

Abstract
Tuberculosis remains the most hazardous bacterial infection worldwide. The causative agent, Mycobacterium tuberculosis, is a facultative intracellular pathogen of resting MΦ. IFN-γ secreted by natural killer, CD4 Th 1 and CD8 T cells upon instruction by IL-12 and -18 activates MΦ to restrict mycobacterial growth. Production of both cytokines is induced by TLR signalling in DC and MΦ. Mice deficient for the TLR adaptor, MyD88, are highly susceptible to M. tuberculosis infection. Shared usage of MyD88 by signalling cascades for TLR and receptors for IL-1 and IL-18 prompted us to revisit the role of IL-18 during experimental infection with M. tuberculosis. We show that mice deficient for IL-18 and MyD88 but not for IL-18 receptor promptly succumbed to M. tuberculosis infection in contrast to WT or TLR-2/-4 double KO mice indicating that lack of IL-18 contributes to the high susceptibility of MyD88 KO mice to M. tuberculosis. Without IL-18, the protective Th1 response was decreased and hence, mycobacterial propagation was favoured. Neutrophil-driven lung immunopathology concomitant with unrestrained growth of tubercle bacilli are most likely responsible for the premature death of IL-18 KO mice. Thus, IL-18 plays a decisive role in protective immunity against tuberculosis.
Keywords: IFN-γ, IL-18, Mouse, Neutrophils, Tuberculosis
Introduction
Despite more than 125 years of research and development, tuberculosis (TB) remains the most hazardous bacterial infection worldwide with approximately 2 million people dying of the disease annually [1]. The risk of disease is increased by immunocompromising conditions such as AIDS emphasizing that T-cell immunity protects latently infected individuals against active TB. The innate immune response to Mycobacterium tuberculosis instructs acquired immunity in the initial stage, and executes effector mechanisms in the chronic stage.
M. tuberculosis is usually transmitted via aerosols and establishes stable infection in the lung. There, M. tuberculosis is engulfed by MΦ and DC, which serve as host cells for mycobacterial survival and propagation. Binding of mycobacterial ligands to TLR-2, -4 and -9 promotes release of chemokines and proinflammatory cytokines, expression of adhesion molecules and attraction of MΦ, DC and PMN. Two crucial MΦ- and DC-derived cytokines, IL-12 and -18, induce NK-cell activity and bias immunity towards a Th1 cell response characterized by profound IFN-γ production, which is considered critical for protection against M. tuberculosis [2]. Activated MΦ express anti-mycobacterial molecules such as nitric oxide synthase (NOS)-2 (also known as inducible NOS) and LRG47 as well as cytokines such as TNF-α, which promotes granuloma formation within the infected tissue to sequester the bacilli from dissemination [2].
Despite the prevailing assumption that resistance to M. tuberculosis infection depends on microbe sensing through TLR, their importance for mounting a protective immune response against M. tuberculosis remains controversial. While some groups found that TLR-mediated signalling is dispensable for protective immunity against M. tuberculosis [3-6], others suggested an essential contribution of TLR to protection [7-9]. The TLR contain a Toll/IL-1 receptor domain, which associates with the adaptor molecule MyD88. MyD88 recruits the IL-1 receptor-associated kinase 1 and/or 4 to trigger downstream signals for NF-κB activation [10]. Although most data support a role for TLR in chronic rather than in acute disease, MyD88-deficient mice are highly susceptible to M. tuberculosis and succumb very rapidly to infection [4, 11]. TLR, however, share the MyD88 signalling cascade with both IL-1 receptor (IL-1R) and IL-18 receptor (IL-18R) [4, 10, 11]. Shared usage of MyD88 by signalling cascades of TLR, IL-1R and IL-18R, therefore raises the question of whether lack of IL-1 or IL-18 signals is responsible for high susceptibility of MyD88 KO mice to M. tuberculosis infection. Fremond et al. recently presented evidence that IL-1R-signalling is important for protection against M. tuberculosis while IL-18R-signalling is not [12]. In contrast to IL-18R-deficient mice, we here demonstrate that mice lacking IL-18 (IL-18 KO) were highly susceptible to M. tuberculosis infection similar to both MyD88 KO mice and those double-deficient in IL-1β and IL-18. IL-18 KO mice succumbed much earlier to experimental infection with M. tuberculosis than WT or TLR-2/-4 double KO (TLR-2/-4 DKO) mice. We thus conclude that the absence of IL-18 signals explains, at least in part, the high susceptibility of MyD88 KO mice to M. tuberculosis infection. In the absence of IL-18, immunity to M. tuberculosis was hampered by decreased Th1 responses, PMN-dominated lung immunopathology concomitant with unrestrained growth of the tubercle bacilli. Thus, in contrast to previous assumptions, IL-18 plays a critical role in protective immunity against M. tuberculosis infection.
Results
IL-18 KO mice are highly susceptible to M. tuberculosis infection
In order to analyse the role of IL-18 in TB, mice deficient for IL-18, IL-1β/IL-18, MyD88, TLR-2/-4 and WT B6 mice were challenged with M. tuberculosis via aerosol infection (100 CFU/lung). Mice deficient for IL-18 alone were highly susceptible to low-dose aerosol infection with virulent M. tuberculosis H37Rv similar to IL-1β/IL-18 DKO and MyD88 KO mice. All three mouse strains succumbed to M. tuberculosis infection at approximately day 25 whereas WT and TLR-2/-4 DKO mice survived for more than 75 days (Fig. 1A). Twenty days post infection (p. i.), acid-fast staining of lungs revealed higher loads of tubercle bacilli in IL-18, IL-1β/IL-18 and MyD88 KO lungs compared with TLR-2/-4 KO and WT lungs (Fig. 1D; and data not shown). As previously published [12] and in contrast to IL-18 KO mice, IL-18R KO mice were able to control M. tuberculosis infection (Fig. 1B). We wondered whether the higher susceptibility of IL-18 DKO mice compared with IL-18R KO mice was due to higher bacterial burden in IL-18 KO mice and determined bacterial loads in lungs by CFU analysis. Early death of IL-18 KO restricted CFU analysis to early time points (we have chosen day 18 p.i.) while IL-18R KO were accessible to CFU analysis at later time points. Lungs of IL-18-deficient mice had significantly higher bacterial numbers as compared with B6 WT lungs on day 18 p.i. (Fig. 1C) in line with the acid fast stain (Fig. 1D). In contrast, bacterial numbers recovered from lungs of IL-18R-deficient mice were only slightly increased but not significantly different from those recovered from WT lungs (Fig. 1C). Thus, the lack of IL-18 but not of IL-18R rendered mice highly susceptible to M. tuberculosis infection allowing unrestrained bacillary growth. We conclude that IL-18 is critically involved in protection against M. tuberculosis challenge.
Figure 1.

IL-18-deficient mice are highly susceptible to M. tuberculosis infection. IL-18 KO and WT B6 mice and IL-1β/IL-18, MyD88, TLR-2/-4 (A) or IL-18R (B) deficient mice were infected by aerosol with M. tuberculosis H37Rv (100 CFUs) and followed for 75 (A) or 90 days (B), respectively. One representative experiment out of three is shown. Statistical analysis of the survival curves was performed using the Log Rank test. Differences in survival kinetics between WT and IL-1β/IL-18 DKO, MyD88 KO and IL-18 KO were statistically significant, respectively, whereas TLR-2/-4 DKO and IL-18R KO did not show impaired survival but were as resistant as WT mice. (A) n = 10; (B) n = 8. (C) Mice were sacrificed after the indicated times post aerosol infection and serial dilutions of lung lysates were plated for CFU determination. Results from one representative experiment out of two independent ones are shown (n = 10 for IL-18 KO, n = 5 for IL-18 R KO). Statistical analysis was performed using the Student’s t-test. ***p = 0.0002. (D) Acid-fast staining revealed high bacterial loads in lungs of MyD88- or IL-18-deficient mice 20 days p.i..
IL-18 KO mice present PMN-dominated inflammation
Lungs of M. tuberculosis-infected mice revealed histopathological sequelae of murine TB though with varying degrees of severity. IL-18 and MyD88 KO mice presented with severe inflammatory lung infiltrates compared with TLR-2/-4 DKO or WT mice. In IL-18 KO mice, lesions were characterized by exacerbated infiltrates and extended necrotic areas (Fig. 2A). On day 18 p.i., immunochemistry revealed that cellular infiltrates were mainly composed of large aggregations of Gr1+ cells in close association with necrotic areas (Fig. 2B) suggesting PMN contribution to pathology in the absence of IL-18. In contrast, Gr1+ cells were less abundant in lungs of infected MyD88 KO mice and almost absent from that of WT and TLR-2/-4 DKO mice. Thus, in the absence of IL-18, M. tuberculosis infection promoted a strong and prolonged attraction of Gr1+ cells to the site of infection in the lung. These pathological sequelae of TB in IL-18 KO mice were distinct from those observed in MyD88 KO mice.
Figure 2.

Strong inflammation accompanied by PMN influx into lungs of IL-18-deficient mice upon M. tuberculosis infection. (A) Representative H&E stain of lungs of IL-18 KO, TLR-2/-4 DKO, MyD88 KO or B6 WT mice 18 days p.i. with M. tuberculosis H37Rv (100 CFU; aerosol). (B) Cellular infiltrates in lungs from IL-18-deficient mice mainly consisted of PMN, which were detected by specific anti-Gr-1 immunostaining on day 18 p.i. with M. tuberculosis H37Rv.
T cells from M. tuberculosis-infected IL-18 KO mice produce IFN-γ
In order to analyse functions and phenotypes of T cells generated in the absence of IL-18 or MyD88, we isolated pulmonary and splenic lymphocytes from IL-18, MyD88 KO, TLR-2/-4 DKO and WT B6 mice. Cells were stimulated for 4 h with a peptide mix (MHC II: Ag85A241–260, Ag85B240–260; MHC I: Mtb32/RV0125309–318) and subsequently stained for CD4, CD8, CD154 (CD40L) and IFN-γ. Twenty days after M. tuberculosis infection, low but similar numbers of peptide-reactive CD4 and CD8 T cells expressing IFN-γ and/or CD40L were detected in spleen cell cultures from all four mouse strains analysed but not in cultures from non-infected mice (data not shown). FACS analyses of lung-derived T cells from M. tuberculosis-infected IL-18, MyD88, TLR-2/-4 KO and WT mice showed significant induction of peptide-specific IFN-γ/CD40L-expressing CD4 and IFN-γ secreting CD8 T cells when compared with unstimulated ones (Fig. 3A and B, black versus white bars). The numbers of peptide-reactive T cells from lungs were similar between all mouse strains analysed. Taken together, peptide-specific CD4 and CD8 T cells were present in M. tuberculosis-infected lungs in the absence of IL-18 or MyD88. Hence, IL-18 was not required to recruit Th1 CD4 and CD8 T cells to the lung tissue.
Figure 3.

T cells are recruited to the lungs and can produce IFN-γ in vitro. Whole lung lysates of IL-18 KO, TLR2/4 DKO, MyD88 KO or B6 WT mice were prepared on day 20 p.i. with M. tuberculosis H37Rv (100 CFU; aerosol), re-stimulated in vitro (black bars; white bars = unstimulated) with a mycobacterial peptide mix and analysed by flow cytometry for the expression of CD4, CD8, CD154 and intracellular IFN-γ as outlined in the Materials and methods. Results for CD4+ (A) and CD8+ (B) T cells from one representative experiment (out of two) are shown as means of five individual mice (bar graphs)+SD. Representative FACS plots for one animal are shown, respectively.
Protective Th1 immune response to M. tuberculosis requires IL-18
In order to more precisely define the immunological basis for the high susceptibility of IL-18 KO mice, we analysed serum and lungs from infected mice for cytokine/chemokine patterns and down-stream effectors. Of all mouse strains analysed, IL-18 KO mice showed the lowest serum levels of both IFN-γ and NO as compared with MyD88 KO, TLR-2/-4 DKO and WT mice at day 19 p.i. (Fig. 4A). Similarly, IFN-γ levels were reduced in lung lysates of IL-18 but also MyD88 KO mice at day 19 p.i. when compared with TLR-2/-4 DKO and WT mice (Fig. 4B). It should be noted that serum and lung levels of IL-12p70 did not differ between IL-18 KO and WT mice, suggesting that decreased IFN-γ production in IL-18 KO mice was not due to impaired IL-12 production (Fig. 4A and B).
Figure 4.

Th1 immune response to M. tuberculosis is hampered in IL-18-deficient mice. NO, cytokine and chemokine levels were measured in serum (A) and lung lysates (B) of IL-18 KO, TLR-2/-4 DKO, MyD88 KO or B6 WT mice on day 19 after M. tuberculosis H37Rv infection (100 CFU; aerosol). Results are shown as means (n = 5)+SD from one representative experiment out of three. Statistical analysis was performed by ANOVA and Tukey’s Multiple Comparison test. *p<0.05, **p<0.01, ***p<0.001. (C) Cytokine mRNA expression in M. tuberculosis H37Rv-infected mice. On day 19 p.i., mice were killed and lungs of each group (n = 5) were pooled. mRNA expression levels of different cytokines and chemokines were determined by qRT PCR as described in the Materials and methods. Triplicates were analysed for each cDNA and primer pair. Results represent fold changes (median+SD of three reactions) of mRNA gene expression for infected KO mice relative to infected B6 WT mice from one representative experiment out of three.
In line with decreased protein levels, quantitative RT PCR (qRT PCR) analyses of mRNA revealed a decrease in IFN-γ mRNA expression in M. tuberculosis-infected lungs of IL-18 and MyD88 KO mice as compared with WT lungs (data not shown). Consequently, mRNA expression of effectors downstream of IFN-γ such as NOS-2 and IDO was reduced in lungs from infected IL-18 and MyD88 KO animals as compared with WT mice (Fig. 4C). TNF-β, which has been shown to promote the local organization of the granulomatous response within the M. tuberculosis-infected tissue [13], was down-regulated in lungs of both IL-18- and MyD88-deficient mice, too (Fig. 4C). Together, these data reveal that IFN-γ responses, which are induced by M. tuberculosis infection in WT mice, were diminished in the absence of IL-18. This finding contrasts with our observation that IL-18 KO mice were generally capable of generating and recruiting IFN-γ producing Th1 cells to the lung. Despite being able to be re-stimulated in vitro (Fig. 3), these cells seem to be functionally hampered in vivo.
We observed enhanced IL-17 and IL-6 expression in lung tissue of IL-18 KO mice at day 18 p.i. with M. tuberculosis as compared with all other mouse strains (Fig. 4B, C). IL-17 protein levels were also elevated in sera of IL-18-deficient mice (Fig. 4A). IL-17 is known to induce pro-inflammatory cytokines, such as IL-6 and various chemokines, thereby triggering PMN influx into inflamed tissues. In line with this, mRNA expression of the PMN-chemoattractants CXCL-1 and CXCL-2 was increased in IL-18 KO lungs upon M. tuberculosis infection (Table 1). Thus, enhanced production of IL-17, CXCL-1 and CXCL-2 is likely responsible for the excessive influx of PMN into infected lungs and the exacerbated histopathological sequelae of IL-18 KO mice.
Table 1.
mRNA expression levels for chemokines in lungs of IL-18-deficient mice after aerosol infection with M. tuberculosis H37Rv compared with respective samples from infected WT micea)
| Chemokine | Increased mRNA expression in IL-18 KO lungs (fold change relative to WT) |
|---|---|
| CXCL-1 | 2.5 |
| 4 | |
| CXCL-2 | 1.2 |
| 19.7 | |
| CCL-2 (MCP-1) | 1.2 |
| 0.5 | |
| CCL-4 (MIP-1b) | 0.8 |
| 8.5 |
mRNA levels were analysed by quantitative RT-PCR as described in the Materials and methods. Data represent fold changes in mRNA expression from two independent infection experiments.
In addition, we found enhanced protein and/or mRNA levels of MIP-1α, MIP-1β and MCP-1 in serum and lung tissue of M. tuberculosis-infected IL-18 mice (Fig. 4B; Table 1). MIP-1α and MIP-1β were also enhanced in lungs of MyD88-deficient mice but to a lesser extent than in IL-18 KO lungs. These chemokines attract MΦ, eosinophils, activated T cells and NK cells and are probably general markers of excessive inflammation in M. tuberculosis infection in the absence of IL-18 or MyD88. However, the finding that PMN were the major inflammatory cell type in lungs of IL-18 KO mice suggests distinct types of immunopathology in IL-18- compared with MyD88-deficient mice probably induced by differential expression of certain chemokines and cytokines as shown above.
While expression of IFN-γ and downstream effectors was reduced, levels of IL-5 were also significantly decreased in both serum and lung samples from IL-18-deficient mice when compared with WT mice. However, levels of IL-4, the hallmark Th2 cytokine, in both sera and lungs did not differ between IL-18 KO and WT mice (Fig. 4A, B). In addition, albeit decreased concentrations of IL-13, another important Th2-associated cytokine, were observed in sera from IL-18 KO mice when compared with WT ones, expression of IL-13 was not affected by IL-18 deficiency at the site of infection. This suggests that decreased production of Th1 cytokines in the presence of unchanged Th2 cytokine levels in IL-18 KO lungs upon M. tuberculosis infection favours a Th2-biased immune response in absence of IL-18 and impaired classical MΦ activation. In line with this, the expression of markers characteristic of alternatively activated MΦ (AAMΦ), arginase-1 and Ym-1, was enhanced in IL-18 KO lungs infected with M. tuberculosis (Fig. 4C). AAMΦ have been shown to be compromised in anti-mycobacterial effector function [14, 15] and promote intracellular survival and replication of M. tuberculosis.
Taken together, these data suggest that M. tuberculosis infection in the absence of IL-18 diminishes Th1 responses, i.e. IFN-γ and its downstream effector molecules such as NOS-2, NO and IDO. In addition, IL-17, CXCL-1 and CXCL-2 cause PMN influx, which exacerbates immunopathology in IL-18 KO mice. This bias provides a functional explanation for the high susceptibility of IL-18 KO mice to M. tuberculosis.
PMN fail to control M. tuberculosis in the absence of IL-18
The influx of PMN into the lungs of M. tuberculosis-infected IL-18 KO mice without apparent effects on mycobacterial growth (Fig. 1A insert) prompted us to analyse the efficacy of PMN against M. tuberculosis. To reveal whether PMN influence mycobacterial load and contribute to protection in vivo, WT or IL-18-deficient mice were infected with M. tuberculosis and subsequently depleted of PMN by i.p. injection of the anti-Gr-1 antibody Rb6-8C5 on days 10 and 16 p.i. In the case of PMN contributing to killing of mycobacteria one would expect higher bacterial numbers upon PMN-depletion. Depletion efficacy was tested by blood smear, and on day 18 p.i. mycobacterial load was determined by CFU count. The bacterial load in lungs of B6 mice was significantly increased after PMN depletion (Fig. 5), indicating that PMN do contribute to control of mycobacteria as already observed earlier [16, 17]. In contrast to B6 mice, mycobacterial load in lungs of IL-18-deficient mice did not increase after PMN depletion but was slightly decreased, suggesting that PMN do not contribute to mycobacterial control in the absence of IL-18. Thus, in the absence of IL-18, PMN were recruited in greater numbers to infected lungs, but failed to restrict growth of M. tuberculosis. From these data we conclude that PMN primarily contributed to pathology of IL-18-deficient animals, probably promoting their premature death.
Figure 5.
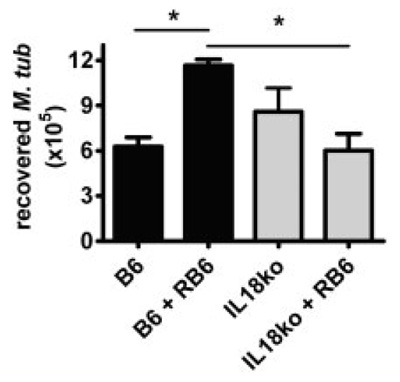
Polymorphonuclear neutrophil depletion has no impact on mycobacterial load in lungs of IL-18 KO mice. B6 WT and IL-18 KO mice were aerosol-infected with a low dose of M. tuberculosis H37Rv and depleted of PMN on day 10 and 16 p.i. by i.p. injection of anti-Gr-1 antibody (Rb6-8C5). On day 18, mice were sacrificed and lung lysates were plated in serial dilutions for CFU determination. Results from one out of three independent experiments are shown as mean numbers of recovered bacteria (n = 5)+SD. Statistical analysis was performed by ANOVA and Tukey’s Multiple Comparison test. *p<0.05.
Discussion
MyD88-deficient mice are highly susceptible to infection with M. tuberculosis [4, 11]. Inconsistencies regarding the importance of TLR [3-6, 8, 9] and shared usage of the adaptor molecule MyD88 by signalling cascades of TLR, IL-1R and IL-18R raised the question as to whether lack of IL-1 or IL-18 signals explain the high susceptibility of MyD88 KO mice to M. tuberculosis infection.
IL-18 induces IFN-γ production and cytotoxicity in T cells and NK cells and promotes surface expression of costimulatory and adhesion molecules as well as generation of reactive nitrogen intermediates (RNI) by MΦ and DC [18-20]. Mice deficient in IL-18 suffer from impaired IFN-γ production and MΦ derived from these animals fail to promote Th1 responses in vitro [21]. Accordingly, IL-18 is involved in protective immunity against a broad range of intracellular pathogens including Leishmania, Toxoplasma, and Salmonella [19]. Here we report that mice lacking IL-18 were highly susceptible to M. tuberculosis infection and succumbed more rapidly than WT B6 mice. Susceptibility correlated with reduced Th1 responses and PMN-dominated pathology. We propose that IL-18 plays a more critical role in promoting protective immunity against TB than previously noted [22].
Our observations of susceptibility of IL-18 KO mice to M. tuberculosis revealed that infection was more pronounced than in earlier reports. Sugawara et al. and Kinjo et al. [22, 23] found that, when compared with WT mice, IL-18 KO mice were slightly more susceptible to M. tuberculosis, but only late in infection. Similarly, IL-18R KO mice showed no striking defects in host resistance to M. tuberculosis [12]. This is in contrast to our result that IL-18 KO mice succumbed to infection with a virulent M. tuberculosis H37Rv strain very rapidly and much earlier than WT mice. Of note is that Sugawara et al. [24] also reported that susceptibility of MyD88 KO mice was only slightly more than in WT mice. This finding was recently revised by two studies [4, 11] demonstrating high susceptibility of MyD88 KO mice to M. tuberculosis. The discrepancy between our observations and earlier ones [22, 23] regarding susceptibility of IL-18 KO mice may be due to several factors including differences in genetic background of the mouse strains and virulence of the M. tuberculosis strains employed. The genetic background of the mice can contribute to differences in susceptibility as suggested by reports on L. major infection in IL-18 KO mice of distinct genetic background [25, 26].
In the absence of IL-18, M. tuberculosis failed to strongly induce IFN-γ as well as downstream effector molecules such as NOS-2, NO and IDO indicating a tissue environment less favourable of classical MΦ activation. MyD88 KO mice also showed reduced expression of these three genes in infected lung tissue whereas serum levels of IFN-γ and NO were comparable to WT and TLR-2/-4 DKO mice. NO is bactericidal against M. tuberculosis and its generation requires l-arginine as substrate [27]. Therefore, NO production is negatively affected by substrate depletion from arginase-1, a hallmark enzyme of AAMΦ. In both MyD88 and IL-18 KO mice, arginase-1 expression was enhanced in M. tuberculosis infected lungs when compared with WT and TLR-2/-4 KO mice. Moreover, unaltered levels of the Th2 cytokines IL-4 and IL-13 in lungs of M. tuberculosis-infected IL-18 KO mice suggest maintenance of Th2 responses while Th1 responses were reduced. However, production of IL-5, another Th2-associated cytokine, is diminished in IL-18 KO mice. This suggests that absence of IL-18 causes a more general suppression of cytokine production in M. tuberculosis-infected lungs and can also affect Th2 cytokines. In sum, the negative impact of IL-18 deficiency on Th1 responses, i.e. diminished production of IFN-γ and downstream effectors, is most relevant for the failure of IL-18 KO mice to control mycobacteria.
Arginase-1 has also been detected in PMN [28, 29]. Thus, the vast number of PMN in IL-18 KO lungs represents an additional or alternative source for arginase-1. Enhanced production of IL-17 in combination with CXCL-1 and CXCL-2, which have been described as strong inducers and/or attractors of PMN [30-32], was most probably responsible for the strong influx of PMN into IL-18 KO lungs upon M. tuberculosis infection. Notably, the pathological outcome of M. tuberculosis infection in IL-18 KO mice is distinct from the disease phenotype observed in MyD88 KO mice. IL-17 and CXCL-1/-2 were down-regulated in MyD88 KO mice. Accordingly, we did not observe a PMN-dominated pathology in their lungs.
The mycobactericidal functions of PMN remain controversial [33] but PMN depletion studies in mice deficient for T, B and NK cells revealed a residual function of PMN in anti-mycobacterial host responses [34]. As shown herein, depletion of PMN during the course of M. tuberculosis infection led to a significant increase in mycobacterial numbers in lungs of immune competent WT mice suggesting a role of PMN in anti-TB defence. However, albeit recruited to lungs of IL-18 KO mice in great numbers, PMN were ineffective against M. tuberculosis in the absence of IL-18 since PMN depletion had no impact on mycobacterial loads in these mice. Hence, in an IL-18-free environment PMN were not able to contribute to the control of M. tuberculosis but rather caused exacerbated immunopathology, which likely resulted in premature death.
Although IL-18 KO mice showed reduced expression of IFN-γ upon M. tuberculosis infection, T cells isolated from IL-18 KO lungs produced IFN-γ after in vitro re-stimulation with M. tuberculosis-derived peptides. This suggests that these T cells were functionally inhibited in the infected tissue probably due to the local cytokine environment in these mice. Both IL-18 and MyD88 KO lungs show diminished expression of TNF-β, which is directly involved in organization of granulomas in mycobacterial infection [13]. Furthermore, up-regulation of arginase-1 could result in a negative nitrogen balance within the lung tissue by depletion of arginine, which is an important micronutrient for T-cell proliferation and cytokine synthesis [29, 35]. The strong influx of metabolically active cells may further deplete micronutrient sources [36]. Therefore, the local environment in IL-18 KO lungs is probably responsible for diminished T-cell functions and cytokine production within infected tissues rather than a more general failure to generate Th1 T cells in IL-18-deficient mice.
MyD88 has been implicated in IFN-γ receptor signalling [37-39]. Recent publications revealed severely impaired IFN-γ responses in MyD88-deficient MΦ, including reduced NOS-2 expression and RNI production upon M. tuberculosis infection [4, 37]. Furthermore, MyD88-deficient MΦ were unable to kill intracellular M. tuberculosis upon IFN-γ activation [4] but supported intracellular replication, which was partially linked to the reduced NO production. The ability to respond to IFN-γ is not impaired in IL-18-deficient MΦ, distinguishing the IL-18- from MyD88-deficient phenotype in MΦ (data not shown). However, given that IFN-γ production was diminished in IL-18 KO mice it is likely that the MΦ were not fully activated in vivo and hence not able to restrict mycobacterial growth. Thus, whereas MyD88-deficient MΦ seem to have an intrinsic activation defect, MΦ in an IL-18- deficient environment are most likely not properly activated due to an unfavourable cytokine environment.
Since IL-18 KO mice were similarly susceptible to M. tuberculosis when compared with IL-1β/IL-18 KO mice, we imply overlapping functions of both cytokines in immunity to TB. This notion could also explain recently published data that revealed enhanced susceptibility of IL-1R KO mice [12] to M. tuberculosis when compared with WT mice. In contrast, mice lacking the IL-18R were able to control infection with M. tuberculosis. The resistance of IL-18R KO mice to M. tuberculosis infection was confirmed in our study. The discrepancy between the outcome of M. tuberculosis infection in mice lacking IL-18R versus those deficient of the cytokine is not understood so far but could be due to redundancy in receptor usage. This needs to be addressed in future experiments and goes beyond the scope of the present study. Thus, we suggest that both innate cytokine signals, IL-1β and IL-18 are essential for instructing protective immunity to M. tuberculosis.
In conclusion, our data demonstrate that IL-18 is important for protective immunity to M. tuberculosis infection.
Materials and methods
Bacterial strains and culture
M. tuberculosis H37Rv was used in all experiments. M. tuberculosis was grown in Middlebrook 7H9 broth (BD Biosciences, UK) supplemented with 0.5% glycerol and Tween 80, and Oleic acid-albumin-dextrose-catalase enrichment medium (BD Biosciences). Bacterial cultures were harvested, aliquoted and frozen at −80°C until later use. Viable cell counts in thawed aliquots were determined by plating serial dilutions of cultures onto Middlebrook 7H11 agar plates followed by incubation at 37°C.
Mice, infection and CFU
All mouse strains (C57BL/6, IL-18 KO, IL-18/IL-1βKO, TLR-2/-4 KO, MyD88 KO) were bred and housed under specific pathogenfree conditions either at the central animal facility of the Bundesinstitut für Risikobewertung (BfR, Berlin, Germany) or the Biological Service Facility of the London School of Hygiene and Tropical Medicine (London, UK). MyD88 KO, TLR-2 KO, TLR-4 KO and IL-18 KO mouse strains were kindly provided by Drs. Kiyoshi Takeda and Shizuo Akira, Osaka University, Japan. IL-1β KO mice were provided by D. Chaplin [40]. IL-18/IL-1β DKO and TLR-2/TLR-4 DKO mice were generated from the respective single KO strains as described [41, 42]. IL-18R KO were purchased from The Jackson Laboratory (USA). All KO strains were backcrossed onto the C57BL/6 genetic background.
In any given experiment, mice were between 6 and 12 wk old and matched for age and sex. For infection experiments, mice were maintained under specific barrier conditions in BSL 3 facilities. All experiments were performed in accordance with German Animal Protection Law or UK Home Office regulations.
For infection of experimental animals, M. tuberculosis stocks were diluted in sterile distilled water/1% v/v Tween-80/1% w/v BSA at a concentration providing an uptake of 100 viable bacilli per lung. Infection was performed via the respiratory route by using an aerosol chamber (Glas-Col, Terre-Haute, IN, USA). Animals were exposed for 60 min to an aerosol generated by nebulising the prepared M. tuberculosis suspension. Numbers of bacteria within the inoculum were determined at day 1 p.i. by plating complete lung homogenates onto Middlebrook 7H11 agar plates. Bacterial loads in lungs were evaluated at the time points indicated by mechanical disruption of the organs in water/1% w/v BSA/1% v/v Tween 80 (WTA), and plating serial dilutions onto Middlebrook 7H11 agar plates. For PMN depletion, mice were treated i.p. with anti-Gr-1 antibodies (Rb6-8C5, [14]) in PBS or with PBS alone on days 10 and 16 p.i.
Multiplex cytokine assays
Sera and lung lysates of infected mice were assayed for cytokine levels using a multiplex bead-based immunoassay kit (Bio-Plex Cytokine Assay, Bio-Rad) according to the manufacturer’s protocol.
qRT PCR
Total RNA was extracted from tissues using TRIzol reagent (Invitrogen, Karlsruhe, Germany) as recommended by the manufacturer. RNA from similarly treated animals was pooled and samples were treated with DNAse I (Gibco, Germany) to eliminate genomic DNA contamination. For semiquantitative real-time RT-PCR, 5 μg of DNAse I-digested RNA-pools were used for reverse transcription using 200 ng random hexamers as primers for Superscript III (Invitrogen) according to the manufacturer’s recommendations. The cDNA samples were loaded onto custom-made Taqman Low Density Arrays, which were then run in the ABI Prism 7900 Sequence Detection System according to manufacturer’s recommendations (Applied Biosystems, Foster city, CA, USA). Quantification was performed at least two times with independent cDNA samples and in duplicates for each cDNA and primer set. Data analysis was performed using the ABI Prism SDS Software (Applied Biosystems) and Excel (Microsoft).
Some samples were analysed in the ABI Prism 7000 Sequence Detection System (Applied Biosystems) using ABI PRISM optical 96-well plates (Applied Biosystems). Primers were designed to span large introns and to produce products sized between 100 and 200 bp (GAPDH 5′-AGGGCTCATGACCACAGTC-3′ and 5′-GGATGCAGGGATGATGTTCT-3′; NOS-2 5′-AGTTTCCAGAAGCAGAATGTGAC-3′ and 5′-GATGCTCCCAGACACTGGA-3′; Arginase 5′-ACCTGCTGGGAAGGAAGAA-3′ and 5′-AAGATAGGCCTCCCAGAACC-3′; IDO 5′-AGGCTGGCAAAGAATCTCCT-3′ and 5′-AATGACAAACTCACGGACTGG-3′; Ym-1 5′-GGACTCCTGGCTTACTATGAGGT-3′ and 5′-CAACTTGAAGCTCCTGACATTG-3′). Reaction mixtures were set up to a final volume of 25 μL using 300 nmol of each primer and 12.5 μL of SYBR Green PCR Master mix (Applied Biosystems). Quantification was performed at least two times with independent cDNA samples and in triplicates for each cDNA and primer pair.
The threshold cycle for 18S RNA (Taqman) or GAPDH RNA (SYBR Green) was determined and subtracted from the threshold cycle of the analysed primerset (correction for different amounts of cDNA). Resulting cycle differences (ΔCt) measured for one cDNA sample were subtracted from cycle difference measured for the cDNA sample defined as standard (WT). The resulting ΔΔCt was used to calculate fold differences relative to expression level of cDNA in one sample compared with untreated (fold difference = 2−ΔCt).
NO assay
NO was determined in sera from infected mice as NO2 following reduction of NO3, using the Griess-reaction described previously [43].
Histology and immunohistochemistry
Organs from M. tuberculosis-infected mice were fixed with 4% w/v Paraformaldehyde for 24 h and embedded in paraffin. Sections (3 μm) were rehydrated by running through xylenes, alcohols of decreasing concentrations and finally water. Sections were stained with H&E. Mycobacteria in lungs were visualized by acid-fast staining (TB Stain Kit K, BD 212522) according to the manufacturer’s protocol. For specific antibody staining, fixed and rehydrated tissue sections were subjected to antigen retrieval via immersion in 10 mM citric acid pH 6.0 for 5 min in a pressure cooker followed by slow cooling. Sections were blocked with goat serum in TBS and exposed to anti-PMN monoclonal antibody (MCA771G, Serotec, 1:100 in TBS), followed by the secondary antibody goat anti-rat-AP (3010-04, Southern Biotechnology, Birmingham, Alabama, USA1:100 in TBS) for 1 h at RT. Alkaline phosphatase was visualized by using fuchsin (DAKO Fuchsin+Substrate-Chromogen System, K0625) as substrate. Sections were counterstained with hematoxylin. Stained sections were thoroughly washed with water and mounted in Aquatex (Merck Darmstadt, Germany).
Purification of cells from different tissues
Single-cell suspensions of spleens were prepared using an iron mesh sieve and erythrocyte lysis. Cells were washed twice with RPMI 1640 medium supplemented with 2 mM glutamine, 1 mM Na-pyruvate, β-mercaptoethanol (50 μM), 1% v/v penicillin, 1% v/v streptomycin and 10% v/v heat-inactivated fetal calf serum (complete RPMI 1640 medium). Lungs were perfused with PBS via the right ventricle, removed and homogenized using an iron mesh sieve. Remaining erythrocytes were lysed and lymphocytes were purified using a 40%/70% v/v Percoll gradient. Subsequently, cells were washed in complete RPMI 1640 medium.
In vitro re-stimulation of cells and flow cytometric determination of cytokine expression
T-cell responses in lung and spleen were determined by intracellular IFN-γ staining [44]. Cells (1–3 × 106) were cultured in a volume of 1 mL complete RPMI medium and stimulated for 5 h with a combination of the peptides Ag85A241–260 (QDAYNAGGGHNGVFDFPDSG) and Ag85B240–260 (FQDAYNAAGG HNAVFNFPPNG) both at 10 μM or with 1 μM of the peptide Mtb32/RV0125309–318 (GAPINSATAM) [45, 46]. During the final 4 h of culture, 10 μg/mL Brefeldin A was added. Cultured cells were washed with cold PBS, and incubated for 5 min in PBS/0.1% BSA with rat serum and anti-CD16/CD32 mAb (1 mg/mL stock, 1:100) to block nonspecific antibody binding. Subsequently, cells were stained with PE-, PerCP- or PECy7-conjugated anti-CD8α mAb and anti-CD4 mAb (all BD Pharmingen BD, Oxford, UK), and after 15 min on ice, cells were washed with PBS and fixed for 15 min at RT with PBS/4% paraformaldehyde. Cells were washed with PBS/0.1% BSA, permeabilized with PBS/0.1%BSA/0.5% saponin, and incubated in this buffer with rat serum and anti-CD16/CD32 mAb. After 5 min, FITC-conjugated anti-IFN-γ mAb and Cy5-conjugated anti-CD154/CD40L mAb were added [47]. After a further 15 min at RT, cells were washed with PBS and fixed with PBS/1% paraformaldehyde. Cells were analysed using a BD FACSCanto flow cytometer and BD FACSDiva software (Becton Dickinson, Mountain View, CA, USA). We routinely acquired 50 000–100 000 lymphocyte-gated CD4+ or CD8+ T cells from each sample.
Anti-CD16/CD32 mAb (2.4G2), anti-IFN-γ mAb (XMG1.2), and anti-CD40L mAb (MR1) were purified from hybridoma supernatants with Protein G sepharose. Antibodies (1 mg/mL) were Cy5- or FITC-conjugated according to standard protocols.
Statistical analysis
Statistical analysis was performed by Student’s t-test or by ANOVA followed by Tukey’s Multiple Comparison Test as described in the figure legends. Statistical analysis of survival curves was performed using the log rank test.
Acknowledgements
The authors would like to thank Dr. John Raynes for critically reading the manuscript. This work was funded through a grant by the Medical Research Council, UK, to U.E.S. (Grant no. G0700163).
Abbreviations
- AAMΦ
alternatively activated MΦ
- DKO
double KO
- NOS
nitric oxide synthase
- p.i.
post infection
- qRT PCR
quantitative RT PCR
- TB
tuberculosis
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.WHO . WHO Report - Global tuberculosis control. World Health Organization; Geneva: 2008. [Google Scholar]
- 2.Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J. Pathol. 2006;208:261–269. doi: 10.1002/path.1906. [DOI] [PubMed] [Google Scholar]
- 3.Shim TS, Turner OC, Orme IM. Toll-like receptor 4 plays no role in susceptibility of mice to Mycobacterium tuberculosis infection. Tuberculosis (Edinb.) 2003;83:367–371. doi: 10.1016/s1472-9792(03)00071-4. [DOI] [PubMed] [Google Scholar]
- 4.Holscher C, Reiling N, Schaible UE, Holscher A, Bathmann C, Korbel D, Lenz I, et al. Containment of aerogenic Mycobacterium tuberculosis infection in mice does not require MyD88 adaptor function for TLR2, -4 and -9. Eur. J. Immunol. 2008;38:680–694. doi: 10.1002/eji.200736458. [DOI] [PubMed] [Google Scholar]
- 5.Shi S, Blumenthal A, Hickey CM, Gandotra S, Levy D, Ehrt S. Expression of many immunologically important genes in Mycobacterium tuberculosis-infected macrophages is independent of both TLR2 and TLR4 but dependent on IFN-alphabeta receptor and STAT1. J. Immunol. 2005;175:3318–3328. doi: 10.4049/jimmunol.175.5.3318. [DOI] [PubMed] [Google Scholar]
- 6.Sugawara I, Yamada H, Li C, Mizuno S, Takeuchi O, Akira S. Mycobacterial infection in TLR2 and TLR6 knockout mice. Microbiol. Immunol. 2003;47:327–336. doi: 10.1111/j.1348-0421.2003.tb03404.x. [DOI] [PubMed] [Google Scholar]
- 7.Drennan MB, Nicolle D, Quesniaux VJ, Jacobs M, Allie N, Mpagi J, Fremond C, et al. Toll-like receptor 2-deficient mice succumb to Mycobacterium tuberculosis infection. Am. J. Pathol. 2004;164:49–57. doi: 10.1016/S0002-9440(10)63095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bafica A, Scanga CA, Feng CG, Leifer C, Cheever A, Sher A. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J. Exp. Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abel B, Thieblemont N, Quesniaux VJ, Brown N, Mpagi J, Miyake K, Bihl F, Ryffel B. Toll-like receptor 4 expression is required to control chronic Mycobacterium tuberculosis infection in mice. J. Immunol. 2002;169:3155–3162. doi: 10.4049/jimmunol.169.6.3155. [DOI] [PubMed] [Google Scholar]
- 10.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 11.Fremond CM, Yeremeev V, Nicolle DM, Jacobs M, Quesniaux VF, Ryffel B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J. Clin. Invest. 2004;114:1790–1799. doi: 10.1172/JCI21027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fremond CM, Togbe D, Doz E, Rose S, Vasseur V, Maillet I, Jacobs M, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J. Immunol. 2007;179:1178–1189. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 13.Roach DR, Briscoe H, Saunders B, France MP, Riminton S, Britton WJ. Secreted lymphotoxin-alpha is essential for the control of an intracellular bacterial infection. J. Exp. Med. 2001;193:239–246. doi: 10.1084/jem.193.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kahnert A, Seiler P, Stein M, Bandermann S, Hahnke K, Mollenkopf H, Kaufmann SH. Alternative activation deprives macrophages of a coordinated defense program to Mycobacterium tuberculosis. Eur. J. Immunol. 2006;36:631–647. doi: 10.1002/eji.200535496. [DOI] [PubMed] [Google Scholar]
- 15.Flesch IE, Kaufmann SH. Activation of tuberculostatic macrophage functions by gamma interferon, interleukin-4, and tumor necrosis factor. Infect. Immun. 1990;58:2675–2677. doi: 10.1128/iai.58.8.2675-2677.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fulton SA, Reba SM, Martin TD, Boom WH. Neutrophil-mediated mycobacteriocidal immunity in the lung during Mycobacterium bovis BCG infection in C57BL/6 mice. Infect. Immun. 2002;70:5322–5327. doi: 10.1128/IAI.70.9.5322-5327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seiler P, Aichele P, Raupach B, Odermatt B, Steinhoff U, Kaufmann SH. Rapid neutrophil response controls fast-replicating intracellular bacteria but not slow-replicating Mycobacterium tuberculosis. J. Infect. Dis. 2000;181:671–680. doi: 10.1086/315278. [DOI] [PubMed] [Google Scholar]
- 18.Reddy P. Interleukin-18: recent advances. Curr. Opin. Hematol. 2004;11:405–410. doi: 10.1097/01.moh.0000141926.95319.42. [DOI] [PubMed] [Google Scholar]
- 19.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001;19:423–474. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- 20.Maxwell JR, Yadav R, Rossi RJ, Ruby CE, Weinberg AD, Aguila HL, Vella AT. IL-18 bridges innate and adaptive immunity through IFN-gamma and the CD134 pathway. J. Immunol. 2006;177:234–245. doi: 10.4049/jimmunol.177.1.234. [DOI] [PubMed] [Google Scholar]
- 21.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 22.Sugawara I, Yamada H, Kaneko H, Mizuno S, Takeda K, Akira S. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 1999;67:2585–2589. doi: 10.1128/iai.67.5.2585-2589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinjo Y, Kawakami K, Uezu K, Yara S, Miyagi K, Koguchi Y, Hoshino T, et al. Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: a comparative study with IL-12p40. J. Immunol. 2002;169:323–329. doi: 10.4049/jimmunol.169.1.323. [DOI] [PubMed] [Google Scholar]
- 24.Sugawara I, Yamada H, Mizuno S, Takeda K, Akira S. Mycobacterial infection in MyD88-deficient mice. Microbiol. Immunol. 2003;47:841–847. doi: 10.1111/j.1348-0421.2003.tb03450.x. [DOI] [PubMed] [Google Scholar]
- 25.Xu D, Trajkovic V, Hunter D, Leung BP, Schulz K, Gracie JA, McInnes IB, Liew FY. IL-18 induces the differentiation of Th1 or Th2 cells depending upon cytokine milieu and genetic background. Eur. J. Immunol. 2000;30:3147–3156. doi: 10.1002/1521-4141(200011)30:11<3147::AID-IMMU3147>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Wei XQ, Niedbala W, Xu D, Luo ZX, Pollock KG, Brewer JM. Host genetic background determines whether IL-18 deficiency results in increased susceptibility or resistance to murine Leishmania major infection. Immunol. Lett. 2004;94:35–37. doi: 10.1016/j.imlet.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Chan ED, Chan J, Schluger NW. What is the role of nitric oxide in murine and human host defense against tuberculosis?Current knowledge. Am. J. Respir. Cell Mol. Biol. 2001;25:606–612. doi: 10.1165/ajrcmb.25.5.4487. [DOI] [PubMed] [Google Scholar]
- 28.Munder M, Mollinedo F, Calafat J, Canchado J, Gil-Lamaignere C, Fuentes JM, Luckner C, et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood. 2005;105:2549–2556. doi: 10.1182/blood-2004-07-2521. [DOI] [PubMed] [Google Scholar]
- 29.Munder M, Schneider H, Luckner C, Giese T, Langhans CD, Fuentes JM, Kropf P, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006;108:1627–1634. doi: 10.1182/blood-2006-11-010389. [DOI] [PubMed] [Google Scholar]
- 30.Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J. Immunol. 2003;170:4665–4672. doi: 10.4049/jimmunol.170.9.4665. [DOI] [PubMed] [Google Scholar]
- 31.Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, Skoogh BE, Linden A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J. Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 32.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 33.Korbel DS, Schneider BE, Schaible UE. Innate immunity in tuberculosis: myths and truth. Microbes Infect. 2008;10:995–1004. doi: 10.1016/j.micinf.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 34.Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, Wynn TA, Sher A. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J. Immunol. 2006;177:7086–7093. doi: 10.4049/jimmunol.177.10.7086. [DOI] [PubMed] [Google Scholar]
- 35.Bernard A, Kasten M, Meier C, Manning E, Freeman S, Adams W, Chang P, et al. Red blood cell arginase suppresses Jurkat (T cell) proliferation by depleting arginine. Surgery. 2008;143:286–291. doi: 10.1016/j.surg.2007.07.037. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, Ochoa AC. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J. Immunol. 2003;171:1232–1239. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 37.Shi S, Nathan C, Schnappinger D, Drenkow J, Fuortes M, Block E, Ding A, et al. MyD88 primes macrophages for full-scale activation by interferon-gamma yet mediates few responses to Mycobacterium tuberculosis. J. Exp. Med. 2003;198:987–997. doi: 10.1084/jem.20030603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han J. MyD88 beyond Toll. Nat. Immunol. 2006;7:370–371. doi: 10.1038/ni0406-370. [DOI] [PubMed] [Google Scholar]
- 39.Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat. Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 40.Shornick LP, De Togni P, Mariathasan S, Goellner J, Strauss-Schoenberger J, Karr RW, Ferguson TA, Chaplin DD. Mice deficient in IL-1beta manifest impaired contact hypersensitivity to trinitrochlorobenzone. J. Exp. Med. 1996;183:1427–1436. doi: 10.1084/jem.183.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weiss DS, Raupach B, Takeda K, Akira S, Zychlinsky A. Toll-like receptors are temporally involved in host defense. J. Immunol. 2004;172:4463–4469. doi: 10.4049/jimmunol.172.7.4463. [DOI] [PubMed] [Google Scholar]
- 43.Bancroft GJ, Collins HL, Sigola LB, Cross CE. Modulation of murine macrophage behavior in vivo and in vitro. Methods Cell Biol. 1994;45:129–146. doi: 10.1016/s0091-679x(08)61849-x. [DOI] [PubMed] [Google Scholar]
- 44.Mittrucker HW, Steinhoff U, Kohler A, Krause M, Lazar D, Mex P, Miekley D, Kaufmann SH. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc. Natl. Acad. Sci. USA. 2007;104:12434–12439. doi: 10.1073/pnas.0703510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Irwin SM, Izzo AA, Dow SW, Skeiky YA, Reed SG, Alderson MR, Orme IM. Tracking antigen-specific CD8 T lymphocytes in the lungs of mice vaccinated with the Mtb72F polyprotein. Infect. Immun. 2005;73:5809–5816. doi: 10.1128/IAI.73.9.5809-5816.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.D’Souza S, Rosseels V, Romano M, Tanghe A, Denis O, Jurion F, Castiglione N, et al. Mapping of murine Th1 helper T-Cell epitopes of mycolyl transferases Ag85A, Ag85B, and Ag85C from Mycobacterium tuberculosis. Infect. Immun. 2003;71:483–493. doi: 10.1128/IAI.71.1.483-493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frentsch M, Arbach O, Kirchhoff D, Moewes B, Worm M, Rothe M, Scheffold A, Thiel A. Direct access to CD4+ T cells specific for defined antigens according to CD154 expression. Nat. Med. 2005;11:1118–1124. doi: 10.1038/nm1292. [DOI] [PubMed] [Google Scholar]