosclerotic plaque (Sluimer et al., 2009) and in animal models of acute lung injury, such as those caused by α-naphthylthiourea (O’Brien et al., 1985), high pressure mechanical ventilation (Dreyfuss et al., 1992), and activation of transient receptor potential vanilloid4 (TRP4) channels (Alvarez et al., 2006). In addition, circulating endothelial cells (CEC) have been reported to be increased in a variety of conditions in which vascular injury might be expected, such as sickle cell anemia with crisis (Sowemimo-Coker et al., 1989), acute coronary syndrome (Boos et al., 2007; Lampka et al., 2010; Quilici et al., 2004), congestive heart failure (Chong et al., 2004), both primary and secondary pulmonary hypertension (Bull et al., 2003), and systemic sclerosis (Del Papa et al., 2004). It is possible that detached CEC are a marker for vascular endothelial cell injury and/or apoptosis (Al-Massarani et al., 2008). Indeed, CEC in blood from patients with obstructive sleep apnea display markers of apoptosis (El Solh et al., 2008). Thus, the demonstration of endothelial detachment from basement membrane (“blebs”) and the presence of CEC indicate that endothelial anoikis does occur during vascular injury.
This review describes the critical role of FAK in the formation and maintenance of focal adhesion complexes and in prevention of endothelial anoikis. We first review molecular mechanisms of apoptosis and anoikis, then discuss how FAK is involved in apoptosis or cell survival. We review our studies of adenosine/homocysteine-induced endothelial apoptosis and how abnormal FAK signaling may contribute to cardiovascular diseases.
1. Molecular mechanisms of apoptosis and anoikis
Apoptosis is an energy-dependent, genetically determined, active form of programmed cell death that eliminates non-functional or injured cells. During apoptosis, cells undergo a series of well-ordered morphologic and molecular alterations, including cell surface exposure of phosphatidylserine, cytoskeletal rearrangement, cell shrinkage, plasma membrane blebbing, nuclear membrane collapse, chromatin condensation, DNA fragmentation, and ultimately formation of apoptotic bodies, which are in turn engulfed and cleared by phagocytes to limit inflammation (Kerr et al., 1972). Apoptosis is a normal physiological response that is important in maintaining tissue homeostasis and remodeling. However, uncontrolled apoptosis plays a fundamental role in pathogenesis of many diseases, such as neurodegenerative diseases, ischemia–reperfusion injury, and heart failure. Apoptosis may also contribute to progression of a number of lung diseases, such as acute lung injury, pulmonary fibrosis, emphysema, and pulmonary artery hypertension, which have been reviewed elsewhere (Lu and Rounds, 2009). Apoptosis is triggered by external and internal stimuli and is mediated through death receptor-dependent extrinsic and mitochondria-dependent intrinsic pathways (Figure 1), reviewed in detail elsewhere (Harrington et al., 2007) and summarized below. In addition, the unfolded protein response and autophagy may result in apoptosis primarily via mitochondria-mediated activation of intrinsic pathway (Groenendyk and Michalak, 2005; Klionsky et al., 2008; Lin et al., 2007; Rutkowski et al., 2006; Szegezdi et al., 2003).
Figure 1.

Schematic representation of molecular mechanisms of apoptosis.
The extrinsic apoptotic pathway is triggered by the ligation of cell surface death receptors, including Fas (CD95), tumor necrosis factor (TNF) receptor (TNFR) 1 and 2, death receptor (DR) 3, 4, and 5, and toll-like receptor (TLR)-4, with their respective death ligands (external stimuli), including Fas ligand (FasL), TNFα, AproL, TNF-related apoptosis-inducing ligand (TRAIL), and lipopolysaccharide (LPS). This results in activation of initiator caspases, caspase-8/-10, which subsequently activate effector caspases3/-6/-7 to execute apoptosis (Figure 1). The external pathway of apoptosis is inhibited by Fas-associated death domain (FADD)-like inhibitory proteins (FLIPs), which inhibit caspase-8 activation (Thome et al., 1997). The death ligands are often released by inflammatory cells; thus, this pathway to apoptosis may be important in the pathogenesis of acute lung injury (Lu and Rounds, 2009). cFLIP-dependent activation of extrinsic apoptotic pathway has also been reported to mediate endothelial anoikis (Aoudjit and Vuori, 2001; Suzuki et al., 2003).
The intrinsic apoptotic pathway is triggered by cellular stressors (such as oxidants, DNA damage, cytokine deprivation, cytotoxic attack, Ca2+ imbalance, and chemotherapeutic agents) (Figure 1). These cause mitochondrial outer membrane permeabilization (MOMP), resulting in release from mitochondria of a number of apoptosis-inducing proteins, including cytochrome c, second mitochondrial-derived activator of caspase (Smac)/direct IAP binding protein with low pI (Diablo), Omi/HtrA2, apoptosis-inducing factor (AIF), and endonuclease G. The released cytochrome c rapidly binds to apoptotic protease activating factor 1 (Apaf-1), leading to caspase-9 activation and subsequent caspase-3/-6/-7 activation, and ultimately apoptosis (Slee et al., 1999; Zou et al., 1999). Cytosolic Smac and Omi activate effector caspases by cleavage of inhibitor of apoptosis proteins (IAPs) (Du et al., 2000; Yang et al., 2003). The released AIF and endonuclease G translocate to the nucleus and initiate chromatin condensation and DNA fragmentation, respectively (Li et al., 2001; Susin et al., 1999). AIF- and endonuclease G -mediated apoptosis is independent of caspase activity. Of note, death receptor-activated caspase-8 can truncate Bid to form tBid, which then translocates to mitochondria and activates the intrinsic pathway (Li et al., 1998).
Bcl-2 family proteins are essential for initiation and regulation of MOMP. In unstimulated cells, pro-apoptotic Bax proteins of Bcl-2 family, including Bax, Bak, and Bok, are sequestered in cytoplasm. Upon stimulation of apoptotic signals, Bax proteins translocate to the mitochondrial outer membrane to form pores, leading to MOMP (Adams and Cory, 1998; Hung and Chow, 2004). The mechanisms underlying mitochondrial membrane translocation of Bax proteins are not well defined. One possibility is that pro-apoptotic, BH3-only family of Bcl-2 proteins, including Bik, Bim, Bmf, Bid, Noxa, and Puma, stimulate homo-oligomerization and translocation of Bax proteins upon receipt of apoptotic signals. Another possible mechanism is due to increased availability of Bax proteins and BH3-only pro-apoptotic Bcl-2 proteins. The anti-apoptotic Bcl-2 family proteins, including Bcl-2, Bcl-xL, Mcl-1, Bfl-1, Bcl-w, and Boo, inhibit MOMP via maintenance of mitochondrial membrane integrity or by direct binding to BH3-only pro-apoptotic Bcl-2 proteins. Overall, MOMP is controlled by the balance of anti-apoptotic and pro-apoptotic Bcl-2 family members (Adams and Cory, 1998; Hung and Chow, 2004). NFκB is an important anti-apoptotic transcription factor. NFκB is activated by TNF-α, AproL, and LPS. Activated NFκB promotes expression of various anti-apoptotic proteins, such as Bcl-2, Bcl-xL, cFLIP, cIAP-1, cIAP-2, and TRAF-1/2, thus attenuating apoptosis (Tamatani et al., 1999; Wang et al., 1998). Though cFLIP-regulated extrinsic pathway may be involved in some conditions (Aoudjit and Vuori, 2001), Anoikis primarily occurs via intrinsic pathway mediated by Bim, Bmf, Bax, Bid, Bcl2, and Bcl-xL (Frisch and Francis, 1994).
2. FAK and apoptosis/aniokis
Focal adhesion kinase (FAK) is a non-receptor protein tyrosine kinase. FAK deficiency is embryonic lethal due to impaired vascular development, indicating that FAK is essential for normal embryo development (Ilic et al., 1995; Ilic et al., 2003). FAK is over-expressed in a variety of invasive human tumors and increased FAK expression is thought to contribute to malignancies (Golubovskaya et al., 2009). Interfering with FAK function is a strategy for development of anti-cancer drugs (Cohen and Guan, 2005; Golubovskaya et al., 2009; McLean et al., 2005). In addition to the role of FAK in tumor cell invasion and proliferation, FAK also provides survival signaling for anchorage-dependent cells. Endothelial cells isolated from FAK-null embryos do not proliferate in culture, but undergo apoptosis (Ilic et al., 1995; Ilic et al., 2003). Endothelium-specific deletion of FAK (Cre/FAKflox) causes vascular defects and is embryonic lethal, associated with increased endothelial cell apoptosis (Braren et al., 2006; Shen et al., 2005). In addition, inhibition of FAK by injection of FAK antibody or a dominant negative integrin peptide into unattached fibroblasts inhibits assembly of F-actin and focal adhesion compleses (FAC) and promotes apoptosis of anchorage-dependent cells (Hungerford et al., 1996). Consistently, over-expression of constitutively active FAK (CD2-FAK) in epithelial cells generates anoikis resistance and transforms epithelial cells into anchorage-independent growth, resulting in tumor formation in nude mice (Frisch et al., 1996). Activation of FAK also protects human ovarian cancer cells from anoikis and promotes cancer progression (Sood et al., 2010). The mechanisms of FAK promotion of cell survival and anoikis resistance are discussed below.
2.1 FERM domain autoinhibits FAK activation
FAK is ubiquitously expressed and highly conserved across species. As diagrammed in Figure 2, FAK contains an N-terminal FERM (band 4.1, ezrin, radixin, moesin homology) domain, a central tyrosine kinase domain (KD), a C-terminal focal adhesion targeting (FAT) domain, a linker region between FERM and KD, and an unstructured proline-rich (PR) region between KD and FAT domains. FERM-truncated FAK mutant displays increased tyrosine phosphorylation and activation but maintains adhesion-mediated signaling, suggesting that the FERM domain auto-inhibits FAK tyrosine kinase activity, but is not required for focal adhesion complex targeting (Jacamo and Rozengurt, 2005). Based on the crystal structure of FAK (Lietha et al., 2007), without engagement of integrins or growth factor receptors, the autophosphorylation site at tyrosine-397 (Y397) in the linker region and the kinase activation sites in the KD of FAK are sequestered by direct FERM/KD interaction. FERM blocks access of Src to the activation domain of FAK. Upon clustering of integrins or growth factor receptors, FERM is displaced thus allowing Y397 autophosphorylation and exposure of kinase activation domain. Subsequently, a nearby PxxP motif of FAK recruits Src via SH2 and SH3 domains of Src. Src is then phosphorylated on Y416 and thus activated. The activated Src contributes to maximal FAK activation by phosphorylation of FAK at Y576 and Y577 in the kinase activation loop and at Y861 and Y925 in the PR region of FAK. The precise mechanism by which integrins and growth factor receptors initiate FERM displacement is not clear. Whether FERM directly interacts with the cytoplasmic domains of activating receptors or additional proteins involved requires further investigation.
Figure 2. Diagram of FAK molecular structure and activation of cell survival signaling.

FERM = Band 4.1, Ezrin Radixin Moesin, KD = kinase domain, PR = proline rich, FAT = focal adhesion targeting, NTS = nuclear target sequence, NES = nuclear export sequence.
2.2 FERM mediates FAK nuclear translocation and promotes cell survival via inhibition of p53
Early studies have revealed nuclear localization of full-length FAK, N-terminal and C-terminal fragments of FAK in a number of cell types, including endothelial cells (Lim et al., 2008b). Recent work suggests that the FERM domain of FAK contains nuclear export signal sequences (NES) (Ossovskaya et al., 2008) and nuclear target sequence (NTS) (Lim et al., 2008b), which may explain nuclear targeting of full-length and N-terminal fragments of FAK. Additional mechanisms have not been defined regarding nuclear import of C-terminal fragments of FAK. FAK nuclear translocation, induced by staurosporine or disruption of cell adhesion, is independent of tyrosine kinase activation (Lim et al., 2008b). Increased nuclear FAK has been noted in various cancers (Murata et al., 2008). Ilic and colleagues have shown that FAK transduces extracellular matrix survival signals to suppress p53-mediated apoptosis (Ilic et al., 1998). They reported that FAK null embryo has increased expression of p53, Mdm2, and p53 target gene, p21(Cip/WAF1). They later on demonstrated that nuclear translocated FAK directly binds to p53 and inhibits p53 transcriptional potential (Lim et al., 2008a). Nuclear FAK also functions as a scaffolding protein for p53-Mdm2 complex and mediates p53 ubiquitination/degradation (Lim et al., 2008a). Consistently, knockdown of FAK by siRNA increases p53 levels in A549 lung and HCT116 colon carcinoma cells, while FERM over-expression reduces p53 levels in HCT116 colon carcinoma cells (Lim et al., 2008b). FERM over-expression also reduces p53-mediated apoptosis (Golubovskaya et al., 2005) and enhances breast cancer survival (Kurenova et al., 2004). These findings suggest that FERM-mediated FAK nuclear translocation promotes cell survival via inhibition of p53 transcriptional activation and enhancement of p53 degradation (Figure 2).
Protein tyrosine kinase 2 (Pyk2) is a cytoplasmic tyrosine kinase related to FAK. Compensatory Pyk2 expression occurs in FAK null mice (Lim et al., 2010). Interestingly, Pyk2 is also translocated into nuclear via FERM and induces p53 ubiquitination and degradation. Thus, Pyk2 inhibition of p53 may serve as an adaptive and intrinsic mechanism facilitating cell proliferation and survival upon inhibition of FAK (Lim et al., 2010).
TAE-226 (Novartis) and PF-562271 (Pfizer), small molecular inhibitors of FAK and Pyk2, have been shown to inhibit tumor growth and invasion and to increase apoptosis in several tumor models (Halder et al., 2007; Roberts et al., 2008). Whether FAK nuclear translocation-dependent p53 inactivation is involved in inhibition of tumor survival by these drugs is not known.
2.3 FAK promotes cell survival via PI3K/Akt pathway
Cell adhesion and integrin engagement causes FAK phosphorylation at Y397, which leads to recruitment of SH2 domain proteins (Schaller et al., 1994), including 85kD subunit of phosphatidylinositol-3-kinase (PI3K), phospholipase C (PLC-γ), and adapter Grb7, to the linker region (Figure 2). The activated PI3K generates PIP3, which stimulates the recruitment of PKB/Akt to membrane through its PH domain, resulting in phosphorylation of PKB/Akt at Thr-308 and Ser-473 (Khwaja et al., 1997). The phosphorylated (active) PKB/Akt promotes survival via phosphorylation and thus inhibition of pro-apoptotic proteins, Bad and pro-caspase-9 (Kennedy et al., 1997). FAK may also promote apoptosis resistance via PI3K/Akt-mediated expression of survivin, a member of inhibitor of apoptotic proteins (IAP) (Hauser and Probst, 1991). We have shown that FAK protects against adenosine/homocysteine-induced endothelial apoptosis via activation of PI3K/Akt signaling (Bellas et al., 2002), described in detail below. FAK also protects against apoptosis via upregulation of the anti-apoptotic NF-κB signaling (Huang et al., 2007).
In addition to effects on endothelial cell permeability and migration, vascular endothelial growth factor (VEGF) promotes endothelial cell survival. Gerber et al (Gerber et al., 1998) demonstrated that this effect of VEGF is mediated via the Flt/KDR receptor and PI3-kinase/Akt signaling. It was subsequently demonstrated that VEGF-induced activation of PI3-kinase (Abedi and Zachary, 1997) is dependent on FAK (Qi and Claesson-Welsh, 2001).
On the other hand, the tumor suppressor PTEN (phosphatase and tensin homologue deleted on chromosome 10) dephosphorylates FAK Y397, thereby inhibiting downstream PI3-kinase/Akt cell survival signaling (Tamura et al., 1999). Thus, PTEN is a negative regulator of FAK in matrix-adhesion dependent cell survival.
2.4 FAK promotes cell survival via Ras GTPase signaling
Integrin and RhoGTPase signaling are intertwined and reciprocal, as reviewed by Schwartz (Schwartz and Shattil, 2000). FAK and recruited Src also affect activation of the Rho family of small GTPases. FAK phosphorylation at Y925 induces recruitment of adaptor Grb2-SOS complex, leading to activation of Ras GTPase and subsequent activation of the Raf1/MEK1/ERK1/2 pathway (Renshaw et al., 1999),, which is important for cell survival and proliferation (Figure 2), reviewed in Mitra and Schlaepfer (Mitra and Schlaepfer, 2006).
2.5 FAK proteolysis and FAC disruption causes aniokis
FAK is targeted to the focal adhesion complexes (FAC) via direct binding to paxillin (Liu et al., 2002) and p130Cas (Polte and Hanks, 1997). Activated Src also binds to the Src-binding domain (SBD) of p130Cas. Multiple interactions between FAK, Src, p130Cas, and paxillin regulate dynamic FAC assembly and disassembly, as reviewed by Playford and Schaller (Playford and Schaller, 2004). Disruption of these interactions causes cell detachment and anoikis, although differing effects may be seen in different cell types (Zouq et al., 2009).
FAK is also cleaved through two caspase cleavage sites within the C-terminal domain. DQTD772 is preferentially cleaved by caspase-3 and to a lesser extent cleaved by caspase-6, whereas VSWD704 is specifically cleaved by caspase-6 (Gervais et al., 1998). FAK is thus cleaved to FAK related non-kinase (FRNK)-like fragments (p35, p40) by caspase-3 and -6 during apoptosis. Similar to FRNK, the FRNK-like fragments inhibit phosphorylation and focal adhesion-localization of endogenous FAK thus promoting apoptosis. Caspase-mediated proteolysis of FAK has been implicated in serum deprivation-induced apoptosis in C-Myc transformed chicken embryo fibroblasts (Crouch et al., 1996). We have shown that caspase-dependent FAK degradation also contributes to endothelial anoikis (Bellas et al., 2002).
FRNK is a natural variant of FAK. It contains a FAT domain and an additional 150 amino acids that are NH2 terminus to FAT. FRNK preserves the FAT domain, binding to integrins or growth factor receptors, but lacks tyrosine kinase activation domain. Thus, FRNK acts as a natural inhibitor for FAK (Ilic et al., 1998). We have shown that over-expression of FRNK inhibits FAK localization to focal adhesion complexes and promotes endothelial apoptosis (Bellas et al., 2002).
3. FAK and small GTPase regulation of adenosine- and adenosine/homocysteine-induced endothelial cell apoptosis
More than a decade ago, we demonstrated that extracellular ATP and adenosine cause cultured pulmonary artery endothelial cell apoptosis via adenosine transporter-mediated intracellular events, rather than through cell surface receptor(s) (Dawicki et al., 1997). We also showed that homocysteine exacerbates adenosine-induced endothelial apoptosis (Rounds et al., 1998). We further noted that adenosine significantly increases protein tyrosine phosphatase (PTPase) activity in pulmonary vascular endothelial cells, and that PTPase inhibition attenuates apoptosis induced by adenosine, adenosine plus homocysteine (Ado/HC), and the SAH hydrolase inhibitor, MDL-28842. In addition, tyrosine kinase inhibitor, genistein, enhances adenosine-induced apoptosis. These findings suggest that adenosine or Ado/HC causes endothelial apoptosis through decreased protein tyrosine phosphorylation (Harrington et al., 2000).
Our subsequent studies demonstrated that Ado/HC-induced endothelial apoptosis is associated with disruption of focal adhesion complexes (FAC) (Figure 3), increased caspase activation, and proteolysis of FAC components, including FAK, paxillin, and p130Cas (Harrington et al., 2001). Either broad caspase inhibitor (z-VAD-FMK) or PTPase inhibitor (sodium orthovanadate) prevents Ado/HC-induced degradation of FAC components and apoptosis. These results suggest that caspase- and PTPase-dependent proteolysis of FAC components is necessary for Ado/HC-induced endothelial apoptosis (Harrington et al., 2001). We also noted that neither caspase inhibitor nor PTPase inhibitor protects against Ado/HC-induced FAC disruption, suggesting that FAC disruption, independent of PTPase or caspase activation, is an early, but not an irrevocable, event in endothelial apoptosis (Harrington et al., 2001).
Figure 3. Ado/HC disrupts focal adhesion complexes in endothelial cells.

Pulmonary artery endothelial cells (PAECs) were incubated in HEPES buffer in the absence (vehicle) or presence of 100 μM adenosine plus 100 μM homocysteine (Ado/HC) for 4 h. The cells were immunofluorescently stained for focal adhesion complex-associated protein, focal adhesion kinase (FAK) and visualized with laser scanning confocal microscopy. Arrows indicate FAK staining. Images are representative of 4 independent experiments. Reproduced by permission of American Physiological Society and originally published in Harrington, E. O., et al., 2001, AJP:Lung Cell Mol Physiol, 280: L342-53.
We further investigated the role of FAK in mediating the effect of adenosine- and Ado/HC-induced endothelial apoptosis and found that adenosine- and Ado/HC-induced endothelial apoptosis is attenuated by over-expression of wild type FAK, but not of paxillin or p130Cas, suggesting that FAK proteolysis plays a critical role in endothelial apoptosis (Bellas et al., 2002). FAT and FRNK fragments of FAK can bind to integrins, but lack the central tyrosine kinase activity of FAK, and thus act as dominant negative FAK (Ilic et al., 1998). We found that over-expression of either FAT or FRNK does not protect, but exacerbates Ado/HC-induced endothelial apoptosis, suggesting that central tyrosine kinase activity is necessary for FAK protection against apoptosis (Bellas et al., 2002). In support of this notion, we documented that adenosine- and Ado/HC-induced endothelial apoptosis was abrogated by over-expression of constitutively active FAK (CD2-FAK) (Figure 4b), but not by the central tyrosine kinase inactive mutant (CD2-FAKK454R) (Bellas et al., 2002). We also demonstrated that neither FAKY397F, which lacks the binding capacity to either PI3K or Src, nor FAKD395A, which is unable to bind to PI3K but maintains Src binding, blunted Ado/HC-induced apoptosis (Bellas et al., 2002) (Figure 4a). In addition, the protective effect of CD2-FAK on adenosine- and Ado/HC-induced endothelial apoptosis was diminished by PI3K inhibitor, LY-294002 (Figure 4b). These results suggest that FAK tyrosine phosphorylation protects against adenosine- and Ado/HC-induced endothelial apoptosis via PI3K signaling (Bellas et al., 2002).
Figure 4. FAK protects against Ado/HC-induced endothelial apoptosis via activation of PI3K signaling.
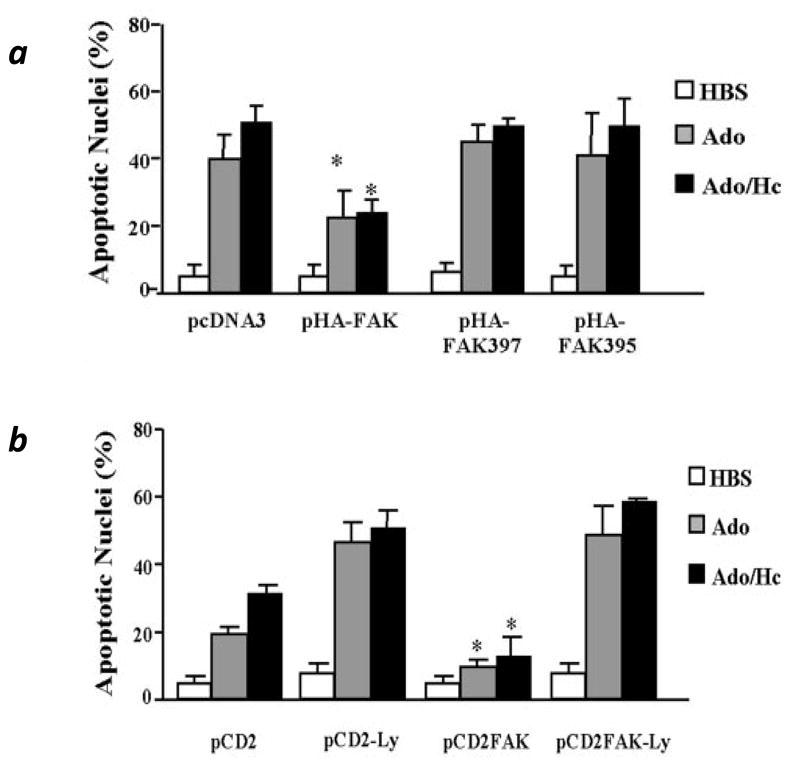
Panel a: Bovine pulmonary artery endothelial cells (PAECs) were transfected with wild type FAK (pHA-FAK) or central kinase inactive FAK mutants (pHA-FAKY397F and pHA-FAKD395A). pcDNA3 was used as a control for transfection. At 24h after transfection, cells were incubated in buffer alone (HBS, vehicle) or with 1 mM adenosine (Ado) or 100 μM adenosine plus 100 μM homocysteine (Ado/HC) for additional 16 h and then apoptosis was assessed by Hoechst staining. Panel b: PAECs were transfected in duplicate with control vector (pCD2) or constitutively active FAK (CD2-FAK) constructs. After 24 h, cells were incubated in buffer alone (HBS, vehicle) or with 0.5 mM Ado or 50 μM Ado plus 50 μM HC (Ado/HC). In addition, cells were incubated in the presence or absence of 25 μM LY-294002 (Ly). After an additional 16 h, apoptosis was assessed by Hoechst staining. Apoptosis was presented as the ratio of apoptotic HA− or CD2 cells to total HA or CD2 expressing cells. p<0.05 for comparison between respective treatments. n=3 (panel a), n=2 (panel b). Reproduced by permission of American Physiological Society and originally published in Bellas et al. 2002. AJP:Lung Cell Mol Physiol. 282: L1135-42.
Endothelial apoptosis caused by extracellular ATP, adenosine, and Ado/HC is associated with a decreased ratio of S-adenosyl-L-methionine (SAM) to S-adenosyl-L-homocysteine (SAH) (Rounds et al., 1998), favoring inhibition of carboxyl methylation. We have demonstrated that Ado/HC inhibits isoprenyl carboxyl methyltransferase (ICMT) activity and decreases carboxyl methylation and activity of ICMT substrates, Ras and RhoA (Kramer et al., 2003; Lu et al., 2004). Moreover, Ado/HC-induced endothelial apoptosis was attenuated by over-expression of either ICMT or wild-type or dominant active H-Ras (Kramer et al., 2003). These findings indicate that inhibition of ICMT by Ado/HC causes endothelial cell apoptosis at least in part by attenuation of Ras GTPase carboxyl methylation and activation (Kramer et al., 2003). Since Ado/HC causes FAK proteolysis via caspase activation and FAK activates Ras GTPase via adaptor Grb2-SOS complex, it is possible that Ado/HC-induced, caspase-dependent FAK proteolysis further decreases Ras activity and exacerbates apoptosis (Figure 5).
Figure 5. Schematic representation of the signal mechanisms of Ado/HC-induced endothelial apoptosis.

Solid lines indicate established pathways and dashed lines indicate suggested pathways.
FAK has been implicated in regulation of Rho GTPase activity. FAK can activate several GTPase-activating proteins (GAPs), such as GRAF (a GAP for Rho), PSGAP1 (a GAP for Arf1), and p190RhoGAP/p120RasGAP (GAPs for RhoA), leading to inactivation of Rho and Arf1 (Schaller, 2010). FAK may also activate RhoA through actions of guanine nucleotide exchange factors (GEFs), PDZRhoGEF and p190RhoGEF (Lim et al., 2008c). Indeed, FAK spatially and temporally alters activity of RhoA thus controlling cytoskeletal changes and cell motility during cell migration (Schaller, 2010). FAK regulation of RhoA activity in the process of anoikis is not known. Whether decreased RhoA carboxyl methylation and activity contribute to Ado/HC-induced endothelial apoptosis remains to be determined. Sustained RhoA activation causes cardiomyocyte apoptosis (Del Re et al., 2007). However, inhibition of RhoA also causes endothelial cell apoptosis (Hippenstiel et al., 2002; Li et al., 2002), suggesting that a basal level of RhoA is important for endothelial survival. Indeed, certain levels of active RhoA attenuate cardiomyocyte apoptosis induced by hydrogen peroxide or glucose deprivation via RhoA kinase–dependent FAK activation and subsequent activation of the PI3K/Akt pathway (Del Re et al., 2008). We demonstrate that FAK activation protects against Ado/HC-induced endothelial apoptosis via activation of PI3K/Akt signaling pathway (Bellas et al., 2002). Thus, we speculate that ICMT-dependent inhibition of RhoA carboxyl methylation and activation may contribute to Ado/HC-induced endothelial apoptosis via suppression of FAK/PI3K/Akt survival signaling (Figure 5).
4. FAK and cardiovascular diseases
FAK integrates and converts signals from integrins and growth factor receptors and thus regulates cell adhesion, mobility, migration, angiogenesis, and proliferation, which are reviewed in other sections of this volume of Microvascular Research. Although epithelial and endothelial apoptosis have been implicated in the pathogenesis of emphysema, it is not known if FAK plays a role in alveolar cell apoptosis in vivo. The role of FAK is also not well understood in lung diseases characterized by abnormal apoptosis, such as emphysema and pulmonary hypertension.
FAK affects the cardiac hypertrophy response, although discrepant results exist between two different groups using cardiomyocyte-specific FAK knockout mice. Peng and colleagues showed that majority of cardiomyocyte-specific FAK knockout mice are embryonic lethal due to cardiac developmental defects (Peng et al., 2008). Surviving mice display spontaneous right ventricular hypertrophy and enhanced cardiac hypertrophy response to Angiotensin II and aortic banding, suggesting that FAK may protect against cardiac hypertrophy (Peng et al., 2006). It is noteworthy that these cardiac defects observed in this FAK inactivation animal model were not associated with apoptosis (Peng et al., 2008). However, DiMichele et al showed that genetic depletion of FAK in cardiomyocytes attenuates pressure overload-induced cardiac hypertrophy, indicating that FAK may promote cardiac hypertrophy (DiMichele et al., 2006). Inhibition of FAK also exacerbates hypoxia/reoxygenation-induced cardiomyocyte cell death in vitro and enhances ischemia/reperfusion (I/R)-induced infarct size and cardiomyocyte apoptosis with reduced left ventricular function (Hakim et al., 2009). The mechanism of this effect was associated with decreased NF-κB survival signaling and reduced Bcl2 and Bcl-xl (Hakim et al., 2009). These results suggest that abnormal FAK signaling may contribute to cardiomyocyte dysfunction and heart failure.
Conclusions and future perspectives
FAK is important in maintenance of normal cell survival. Disruption of FAK signaling leads to loss of substrate adhesion and abnormal cell death. Overexpression of FAK is associated with tumorigenesis. FAK also appears to be important in cardiomyocyte hypertrophy and hypoxia/reoxygenation-induced cell death. However, little is known regarding the role of FAK in lung endothelial and epithelial cell survival and death in vivo. There is a need to better understand the role of FAK in the pathogenesis of lung vascular diseases characterized by abnormal cell death and proliferation, such as emphysema and pulmonary hypertension. Drug discovery is revealing small molecule FAK inhibitors effective in preventing abnormal cell proliferation. Thus, lung endothelial cell specific FAK transgenic or knockout mice and small molecule FAK inhibitors may be effective tools to study the role of FAK in the pathogenesis of lung vascular diseases.
Research Highlights.
Focal Adhesion Kinase (FAK) is a key component of Focal Adhesion Complexes, in anchorage dependent cells, such as endothelial cells.
Detachment of endothelial cells from underlying basement membrane results in apoptosis, or programmed cell death.
FERM-mediated FAK translocation promotes cell survival via inhibition of p53.
The tyrosine kinase activity of FAK is necessary for protection against apoptosis.
FAK promotes cell survival via the PI3K/Akt pathway.
Acknowledgments
This work was supported with resources and the use of facilities at the Providence VA Medical Center and supported by VA Merit Review (Rounds), HL64936 (Rounds), and American Thoracic Society/Pulmonary Hypertension Association research grant (Lu).
This work was supported by VA Merit Review (Rounds), HL64936 (Rounds), and American Thoracic Society/Pulmonary Hypertension Association research grant (Lu).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abedi H, Zachary I. Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J Biol Chem. 1997;272:15442–51. doi: 10.1074/jbc.272.24.15442. [DOI] [PubMed] [Google Scholar]
- 2.Adams J, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 3.Al-Massarani G, et al. Impact of immunosuppressive treatment on endothelial biomarkers after kidney transplantation. Am J Transplant. 2008;8:2360–7. doi: 10.1111/j.1600-6143.2008.02399.x. [DOI] [PubMed] [Google Scholar]
- 4.Albelda SM, et al. Identification and characterization of cell-substratum adhesion receptors on cultured human endothelial cells. J Clin Invest. 1989;83:1992–2002. doi: 10.1172/JCI114109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez DF, et al. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res. 2006;99:988–95. doi: 10.1161/01.RES.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aoudjit F, Vuori K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: a role for c-FLIP and implications for anoikis. J Cell Biol. 2001;152:633–643. doi: 10.1083/jcb.152.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellas RE, et al. FAK blunts adenosine-homocysteine-induced endothelial cell apoptosis: requirement for PI 3-kinase. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1135–42. doi: 10.1152/ajplung.00174.2001. [DOI] [PubMed] [Google Scholar]
- 8.Boos CJ, et al. Relationship between circulating endothelial cells and the predicted risk of cardiovascular events in acute coronary syndromes. Eur Heart J. 2007;28:1092–101. doi: 10.1093/eurheartj/ehm070. [DOI] [PubMed] [Google Scholar]
- 9.Braren R, et al. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006;172:151–62. doi: 10.1083/jcb.200506184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bull TM, et al. Circulating endothelial cells in pulmonary hypertension. Thromb Haemost. 2003;90:698–703. doi: 10.1160/TH03-04-0251. [DOI] [PubMed] [Google Scholar]
- 11.Cheng YF, Kramer RH. Human microvascular endothelial cells express integrin-related complexes that mediate adhesion to the extracellular matrix. J Cell Physiol. 1989;139:275–86. doi: 10.1002/jcp.1041390209. [DOI] [PubMed] [Google Scholar]
- 12.Chong AY, et al. Endothelial dysfunction and damage in congestive heart failure: relation of flow-mediated dilation to circulating endothelial cells, plasma indexes of endothelial damage, and brain natriuretic peptide. Circulation. 2004;110:1794–8. doi: 10.1161/01.CIR.0000143073.60937.50. [DOI] [PubMed] [Google Scholar]
- 13.Cohen LA, Guan JL. Mechanisms of focal adhesion kinase regulation. Curr Cancer Drug Targets. 2005;5:629–43. doi: 10.2174/156800905774932798. [DOI] [PubMed] [Google Scholar]
- 14.Crouch DH, et al. Targeted proteolysis of the focal adhesion kinase pp125 FAK during c-MYC-induced apoptosis is suppressed by integrin signalling. Oncogene. 1996;12:2689–96. [PubMed] [Google Scholar]
- 15.Dawicki DD, et al. Extracellular ATP and adenosine cause apoptosis of pulmonary artery endothelial cells. Am J Physiol. 1997;273:L485–94. doi: 10.1152/ajplung.1997.273.2.L485. [DOI] [PubMed] [Google Scholar]
- 16.Dejana E, et al. Fibronectin and vitronectin regulate the organization of their respective Arg-Gly-Asp adhesion receptors in cultured human endothelial cells. J Cell Biol. 1988;107:1215–23. doi: 10.1083/jcb.107.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Papa N, et al. Circulating endothelial cells as a marker of ongoing vascular disease in systemic sclerosis. Arthritis Rheum. 2004;50:1296–304. doi: 10.1002/art.20116. [DOI] [PubMed] [Google Scholar]
- 18.Del Re DP, et al. RhoA/Rho kinase up-regulate Bax to activate a mitochondrial death pathway and induce cardiomyocyte apoptosis. J Biol Chem. 2007;282:8069–78. doi: 10.1074/jbc.M604298200. [DOI] [PubMed] [Google Scholar]
- 19.Del Re DP, et al. Focal adhesion kinase as a RhoA-activable signaling scaffold mediating Akt activation and cardiomyocyte protection. J Biol Chem. 2008;283:35622–9. doi: 10.1074/jbc.M804036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DiMichele LA, et al. Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circ Res. 2006;99:636–45. doi: 10.1161/01.RES.0000240498.44752.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dreyfuss D, et al. Spontaneous resolution of pulmonary edema caused by short periods of cyclic overinflation. J Appl Physiol. 1992;72:2081–9. doi: 10.1152/jappl.1992.72.6.2081. [DOI] [PubMed] [Google Scholar]
- 22.Du C, et al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 23.El Solh AA, et al. Hemostatic implications of endothelial cell apoptosis in obstructive sleep apnea. Sleep Breath. 2008;12:331–7. doi: 10.1007/s11325-008-0182-x. [DOI] [PubMed] [Google Scholar]
- 24.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–26. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frisch SM, et al. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–9. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geiger B, et al. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 27.Gerber HP, et al. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–43. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 28.Gervais FG, et al. Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J Biol Chem. 1998;273:17102–8. doi: 10.1074/jbc.273.27.17102. [DOI] [PubMed] [Google Scholar]
- 29.Golubovskaya VM, et al. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–21. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 30.Golubovskaya VM, et al. Focal adhesion kinase and cancer. Histol Histopathol. 2009;24:503–10. doi: 10.14670/HH-24.503. [DOI] [PubMed] [Google Scholar]
- 31.Groenendyk J, Michalak M. Endoplasmic reticulum quality control and apoptosis. Acta Biochim Pol. 2005;52:381–395. [PubMed] [Google Scholar]
- 32.Guan JL, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. 1992;358:690–2. doi: 10.1038/358690a0. [DOI] [PubMed] [Google Scholar]
- 33.Guan JL, et al. Fibronectin/integrin interaction induces tyrosine phosphorylation of a 120-kDa protein. Cell Regul. 1991;2:951–64. doi: 10.1091/mbc.2.11.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hakim ZS, et al. FAK regulates cardiomyocyte survival following ischemia/reperfusion. J Mol Cell Cardiol. 2009;46:241–8. doi: 10.1016/j.yjmcc.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halder J, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67:10976–83. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- 36.Harrington EO, et al. Endothelial Cell Apoptosis. In: Aird WC, editor. Endothelial Biomedicine. Cambridge University Press; New York, NY: 2007. pp. 1081–1097. [Google Scholar]
- 37.Harrington EO, et al. Protein tyrosine phosphatase-dependent proteolysis of focal adhesion complexes in endothelial cell apoptosis. Am J Physiol Lung Cell Mol Physiol. 2001;280:L342–53. doi: 10.1152/ajplung.2001.280.2.L342. [DOI] [PubMed] [Google Scholar]
- 38.Harrington EO, et al. Adenosine induces endothelial apoptosis by activating protein tyrosine phosphatase: a possible role of p38alpha. Am J Physiol Lung Cell Mol Physiol. 2000;279:L733–42. doi: 10.1152/ajplung.2000.279.4.L733. [DOI] [PubMed] [Google Scholar]
- 39.Hauser R, Probst R. The influence of systematic primary-tone level variation L2-L1 on the acoustic distortion product emission 2f1-f2 in normal human ears. J Acoust Soc Am. 1991;89:280–6. doi: 10.1121/1.400511. [DOI] [PubMed] [Google Scholar]
- 40.Hippenstiel S, et al. Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;283:L830–8. doi: 10.1152/ajplung.00467.2001. [DOI] [PubMed] [Google Scholar]
- 41.Huang D, et al. Focal adhesion kinase mediates cell survival via NF-kappaB and ERK signaling pathways. Am J Physiol Cell Physiol. 2007;292:C1339–52. doi: 10.1152/ajpcell.00144.2006. [DOI] [PubMed] [Google Scholar]
- 42.Hung R, Chow A. Dissecting the “end game”: clinical relevance, molecular mechanisms and laboratory assessment of apoptosis. Clin Invest Med. 2004;27:324–344. [PubMed] [Google Scholar]
- 43.Hungerford JE, et al. Inhibition of pp125FAK in cultured fibroblasts results in apoptosis. J Cell Biol. 1996;135:1383–90. doi: 10.1083/jcb.135.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 45.Ilic D, et al. Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. J Cell Biol. 1998;143:547–60. doi: 10.1083/jcb.143.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ilic D, et al. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–44. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 47.Ilic D, et al. Focal adhesion kinase is required for blood vessel morphogenesis. Circ Res. 2003;92:300–7. doi: 10.1161/01.res.0000055016.36679.23. [DOI] [PubMed] [Google Scholar]
- 48.Jacamo RO, Rozengurt E. A truncated FAK lacking the FERM domain displays high catalytic activity but retains responsiveness to adhesion-mediated signals. Biochem Biophys Res Commun. 2005;334:1299–304. doi: 10.1016/j.bbrc.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 49.Kennedy SG, et al. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–13. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 50.Kerr J, et al. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. British Journal of Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khwaja A, et al. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J. 1997;16:2783–93. doi: 10.1093/emboj/16.10.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kramer K, et al. Isoprenylcysteine carboxyl methyltransferase activity modulates endothelial cell apoptosis. Mol Biol Cell. 2003;14:848–57. doi: 10.1091/mbc.E02-07-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kurenova E, et al. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24:4361–71. doi: 10.1128/MCB.24.10.4361-4371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lampka M, et al. Circulating endothelial cells in coronary artery disease. Kardiol Pol. 2010;68:1100–5. [PubMed] [Google Scholar]
- 56.Li H, et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas-pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 57.Li L, et al. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 58.Li X, et al. Inhibition of protein geranylgeranylation and RhoA/RhoA kinase pathway induces apoptosis in human endothelial cells. J Biol Chem. 2002;277:15309–16. doi: 10.1074/jbc.M201253200. [DOI] [PubMed] [Google Scholar]
- 59.Lietha D, et al. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–87. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim ST, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008a;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lim ST, et al. FERM control of FAK function: implications for cancer therapy. Cell Cycle. 2008b;7:2306–14. doi: 10.4161/cc.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lim ST, et al. Pyk2 inhibition of p53 as an adaptive and intrinsic mechanism facilitating cell proliferation and survival. J Biol Chem. 2010;285:1743–53. doi: 10.1074/jbc.M109.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim Y, et al. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008c;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin JH, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu G, et al. Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol Cell Biol. 2002;22:2751–60. doi: 10.1128/MCB.22.8.2751-2760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lu Q, et al. Isoprenylcysteine carboxyl methyltransferase modulates endothelial monolayer permeability: involvement of RhoA carboxyl methylation. Circ Res. 2004;94:306–15. doi: 10.1161/01.RES.0000113923.85084.C1. [DOI] [PubMed] [Google Scholar]
- 67.Lu Q, Rounds S. Pulmonary endothelial cell death: implications for lung disease pathogenesis. In: Voelkel NF, Rounds S, editors. The Pulmonary Endothelium: Function in health and disease. John Wiley & Sons, Ltd; Chichester, UK: 2009. pp. 243–260. [Google Scholar]
- 68.McLean GW, et al. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–15. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 69.Mechtersheimer G, et al. In situ expression of beta 1, beta 3 and beta 4 integrin subunits in non-neoplastic endothelium and vascular tumours. Virchows Arch. 1994;425:375–84. doi: 10.1007/BF00189575. [DOI] [PubMed] [Google Scholar]
- 70.Meredith JE, Jr, et al. The extracellular matrix as a cell survival factor. Mol Biol Cell. 1993;4:953–61. doi: 10.1091/mbc.4.9.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 72.Murata T, et al. Localization of FAK is related with colorectal carcinogenesis. Int J Oncol. 2008;32:791–6. [PubMed] [Google Scholar]
- 73.O’Brien RF, et al. Studies on the mechanism of decreased angiotensin I conversion in rat lungs injured with alpha-naphthylthiourea. Exp Lung Res. 1985;8:243–59. doi: 10.3109/01902148509087807. [DOI] [PubMed] [Google Scholar]
- 74.Ossovskaya V, et al. FAK nuclear export signal sequences. FEBS Lett. 2008;582:2402–6. doi: 10.1016/j.febslet.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 76.Peng X, et al. Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–27. doi: 10.1172/JCI24497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Peng X, et al. Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proc Natl Acad Sci U S A. 2008;105:6638–43. doi: 10.1073/pnas.0802319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pierschbacher MD, Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309:30–3. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- 79.Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23:7928–46. doi: 10.1038/sj.onc.1208080. [DOI] [PubMed] [Google Scholar]
- 80.Polte TR, Hanks SK. Complexes of focal adhesion kinase (FAK) and Crk-associated substrate (p130(Cas)) are elevated in cytoskeleton-associated fractions following adhesion and Src transformation. Requirements for Src kinase activity and FAK proline-rich motifs. J Biol Chem. 1997;272:5501–9. doi: 10.1074/jbc.272.9.5501. [DOI] [PubMed] [Google Scholar]
- 81.Qi JH, Claesson-Welsh L. VEGF-induced activation of phosphoinositide 3-kinase is dependent on focal adhesion kinase. Exp Cell Res. 2001;263:173–82. doi: 10.1006/excr.2000.5102. [DOI] [PubMed] [Google Scholar]
- 82.Quilici J, et al. Circulating endothelial cell count as a diagnostic marker for non-ST-elevation acute coronary syndromes. Circulation. 2004;110:1586–91. doi: 10.1161/01.CIR.0000142295.85740.98. [DOI] [PubMed] [Google Scholar]
- 83.Renshaw MW, et al. Focal adhesion kinase mediates the integrin signaling requirement for growth factor activation of MAP kinase. J Cell Biol. 1999;147:611–8. doi: 10.1083/jcb.147.3.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roberts WG, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935–44. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 85.Rounds S, et al. Mechanism of extracellular ATP- and adenosine-induced apoptosis of cultured pulmonary artery endothelial cells. Am J Physiol. 1998;275:L379–88. doi: 10.1152/ajplung.1998.275.2.L379. [DOI] [PubMed] [Google Scholar]
- 86.Ruoslahti E, Reed JC. Anchorage dependence, integrins, and apoptosis. Cell. 1994;77:477–8. doi: 10.1016/0092-8674(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 87.Rutkowski DT, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123:1007–13. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 89.Schaller MD, et al. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–8. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schwartz MA, Shattil SJ. Signaling networks linking integrins and rho family GTPases. Trends Biochem Sci. 2000;25:388–91. doi: 10.1016/s0968-0004(00)01605-4. [DOI] [PubMed] [Google Scholar]
- 91.Shen TL, et al. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005;169:941–52. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Slee E, et al. Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspase-2, -3, -6, -7, and -10 in a caspase-9-dependent manner. J Cell Biol. 1999;144:281–292. doi: 10.1083/jcb.144.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sluimer JC, et al. Thin-walled microvessels in human coronary atherosclerotic plaques show incomplete endothelial junctions relevance of compromised structural integrity for intraplaque microvascular leakage. J Am Coll Cardiol. 2009;53:1517–27. doi: 10.1016/j.jacc.2008.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sood AK, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest. 2010;120:1515–23. doi: 10.1172/JCI40802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sowemimo-Coker SO, et al. Increased circulating endothelial cells in sickle cell crisis. Am J Hematol. 1989;31:263–5. doi: 10.1002/ajh.2830310409. [DOI] [PubMed] [Google Scholar]
- 96.Susin S, et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 97.Suzuki T, et al. Eicosapentaenoic acid protects endothelial cells against anoikis through restoration of cFLIP. Hypertension. 2003;42:342–8. doi: 10.1161/01.HYP.0000084602.06114.AD. [DOI] [PubMed] [Google Scholar]
- 98.Szegezdi E, et al. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–94. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- 99.Tamatani M, et al. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- 100.Tamura M, et al. PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem. 1999;274:20693–703. doi: 10.1074/jbc.274.29.20693. [DOI] [PubMed] [Google Scholar]
- 101.Thome M, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- 102.Wang C, et al. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 103.Yang Q, et al. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis protein (IAP) irreversibly inactivates IAPs and faciliatates caspase activity in apoptosis. Genes Dev. 2003;17:1487–1496. doi: 10.1101/gad.1097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114:3583–90. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 105.Zou H, et al. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 106.Zouq NK, et al. FAK engages multiple pathways to maintain survival of fibroblasts and epithelia: differential roles for paxillin and p130Cas. J Cell Sci. 2009;122:357–67. doi: 10.1242/jcs.030478. [DOI] [PMC free article] [PubMed] [Google Scholar]