

Abstract
Caspases are a powerful class of cysteine proteases. Introduction of activated caspases in healthy or cancerous cells results in induction of apoptotic cell death. In this study, we have designed and characterized a version of caspase-7 that can be inactivated under oxidizing extracellular conditions and then reactivated under reducing intracellular conditions. This version of caspase-7 is allosterically inactivated when two of the substrate-binding loops are locked together via an engineered disulfide. When this disulfide is reduced, the protein regains its full function. The inactive loop-locked version of caspase-7 can be readily observed by immunoblotting and mass spectrometry. The reduced and reactivated form of the enzyme observed crystallographically is the first caspase-7 structure in which the substrate-binding groove is properly ordered even in the absence of an active-site ligand. In the reactivated structure, the catalytic-dyad cysteine–histidine are positioned 3.5 Å apart in an orientation that is capable of supporting catalysis. This redox-controlled version of caspase-7 is particularly well suited for targeted cell death in concert with redox-triggered delivery vehicles.
Keywords: apoptosis, caspase activation, loop conformation, disulfide, engineered cysteine, induced cell death, allostery
Introduction
Caspases are a class of cysteine proteases that are involved in biological cascades ranging from apoptosis1,2 to inflammation,3 to keratinocyte differentiation,4 and to innate immunity.5 The apoptotic caspases are of particular interest since their activity controls the delicate balance between cellular survival and death. The downstream executioner caspases are typically activated by upstream initiator caspases.6 Executioner caspase activity is antagonized by the inhibitor of apoptosis protein (IAP) family.7 Likewise, the activity of the IAPs is regulated by proapoptotic Smac/Diablo and Bcl family members.8,9 Thus, appropriate regulation of caspase activity is complex and is of paramount importance for appropriate cell survival or cell death. The activity of the executioner caspases is tightly controlled because erroneous activation leads to unwanted cell death. In cancer cells, the balance between cellular survival and death has been disrupted. Part of the transformation of healthy cells to cancerous cells includes overexpression of antiapoptotic factors, which block apoptosis-inducing signals.10,11 Because they are the final step in a tightly controlled cascade, induction of apoptosis is most direct by delivery or production of activated caspases.
The molecular details of activation of various classes of caspases differ; however, the apoptotic executioner caspases, including caspase-3 and caspase-7, share similar mechanisms of activation. The executioner caspases are produced as inactive proenzymes (zymogens), which dimerize with a low-nanomolar Kd for dimerization.12 Executioner caspases must be cleaved to become active. Cleavage at the intersubunit linker is the activating event, but executioner caspases are also cleaved to remove the N-terminal prodomain. Cleavage of the intersubunit linker generates an active heterotetramer composed of two large and two small subunits. The two caspase active sites are each composed of four mobile loops. Three loops (L2, L3, and L4) from one-half of the caspase must interact with the L2′ loop from the opposite half of the caspase to form a competent substrate-binding groove. This observation explains the obligatory dimerization of the executioners.
The loops that form the caspase substrate-binding groove are extraordinarily mobile and controlling their conformation controls the caspase function. Among the caspases, the complete molecular descriptions of activation, regulation, and inhibition have been observed in caspase-7. In the procaspase-7 zymogen, the active-site loops are disordered and incompatible with substrate binding.13 On zymogen activation, the active-site loops are partially ordered, but the L2′ loop remains in an inactive, down conformation, as is shown in Figure 1. From this state, caspase-7 is competent to bind substrate or allosteric inhibitors, mutually exclusively. When substrate binds, L2′ moves to the up conformation, forming a foundation or buttress beneath the L2 loops.13–16 When caspase-7 binds allosteric inhibitors in a cavity at the dimer interface, the L2′ loops are held in the down conformation and the enzyme becomes inactive.17,18 Thus, controlling caspase-7 loop conformations is an effective way to modulate activity.
Figure 1.
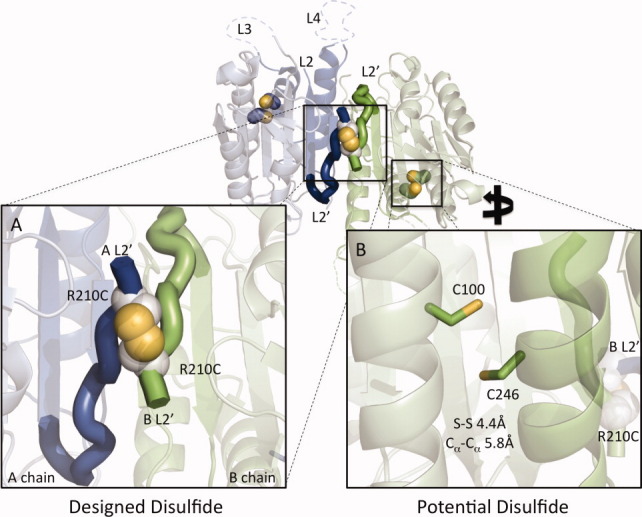
Design of a reductant activatable caspase-7. The allosterically inactivated caspase-7 dimer consists of two chains (A chain, green; B chain, blue). (A) An introduced disulfide between R210C residues on the two chains (white and yellow spheres) is designed to lock the protein into the inactive conformation by keeping the L2′ loops in the down conformation. Formation of this disulfide would result in a covalently crosslinked small-subunit homodimeric species. (B) Pairs of native cysteines (C100 and C246, blue and yellow sticks) are computationally predicted to form disulfides on each side of the dimer. These disulfides would drive formation of a large subunit–small-subunit crosslinked species.
Caspases are found only intracellularly and must function inside the cell to activate apoptosis. Caspases make use of a cysteine–histidine active-site dyad for proteolysis. The catalytic cysteine nucleophile must be maintained in a reduced state to function. Glutathione is the most prevalent cellular redox component. Intracellular glutathione concentrations in healthy cells range from 1 to 5 mM, with cancerous cells showing concentrations up to sevenfold higher,19 whereas extracellular concentrations are 1–4 μM.20 Thus, differences in redox environment provide an intriguing means of differentially controlling intracellular and extracellular activities.
Caspases are exquisitely useful cellular players. Once activated, they can single-handedly induce the death of a cell. There is a great deal of interest in small-molecule activation of caspases; however, this has been challenging to achieve.21–24 Another option for selective killing of cells therapeutically is the use of preactivated caspases. Injecting activated caspases, or any activated protease, into circulation is likely to produce many unwanted and deleterious effects. To avoid these complications while still taking advantage of the cell-killing properties of activated caspases, we aim to engineer a version of the executioner caspase-7 that can be held in an inactive conformation in the oxidizing, extracellular conditions, but regains its full activity in the reducing, intracellular conditions. Targeted delivery of this type of engineered caspase could potentially provide a safe and effective mechanism of selectively killing unwanted cell types.
Results
Rational design of a redox-activatable caspase-7
The aim of this study is to control the conformation of the L2′ loops to allosterically regulate caspase-7 function. In the crystal structure of caspase-7 bound to allosteric inhibitors FICA (5-fluoro-1H-indole-2-carboxylic acid (2-mercapto-ethyl)-amide) or DICA (2-(2,4-dichlorophenoxy)-N-(2-mercapto-ethyl)-acetamide), caspase-7 attains a conformation that is distinct from the mature or substrate-bound forms. We have previously shown that caspase-7 likewise exists in two distinct conformations in solution25: a bound active state and an unbound inactive state. In the inactive state with no substrate present, the L2′ loops rest over the allosteric cavity and do not contribute to the formation of the active-site loop bundle. The L2′ loops from the opposite sides of the dimer form an antiparallel structure with a nearly 90° crossing angle. The residues with the closest approach are the two R210 residues, with a Cα–Cα distance of 5.5 Å [Fig. 1(A)]. We modeled the pairwise mutations to cysteine for all residues in the L2′ loops and then used the MODIP26 server to score the probability of forming various disulfides. MODIP scored an R210C–R210C disulfide in the C category on a scale of A (most favorable) to F (least favorable). This intermediate score may be related to the fact that all Cα positions are fixed during MODIP modeling. The L2′ loop is necessarily flexible, as it undergoes significant rearrangement during the caspase lifecycle; thus, a score of C may still indicate a favorable interaction. In contrast, a second potential native disulfide, which we subsequently observed crystallographically (unpublished data, Supporting Information Fig. S1), was scored as an A. Our first design attempt was the substitution of R210C. Purification of the single R210C variant was not successful due to low protein yields. We speculated that the formation of mixed disulfides may have contributed to low protein yields.
We found that the removal of one native cysteine, which improved protein yields and eased analysis, was essential. Caspase-7 is unique among caspases in that it contains a potential disulfide interaction between residues 100 (large subunit) and 246 [small subunit; Fig. 1(B)]. We engineered out the native C100–C246 disulfide by making the C246S substitution. C246S had similar catalytic parameters to wild type (WT; Table I). We combined this with the disulfide locking R210C substitution. The doubly substituted variant (R210C/C246S) had good catalytic activity and exhibited KM and kcat very similar to WT (Table I).
Table I.
Kinetic Parameters for Caspase-7 Variantsa
| 5 mM BME | 10 mM DTT | ||||
|---|---|---|---|---|---|
| WT | C246S | R210C/C246S | WT | R210C/C246S | |
| kcat (s−1) | 0.36 ± 0.01 | 0.33 ± 0.01 | 0.40 ± 0.01 | 2.42 ± 0.07 | 2.46 ± 0.06 |
| KM (μM) | 24.0 ± 2.0 | 21.5 ± 2.8 | 22.7 ± 2.9 | 41.9 ± 4.2 | 39.4 ± 3.4 |
Kinetic parameters are based on substrate titrations measured from independent triplicate dilutions of substrate on two days.
Observation of the designed disulfide dimer
The variant R210C/C246S is designed such that mildly oxidizing conditions should drive formation of a disulfide, locking the two L2′ loops together across the dimer interface and allosterically inhibiting caspase-7. Because the L2′ loops are the N-termini of the small subunits, formation of the inactivating R210C–R210C disulfide will generate covalent small-subunit dimers. To measure the formation of small-subunit dimers, we exposed WT or R210C/C246S caspase-7 to native (reducing conditions) or mildly oxidizing conditions (300 μM cystamine). The bands on a denaturing acrylamide gel were unambiguously identified by a combination of mass spectrometry and immunoblotting with antibodies specific for the large or small subunits of caspase-7 (Figs. 2 and 3). Caspase-7 has two major auto-activating cleavage sites in the intersubunit linker at residues 198 and 206 (Supporting Information Fig. S2). In WT caspase-7, the intersubunit linker (residues 199–206) is removed, so the prodomain-deleted large subunit is composed of residues 24–198 and the small subunit consists of residues 207–311. For our designs, we used a construct that allows constitutive expression of the two-chain (active) form of caspase-7. R210C/C246S caspase-7 was produced as two polypeptides of residues 1–198 and 199–311. In addition to removal of the prodomain, the large subunit was also processed at a minor cleavage site (residue 192, which has been extensively probed by Denault and coworkers27) generating some large subunits composed of residues 24–192. The small subunit of R210C/C246S appears in two forms (Fig. 2, Supporting Information Fig. S2, and Fig. 3), both with the intersubunit linker (residues 199–311) and without the intersubunit linker (residues 207–311). Under oxidizing conditions, the C100–C246 disulfide can be observed in WT caspase-7 as a large–small dimer, which is a minor product. Importantly, the expected small-subunit (small–small) dimer band is strongly formed in R210C/C246S, but it is not observed in WT caspase-7. This demonstrates that the designed dimer forms as expected, but spurious and random disulfides do not form. The small–small dimer could also be readily observed by mass spectrometry (Fig. 3 and Supporting Information Fig. S3). Under reducing conditions, only monomeric large and small subunits of R210C/C246S were observed. Under oxidizing conditions, small–small dimers were readily observed, further confirming the success of the design. Interestingly, both homodimers and heterodimers of the two small subunit cleavage variants are observed. For this design to be fully successful, it is essential that small–small dimers are strongly formed, inactivating R210C/C246S, and subsequently be reduced, resulting in the return of full activity.
Figure 2.
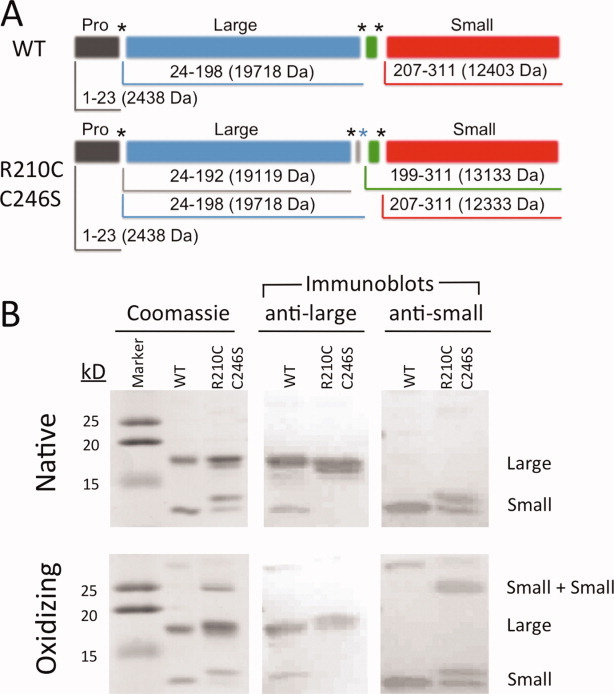
Incubation of caspase-7 R210C/C246S under oxidizing condition results in formation of a unique disulfide between the two small subunits of caspase-7. (A) Domain structure and molecular weights of WT or R210C/C246S caspase-7. (B) WT and R210C/C246S caspase-7 were incubated in native conditions (2 mM DTT) or in oxidizing conditions (300 μM cystamine), and then analyzed on a Coomassie-stained SDS gel or by Western blot with a caspase-7 antilarge subunit or antismall-subunit antibody. The two small-subunit bands in the native conditions are alternatively cleaved versions of caspase-7, with cleavages at naturally occurring residues 199 or 207. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 3.
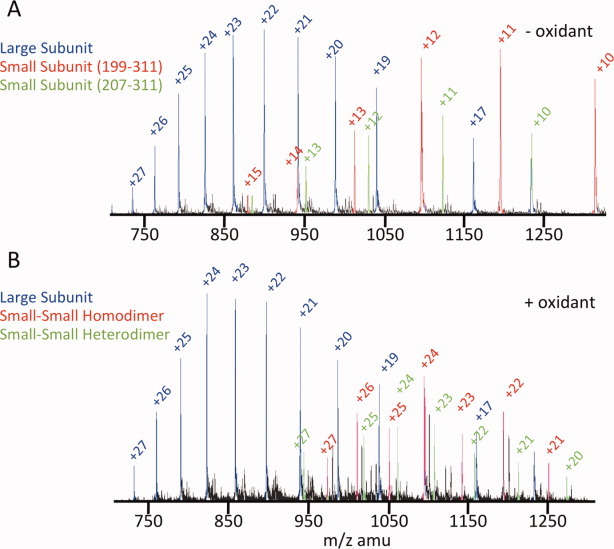
The small-small subunit dimer can be observed by mass spectrometry. R210C/C246S was incubated in the presence or absence of oxidant (1.5 mM cystamine) and then analyzed by mass spectrometry. (A) In the absence of oxidant, clear ions for the large subunit (blue) and the two cleavage variants of the small subunit (red, residues 199–311; green, 207–311) were observed. (B) In the presence of oxidant, identical ions were observed for the large subunit (blue). For the small subunits, twice as many ions were observed indicating a doubling of the molecular weight due to dimerization of the 199–311 species (red). A hybrid small-subunit dimer between the 199–311 and the 207–311 species (green) is also observed. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Inactivating disulfide forms reversibly
R210C/C246S can be completely inhibited under mild oxidizing conditions (Fig. 4) in a manner that is consistent with observation of the small-subunit dimers (Figs. 2 and 3). Inactivation of oxidized R210C/C246S can be monitored either directly by assaying the cleavage of a fluorescence substrate (Fig. 4) or indirectly by observing that cleavage at the 192 site is slowed only under oxidizing conditions (Fig. 2). We tested the reversibility of R210C/C246S inhibition by incubation in reductant at a roughly intracellular redox potential. R210C/C246S was able to fully recover from oxidation-induced inhibition. The remaining 40–50% activity in the oxidized R210C/C246S variant (Fig. 4) is an artifact of the activity assay. It is impossible to measure caspase activity in the absence of reductant due to the requirement that the catalytic cysteine thiol remain reduced. We found it necessary to use caspase assay buffer containing 5 mM 2-mercaptoethanol to observe full caspase activity for the WT protein. Thus, the reductant intrinsic to the buffer in which activity is measured accounts for the partial reactivation of R210C/C246S under mild reducing conditions and the apparent 40–50% remaining activity. Inhibition of WT caspase-7 is likely the result of mixed disulfide formation between the catalytic cysteine and the cystamine oxidant, which has previously been reported28 and is also fully reversible under these conditions.
Figure 4.
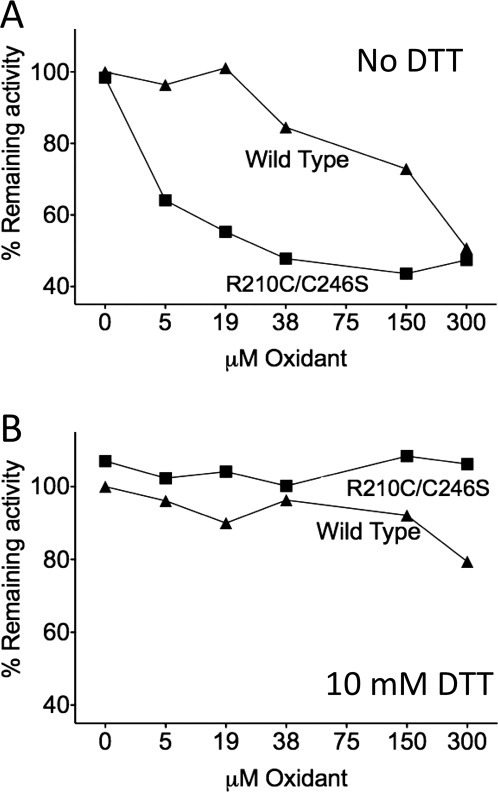
Disulfide bond formation induces the allosterically inactive state of caspase-7, which can be reactivated by the presence of reductant. Caspase-7 proteolytic activity was monitored in the presence of increasing concentrations of oxidant (cystamine). (A) Oxidation of the R210C disulfide is capable of inactivating R210C/C246S at much lower concentrations than the active site and can be oxidized in WT caspase-7. (B) In the presence of reductant (10 mM DTT), full activity is recovered. Data are based on activity assays measured from independent duplicate data sets on two separate days.
We solved the structure of R210C/C246S in two independent crystals (Table II). We observed that caspase-7 R210C/C246S not in the oxidized loop-locked conformation as expected, but in an active conformation in the crystal structure. From the structures of both the crystals, it was clear that the R210C disulfide had been reduced at some point after the crystallization experiment was initiated. We were pleased to see that the R210C–R210C disulfide could be reversed allowing the L2′ loop to adopt the active conformation [Fig. 5(A)].
Table II.
Crystallographic and Refinement Statistics for Caspase-7 R210C/C246S
| Native data set 1 | Native data set 2a | |
|---|---|---|
| Diffraction data | ||
| Wavelength (Å) | 0.9792 | 0.9792 |
| Resolution range (Å) | 48–2.86 | 48–3.1 |
| Oscillation range per frame (°) | 1.0 | 1.5 |
| Measured reflection (n) | 106,848 | 28,496 |
| Unique reflections | 20,763 | 17,353 |
| Completeness (%) | 99.2 (97.2) | 83.6 (87.9) |
| Redundancy | 5.2 (4.7) | 1.6 (1.6) |
| <I/σI> | 16.0 (2.7) | 16.4 (2.0) |
| Space group | P3221 | P3221 |
| a = b (Å) | 89.85 | 89.98 |
| c (Å) | 185.91 | 185.11 |
| α = β (°) | 90 | 90 |
| γ (°) | 120 | 120 |
| Rsym | 12.7 (69.6) | 9.1 (69.4) |
| Refinement statistics | ||
| Atoms (n) | 3900 | |
| Water molecules | 103 | |
| Rwork (%) | 18.81 | |
| Rfree (%) | 25.13 | |
| RMSD bond length (Å) | 0.010 | |
| RMSD bond angle (°) | 1.197 | |
| Average B-factor (Å2) | 58.93 | |
Collected on a second crystal, with minimal X-ray exposure minimize radiation damage. Numbers in parentheses indicate values for highest resolution shell.
Figure 5.
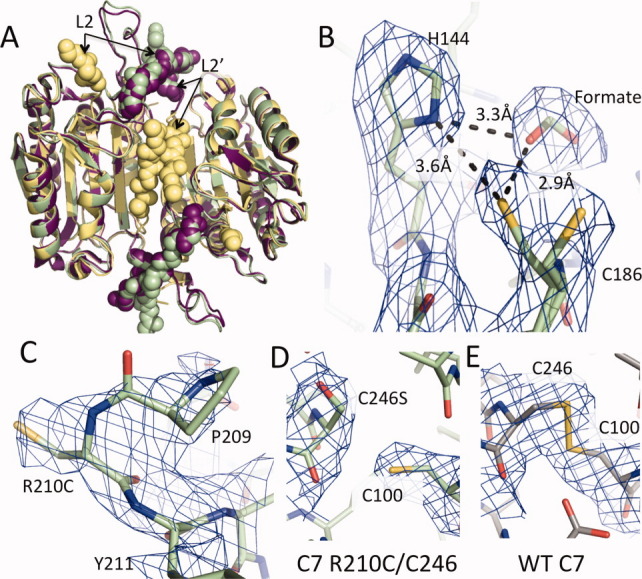
Caspase-7 R210C/C246S can attain the active conformation. (A) Oxidized R210C/C246S was crystallized but was reduced prior to or during data collection. Reduced caspase-7 R210C/C246S (3R5K, green) attains a conformation like that of WT caspase-7 bound to substrate (1F1J, purple) with L2 and L2′ in the up conformation interacting across the dimer interface. This is in contrast to the allosterically inhibited conformation of caspase-7 (1SHJ, yellow) in which the L2′ loop is folded over the allosteric site and the two L2′ loops make contact with one another. (B) The catalytic residues H144 and C186 are in a conformation that is primed for catalysis. Additional 2Fo − Fc density contoured at 1σ was observed in the S1 pocket was modeled as a formate molecule from the crystallization solution. The density around the catalytic cysteine (C186) was larger than one cysteine residue. It was best modeled as two cysteine conformations each with 50% occupancy. (C) 2Fo − Fc density contoured at 0.8σ for R210C. (D, E) 2Fo − Fc density contoured at 1σ for R210C/C246S (D) and WT (E) caspase-7.
There are two possible explanations for why the oxidized form of the enzyme was not observed. We observed by mass spectrometry that 1.5 mM cystamine oxidant was able to fully oxidize the R210C–R210C disulfide in R210C/C246S within a 5-min incubation (Fig. 3). Thus, we are certain that the sample we set up for crystallization was fully oxidized. One possibility for the reduction of the disulfide is that the residual presence of dithiothreitol (DTT; 2 mM) from the purification, which should have been highly oxidized since it was more than 1-month old, competed with 1.5 mM oxidant so that reduction of the disulfide occurred during the crystallization process. We consider this to be somewhat unlikely given that these same samples were fully oxidized as assessed by mass spectrometry. A second possibility is that the disulfide was reduced by synchrotron radiation. It would not be surprising to us that the reduction of the R210C–R210C disulfide occurred during the diffraction experiment, as ionizing radiation has been reported to reduce disulfides.29 In an attempt to observe the disulfide-containing form of R210C/C246S, we limited X-ray exposure to 1 s with the APS 24-ID-C X-ray beam attenuated to 7% and solved the structure using the minimal data necessary (30° of data, 20 frames, 1 s/frame, 83% completeness). Even with only 20-s total exposure to the crystal, we were unable to observe any oxidized R210C disulfides, suggesting that the inactivating disulfide is quite labile. We wonder whether it is possible to estimate the lability of the R210C disulfide by comparison with other caspase-7 disulfides that do withstand X-ray interrogation. DICA and FICA, which bind to caspase-7 via disulfides with C290, withstand X-ray radiation and are visible in the crystal structure.30 The disulfide linking the large and small subunits of caspase-7 (C100–C246) also withstands X-ray radiation under certain conditions (unpublished data, Supporting Information Fig. S1). Thus, of the three types of disulfides observed to date in caspase-7, the R210C disulfide may be the most labile. All caspase-7 structures observed to date show the same crystal packing arrangement. Regardless of the loop conformation, the active-site loops always point toward the large solvent channels. Thus, it would not be surprising if the loop reorganization we observed between the oxidized, disulfide-locked R210C/C246S and the reduced, unlocked form can be accommodated and interconverted within the same crystal.
Catalytic site geometry
The reduced form of R210C/C246S is in a conformation that is primed for substrate binding [Fig. 5(B)]. Typically, in the absence of an active-site ligand, the L2′ loop is in a catalytically compromised conformation13 similar to the allosterically inhibited form30 [Fig. 5(A), yellow spheres]. In our structure, the presence of formate at high concentrations (2.1M) in the crystallization buffer is sufficient to nucleate formation of the substrate-binding conformation [Fig. 5(A), green and purple spheres]. Here, formate serves as a surrogate for the aspartic acid that normally occupies the S1 pocket [Fig. 5(B)]. A similar phenomenon has been observed for caspase-1, where malonate assists in refolding caspase-1 and serves as a mimic for aspartic acid bound in the active site.31
In R210C/C246S, the catalytic dyad (H144 and C186) are in an optimal arrangement for catalysis (3.6 Å)32,33 that has not been observed before in caspase-7. All previous caspase-7 structures in which the substrate-binding loops are ordered relied on covalent modification of the catalytic cysteine13–16,25,30,34 and therefore had H144–C186 distances that were too great (typically ∼ 5.5 Å) to support necessary proton abstraction. The density for the catalytic thiol is ambiguous. We have modeled this density in several ways including positioning a water molecule in one lobe of the density. The unfavorable angle of the hydrogen bonds for water indicated that this is not a water molecule. Therefore, we concluded that the two conformations of the catalytic thiol may be a more accurate representation. Thus, we have modeled the density as a 50% mixture of two thiol conformations [Fig. 5(B)]. R210C is near the N-terminus of the small subunit. In caspase-7, it is typically only possible to observe residues starting at residue 212, due to disorder of the N-termini. It is, therefore, not a surprise that the density is weak, nevertheless we were able to visualize and build R210C [Fig. 5(C)]. It was also possible to visualize the substitution in our design, C246S [Fig. 5(D)]. C246S is in a different conformation than C246 locked into a disulfide by an uncleavable peptide [Fig. 5(E)].
Discussion
The disulfide bond is a useful protein-engineering tool since it can form covalent, redox-controllable bonds at discrete locations using naturally occurring amino acids. Since the advent of site-directed mutagenesis, introduction of disulfide bonds has been a well-used tool for controlling protein conformation. Engineered disulfides have been used to examine functional mechanisms,35,36 stabilize proteins,37,38 understand allosteric coupling,39,40 and even to trap transition states.41 Redox reversible switches have been developed in a number of proteins including measles virus F protein,42 malate dehydrogenase,43 integrins,44 kinesin,45 and the OxyR transcription factor,46 among many others. We sought to build upon this rich history and design allosterically controlled caspases, a class of proteins capable of regulating survival and death of cells.
The criteria that guided the rational design of redox-activatable caspase-7 included the following: (1) motion of the L2′ loops should be restricted by disulfide bond formation, (2) the catalytic efficiency of the untreated protein should mirror that of the WT enzyme, (3) full oxidation of the disulfide should result in full inhibition of caspase-7 under extracellular redox environments, and (4) introduction into intracellular reducing conditions should result in full recovery of proteolytic activity. Caspase-7 is a dynamic protein in which loop arrangement plays a key role in the activation of the enzyme. In our designed variant R210C/C246S, we placed the inactivation switch at the heart of a critical, active-site loop bundle. In the down conformation, with the L2′ loop pinned over the allosteric cavity, the substrate-binding groove is not formed, so the protein is unable to bind the substrate or catalyze the peptide cleavage reaction. By placing the disulfide over the allosteric site at the intersection of the L2′ loops, we can enforce a naturally occurring inactive state using native-like redox conditions. In R210C/C246S, we fulfilled all of the design criteria. In vitro activity results and the observation that oxidation of the R210C disulfide could also prevent self-processing at the minor D192 processing site both suggest ready inhibition by disulfide formation. The crystal structure of R210C/C246S that was first fully oxidized and then re-reduced in the crystal demonstrates the successful reversibility of our design. Finally, the observation of small-subunit dimers points to the inhibition mechanism by covalent locking of the L2′ loops.
Engineering in redox switches in a critical region like the active-site loops bundle could be a risky proposition. We had previously interrogated residues 211–215 in the L2′ loop and found that they were amenable to substitution.25 By extension, we hoped that R210 would likewise be amenable to substitution without negative functional effects. Fortunately, this was the case. All caspases have mobile active-site loops. Although this endeavor used caspase-7 as a template, the approach of pinning the L2′ loops should be applicable for any caspase since the active-site loops of all caspases are mobile prior to the binding of the substrate. Residue 210 is not highly conserved, so this position should be amenable to this substitution across the family. Intriguingly, residue 210 is natively a cysteine in caspase-3, begging the question of whether caspase-3 uses formation of this potential disulfide as part of its unique regulatory mechanisms. In a very high-resolution structure of caspase-3 bound to active-site ligand, the L2′ loop is seen to wrap around the opposite half of the dimer with C210 attaining two solvent accessible conformations.47 Because caspase-3 has not been crystallized in the absence of a substrate-mimic or in the allosterically inhibited conformation, the best models of what this region should look like are the allosterically inhibited structures of caspase-7, where the 210 residues make the closest approach. Thus, it may be possible to make a loop-locked version of caspase-3. It is intriguing to envision controlling the activity of an engineered, constitutively active and uninhibitable version of the caspase-3 zymogen48 using our loop-locking design. Clearly, there are many possibilities for designing loop-locked caspases. Induction of apoptosis by treatment with a variety of allosterically loop-locked caspases should, therefore, be possible given appropriate means of delivery.
Breakthroughs in targeted nanoparticle and polymer design have begun to make delivery of a much wider range of protein-based drugs feasible (for review49). Active targeting of nanoparticles, polymers, dendrimers, and nanogels can be achieved using disulfide functionalization, with the prospect of delivering encapsulated payloads including both small molecules and proteins. The combination of caspase-7 R210C/C246S with a redox-triggered delivery vehicle is, therefore, an intriguing prospect. If R210C/C246S was encapsulated in a redox-activable nanocarrier, in the relatively oxidizing extracellular environment R210C/C246S should remain inactive and inaccessibly encapsulated. Upon introduction into the reducing cytosolic environment, the redox-sensitive delivery device would release R210C/C246S (Fig. 6). Liberated R210C/C246S could then likewise be reduced, regain function, and activate apoptosis, leading to cell death. This scheme is particularly promising in cancers and proliferative disorders, where suppression of apoptosis is nearly always a contributor to the evasion of cell death pathways.
Figure 6.
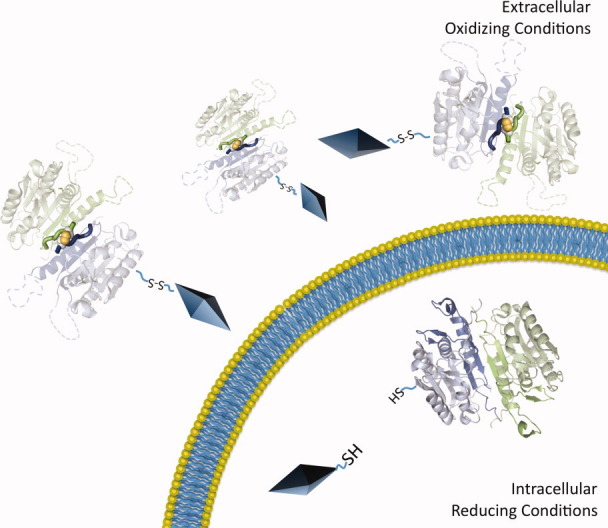
Delivery scheme for caspase-7 R210C/C246S. Capase-7 R210C/C246S is amenable for use with delivery vehicles (diamonds). We have designed the R210C/C246S version of caspase-7 so that under oxidizing extracellular conditions, caspase-7 is inactive. Upon entry into the cell, reducing intracellular conditions break the R210C disulfide yielding active caspase-7, which can cleave intracellular targets leading to apoptotic cell death.
Materials and Methods
Caspase-7 mutant generation, expression, and purification
WT caspase-7 was expressed from a construct encoding residues 1–303 plus codons for the two amino acids LE, then a six-Histidine tag contained in the plasmid pET23-b (AmpR) plasmid.50 Expression of caspase-7 variant R210C/C246S was carried out in a constitutive two-chain expression system.51 Residues 1–198 were followed by a TAA stop codon, a second ribosome-binding sequence, and residues 199–303, plus codons for the two amino acids LE and a six-Histidine tag contained in the plasmid pET23-b (AmpR). Variants in the protein were generated using QuikChange (Stratagene) site-directed mutagenesis. The recombinant protein was expressed in Escherichia coli in 2 × YT media grown for 18 h after induction with 1 mM isopropyl β-d-1-thiogalactopyranoside at an OD600 of 0.6.52 WT and mutant caspase-7 variants were purified using Ni-affinity liquid chromatography (HiTrap Chelating HP, GE). After binding of protein to the affinity column, the protein was eluted with a step gradient from 2 mM imidazole to 250 mM imidazole. Protein was diluted to 50 mM NaCl and then purified using a Macro-Prep High Q ion exchange column (Bio-Scale Mini 5 mL, Bio-Rad) with a linear gradient from 50 to 500 mM NaCl in 20 mM Tris buffer pH 8.0, with 2 mM DTT. Protein eluted in 120 mM NaCl and 20 mM Tris buffer, pH 8.0, was assessed for purity by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and found to be greater than 98% pure and was stored in elution buffer at –80°C.
Caspase activity assays
Enzyme concentrations were determined by active-site titration with covalent substrate DEVD-CHO (N-acetyl-Asp-Glu-Val-Asp-aldehyde; Enzo Life Sciences) in caspase activity buffer containing 100 mM HEPES pH 7.0, 10% polyethylene glycol 400, 0.1% CHAPS, 5 mM 2-mercaptoethanol, and 5 mM CaCl2 using the flourogenic substrate, DEVD-AMC (N-acetyl-Asp-Glu-Val-Asp-(7-amino-4-methylcoumarin)); Enzo Life Sciences), Ex365/Em495. Active-site titration samples were incubated over a period of 2 h in 120 mM NaCl and 20 mM Tris buffer, pH 8.0, at nanomolar concentrations. Optimal labeling was observed when protein was added to DEVD-CHO solvated in DMSO in 96-well V-bottom plates, sealed with tape, and incubated at room temperature. About 22.5 and 90 μL of aliquots were transferred to black-well plates in duplicate and assayed with 50-fold molar excess of substrate. For kinetic measurements, 100 nM protein was assayed with the range of 0–500 μM DEVD-AMC over the course of 7 min. Assays were performed at 37°C in 25 and 100 μL volumes in 96-well or 384-well microplate format using a Molecular Devices Spectramax M5 spectrophotometer. Initial velocities versus substrate concentration were fit using Prism software (Graphpad Software) to determine kinetic parameters Km and kcat.
Enzyme activity recovery assays were carried out using WT and R210C/C246S mutant protein buffer exchanged to remove DTT into pH 7.5 10 mM sodium phosphate buffer using Viva Spin 3K concentrators (Sartorius Stedim Biotech). Working at intracellular reduction conditions in vitro proved challenging, due to the propensity of caspase-7 active site to form mixed disulfides with glutathione. As a surrogate, we selected cystamine as the oxidant and DTT or 2-mercaptoethanol as reductants. The reduction potentials of these four redox systems have been shown to be similar.53,54 Protein was incubated in 96-well format in a range of cystamine concentrations (300–5 μM; 0 μM control) for a period of 1 h. Half the volume was subsequently transferred to conditions with a final concentration of 10 mM DTT and assayed for activity. The redox chemistry of DTT is at equilibrium with cystamine at pH 7.9, supporting our use of 10 mM DTT as an adequate substitute for a highly reducing intracellular environment.
A similar ratio of oxidant to protein was used to treat caspase samples for caspase activity assays, immunoblot detection, mass spectrometry, and crystallography. Caspases were then diluted to the concentration appropriate for the individual type of analysis.
Gel electrophoresis and immunoblot detection
WT and R210C/C246S protein were buffer exchanged using the above-mentioned method into pH 7.5 10 mM sodium phosphate buffer, and then divided into two samples and incubated in the presence or absence of 300 μM cystamine for 2 h. Samples for nonreducing gel electrophoresis were boiled for 10 min in gel-loading buffer (New England Biolabs) lacking DTT and loaded onto 16% polyacrylamide gels. Immunoblots of the subsequently separated proteins were performed using antibodies specific for caspase-7 large subunit (SIGMA Monoclonal Anti-caspase-7 C1104) and small subunit (SIGMA Anti-caspase-7 PRS3465) and compared against known size standards (Kaleidoscope protein marker; Bio-Rad).
Protein mass spectrometry
Protein for mass spectrometry was prepared at 18 μM in 120 mM NaCl, 20 mM Tris buffer pH 8.0, and 2 mM DTT with or without 1.5 mM cystamine. Mass spectra were acquired on a QStar-XL (MDS Sciex, Toronto, Canada) hybrid quadrupole/time-of-flight instrument. Reverse-phase liquid chromatography (Agilent 1100 LC) using a C4 column was utilized to desalt and separate excess cystamine. Column output was infused directly into the electrospray ionization (ESI) source at a rate of 0.5 mL/min using N2 as a nebulizing gas. Protein was eluted over a 10-min gradient from 0 to 100% acetonitrile and monitored using UV and total ion current plot. A single-protein peak was eluted between 17 and 18 min at the end of the gradient. Data were analyzed using Analyst QS (Applied Biosystems) with BioAnalyst extensions. All spectra deconvolutions were gated for proteins between 10 and 30 kDa, with m/z ratios between 700 and 1700. The mass spectrometers are housed in the Mass Spectrometry Center of the University of Massachusetts at Amherst.
Caspase-7 R210C/C246S crystallization and X-ray data collection
To prepare R210C/C246S crystals, protein in a buffer containing 120 mM NaCl and 20 mM Tris buffer, pH 8.0, was concentrated using Millipore Ultrafree 5K nominal molecular weight limit (NMWL) membrane concentrators (Millipore) to 6.3 mg/mL as assessed by absorbance at 280 nm. Cystamine was added to a final concentration of 1.25 mM. Crystal trays were setup at room temperature and grown in 3 μL hanging drop with mother liquor consisting of 2.1M sodium formate and 300 mM sodium citrate, pH 5.0, in a 2:1 ratio of protein to mother liquor. Crystals grew to a maximum of 180 μM in 3 days at 20°C. Crystals were cryoprotected in 20% ethylene glycol in mother liquor with a 60-s incubation, and then frozen by rapid immersion in liquid N2. Data were collected using synchrotron radiation at APS 24-ID-C over 90 frames with 1° oscillations and showed diffraction data to 2.86 Å. Indexing and integration were carried out with MAR XDS.55 Data were scaled using SCALA.56 Data for the low-radiation damage crystal were collected using with 1.5° oscillations. These data were indexed and integrated using HKL2000,57 and the data were scaled using SCALA.56
Structure determination
Phase information was obtained by molecular replacement using 1F1J as the search model with residues corresponding to L2′ omitted during searches with PHASER.58 Removed residues were rebuilt into unambiguous density using COOT,59 and then refined using Refmac60-restrained refinement. Water and formate molecules were inserted manually and checked for stereochemical appropriateness. The final refined model contains residues 57–196 and 210–303 for chain A and 357–496 and 510–605 for chain B. A total of 103 water molecules were assigned chain O, and seven formates were assigned chain F.
Acknowledgments
The authors thank Stephen Eyles of the UMass Amherst Mass Spectrometry Facility for assistance with mass spectrometric analysis and Elih M. Velázquez-Delgado for assistance in crystal data collection.
Supplementary material
References
- 1.Orth K, Chinnaiyan AM, Garg M, Froelich CJ, Dixit VM. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J Biol Chem. 1996;271:16443–16446. [PubMed] [Google Scholar]
- 2.Fraser AG, Evan GI. Identification of a Drosophila melanogaster ICE/CED-3-related protease, drICE. EMBO J. 1997;16:2805–2813. doi: 10.1093/emboj/16.10.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9678. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 4.Eckhart L, Ban J, Fischer H, Tschachler E. Caspase-14: analysis of gene structure and mRNA expression during keratinocyte differentiation. Biochem Biophys Res Commun. 2000;277:655–659. doi: 10.1006/bbrc.2000.3698. [DOI] [PubMed] [Google Scholar]
- 5.Wang P, Arjona A, Zhang Y, Sultana H, Dai J, Yang L, LeBlanc PM, Doiron K, Saleh M, Fikrig E. Caspase-12 controls West Nile virus infection via the viral RNA receptor RIG-I. Nat Immunol. 2010;11:912–919. doi: 10.1038/ni.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 7.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 8.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 9.Srinivasula SM, Datta P, Fan XJ, Fernandes-Alnemri T, Huang Z, Alnemri ES. Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J Biol Chem. 2000;275:36152–36157. doi: 10.1074/jbc.C000533200. [DOI] [PubMed] [Google Scholar]
- 10.Boise LH, Gonz·lez-Garcìa M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X. Bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 11.Dohlman W, Caroti M, Leflowitz R, Nathans J, Kubo T, Bonner T, Buckley N, Young A, Brann M, Julius D. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 12.Pop C, Chen YR, Smith B, Bose K, Bobay B, Tripathy A, Franzen S, Clark AC. Removal of the pro-domain does not affect the conformation of the procaspase-3 dimer. Biochemistry. 2001;40:14224–14235. doi: 10.1021/bi011037e. [DOI] [PubMed] [Google Scholar]
- 13.Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y. Crystal structure of a procaspase-7 zymogen: mechanisms of activation and substrate binding. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]
- 14.Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W. Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci USA. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei Y, Fox T, Chambers SP, Sintchak J, Coll JT, Golec JM, Swenson L, Wilson KP, Charifson PS. The structures of caspases-1, -3, -7 and -8 reveal the basis for substrate and inhibitor selectivity. Chem Biol. 2000;7:423–432. doi: 10.1016/s1074-5521(00)00123-x. [DOI] [PubMed] [Google Scholar]
- 16.Agniswamy J, Fang B, Weber IT. Plasticity of S2–S4 specificity pockets of executioner caspase-7 revealed by structural and kinetic analysis. FEBS J. 2007;274:4752–4765. doi: 10.1111/j.1742-4658.2007.05994.x. [DOI] [PubMed] [Google Scholar]
- 17.Hardy JA, Wells JA. Searching for new allosteric sites in enzymes. Curr Opin Struct Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Hardy JA, Wells JA. Dissecting an allosteric switch in caspase-7 using chemical and mutational probes. J Biol Chem. 2009;284:26063–26069. doi: 10.1074/jbc.M109.001826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo A, DeGraff W, Friedman N, Mitchell JB. Selective modulation of glutathione levels in human normal versus tumor cells and subsequent differential response to chemotherapy drugs. Cancer Res. 1986;46:2845–2848. [PubMed] [Google Scholar]
- 20.Jones DP, Brown LAS, Sternberg P. Variability in glutathione-dependent detoxification in vivo and its relevance to detoxication of chemical mixtures. Toxicology. 1995;105:267–274. doi: 10.1016/0300-483x(95)03221-z. [DOI] [PubMed] [Google Scholar]
- 21.Denault JB, Drag M, Salvesen GS, Alves J, Heidt AB, Deveraux Q, Harris JL. Small molecules not direct activators of caspases. Nat Chem Biol. 2007;3:519. doi: 10.1038/nchembio0907-519. author reply 520. [DOI] [PubMed] [Google Scholar]
- 22.Peterson QP, Goode DR, West DC, Ramsey KN, Lee JJ, Hergenrother PJ. PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J Mol Biol. 2009;388:144–158. doi: 10.1016/j.jmb.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon JT, Hwang SK, Jin H, Churchwell MI, Cho MH, Doerge DR, Helferich WG, Hergenrother PJ. Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy. Nat Chem Biol. 2006;2:543–550. doi: 10.1038/nchembio814. [DOI] [PubMed] [Google Scholar]
- 24.Wolan DW, Zorn JA, Gray DC, Wells JA. Small-molecule activators of a proenzyme. Science. 2009;326:853–858. doi: 10.1126/science.1177585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witkowski WA, Hardy JA. L2′ loop is critical for caspase-7 active site formation. Protein Sci. 2009;18:1459–1468. doi: 10.1002/pro.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sowdhamini R, Srinivasan N, Shoichet B, Santi DV, Ramakrishnan C, Balaram P. Stereochemical modeling of disulfide bridges. Criteria for introduction into proteins by site-directed mutagenesis. Protein Eng. 1989;3:95–103. doi: 10.1093/protein/3.2.95. [DOI] [PubMed] [Google Scholar]
- 27.Boucher D, Blais V, Drag M, Denault JB. Molecular determinants involved in activation of caspase 7. Biosci Rep. 2011;31:283–294. doi: 10.1042/BSR20100111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesort M, Lee M, Tucholski J, Johnson GVW. Cystamine inhibits caspase activity. J Biol Chem. 2003;278:3825–3830. doi: 10.1074/jbc.M205812200. [DOI] [PubMed] [Google Scholar]
- 29.Weik M, Ravelli RB, Kryger G, McSweeney S, Raves ML, Harel M, Gros P, Silman I, Kroon J, Sussman JL. Specific chemical and structural damage to proteins produced by synchrotron radiation. Proc Natl Acad Sci USA. 2000;97:623–628. doi: 10.1073/pnas.97.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardy JA, Lam J, Nguyen JT, O'Brien T, Wells JA. Discovery of an allosteric site in the caspases. Proc Natl Acad Sci USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romanowski MJ, Scheer JM, O'Brien T, McDowell RS. Crystal structures of a ligand-free and malonate-bound human caspase-1: implications for the mechanism of substrate binding. Structure (Camb) 2004;12:1361–1371. doi: 10.1016/j.str.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 32.Podobnik M, Kuhelj R, Turk V, Turk D. Crystal structure of the wild-type human procathepsin B at 2.5 A resolution reveals the native active site of a papain-like cysteine protease zymogen1. J Mol Biol. 1997;271:774–788. doi: 10.1006/jmbi.1997.1218. [DOI] [PubMed] [Google Scholar]
- 33.Strajbl M, Florian J, Warshel A. Ab initio evaluation of the free energy surfaces for the general base/acid catalyzed thiolysis of formamide and the hydrolysis of methyl thiolformate: a reference solution reaction for studies of cysteine proteases. J Phys Chem B. 2001;105:4471–4484. [Google Scholar]
- 34.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Dataa P, Alnemri ES, Shi Y. Structural basis of caspase-7 inhibition by XIAP. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 35.Seeger MA, Von Ballmoos C, Eicher T, Brandstotter L, Verrey F, Diederichs K, Pos KM. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat Struct Mol Biol. 2008;15:199–205. doi: 10.1038/nsmb.1379. [DOI] [PubMed] [Google Scholar]
- 36.Ma K, Temiakov D, Anikin M, McAllister WT. Probing conformational changes in T7 RNA polymerase during initiation and termination by using engineered disulfide linkages. Proc Natl Acad Sci USA. 2005;102:17612–17617. doi: 10.1073/pnas.0508865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong MY, Kim S, Yun CW, Choi YJ, Cho SG. Engineering a de novo internal disulfide bridge to improve the thermal stability of xylanase from Bacillus stearothermophilus No. 236. J Biotechnol. 2007;127:300–309. doi: 10.1016/j.jbiotec.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Mitchinson C, Wells JA. Protein engineering of disulfide bonds in subtilisin BPN. Biochemistry. 1989;28:4807–4815. doi: 10.1021/bi00437a043. [DOI] [PubMed] [Google Scholar]
- 39.Falcon CM, Swint-Kruse L, Matthews KS. Designed disulfide between N-terminal domains of lactose repressor disrupts allosteric linkage. J Biol Chem. 1997;272:26818. doi: 10.1074/jbc.272.43.26818. [DOI] [PubMed] [Google Scholar]
- 40.Qureshi SH, Yang L, Manithody C, Iakhiaev AV, Rezaie AR. Mutagenesis studies toward understanding allostery in thrombin. Biochemistry. 2009;48:8261–8270. doi: 10.1021/bi900921t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shandiz AT, Capraro BR, Sosnick TR. Intramolecular cross-linking evaluated as a structural probe of the protein folding transition state. Biochemistry. 2007;46:13711–13719. doi: 10.1021/bi701042e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JK, Prussia A, Snyder JP, Plemper RK. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J Virol. 2007;81:8821. doi: 10.1128/JVI.00754-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muslin EH, Li D, Stevens FJ, Donnelly M, Schiffer M, Anderson LE. Engineering a domain-locking disulfide into a bacterial malate dehydrogenase produces a redox-sensitive enzyme. Biophys J. 1995;68:2218–2223. doi: 10.1016/S0006-3495(95)80430-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimaoka M, Lu C, Palframan RT, Von Andrian UH, McCormack A, Takagi J, Springer TA. Reversibly locking a protein fold in an active conformation with a disulfide bond: integrin L I domains with high affinity and antagonist activity in vivo. Proc Natl Acad Sci USA. 2001;98:6009–6014. doi: 10.1073/pnas.101130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tomishige M, Vale RD. Controlling kinesin by reversible disulfide cross-linking. Identifying the motility-producing conformational change. J Cell Biol. 2000;151:1081–1092. doi: 10.1083/jcb.151.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 47.Ganesan R, Mittl PR, Jelakovic S, Grutter MG. Extended substrate recognition in caspase-3 revealed by high resolution X-ray structure analysis. J Mol Biol. 2006;359:1378–1388. doi: 10.1016/j.jmb.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 48.Walters J, Pop C, Scott FL, Drag M, Swartz P, Mattos C, Salvesen GS, Clark AC. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem J. 2009;424:335–345. doi: 10.1042/BJ20090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh R, Lillard JW., Jr Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86:215–223. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. Target protease specificity of the viral serpin CrmA. J Biol Chem. 1997;272:7797–7800. doi: 10.1074/jbc.272.12.7797. [DOI] [PubMed] [Google Scholar]
- 51.Vaidya S, Velazquez-Delgado EM, Abbruzzese G, Hardy JA. Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J Mol Biol. 2011;406:75–91. doi: 10.1016/j.jmb.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stennicke HR, Salvesen GS. Caspases: preparation and characterization. Methods. 1999;17:313–319. doi: 10.1006/meth.1999.0745. [DOI] [PubMed] [Google Scholar]
- 53.Cleland W. Dithiothreitol, a new protective reagent for SH groups. Biochemistry. 1964;3:480–482. doi: 10.1021/bi00892a002. [DOI] [PubMed] [Google Scholar]
- 54.Millis KK, Weaver KH, Rabenstein DL. Oxidation/reduction potential of glutathione. J Org Chem. 1993;58:4144–4146. [Google Scholar]
- 55.Kabsch W. XDS. Acta Crystallogr D: Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 57.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 58.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.