

Abstract
The mechanism of channel opening for voltage-gated calcium channels is poorly understood. The importance of a conserved isoleucine residue in the pore-lining segment IIS6 has recently been highlighted by functional analyses of a mutation (I745T) in the CaV1.4 channel causing severe visual impairment (Hemara-Wahanui, A., Berjukow, S., Hope, C. I., Dearden, P. K., Wu, S. B., Wilson-Wheeler, J., Sharp, D. M., Lundon-Treweek, P., Clover, G. M., Hoda, J. C., Striessnig, J., Marksteiner, R., Hering, S., and Maw, M. A. (2005) Proc. Natl. Acad. Sci. U. S. A. 102, 7553–7558). In the present study we analyzed the influence of amino acids in segment IIS6 on gating of the CaV1.2 channel. Substitution of Ile-781, the CaV1.2 residue corresponding to Ile-745 in CaV1.4, by residues of different hydrophobicity, size and polarity shifted channel activation in the hyperpolarizing direction (I781P > I781T > I781N > I781A > I781L). As I781P caused the most dramatic shift (−37 mV), substitution with this amino acid was used to probe the role of other residues in IIS6 in the process of channel activation. Mutations revealed a high correlation between the midpoint voltages of activation and inactivation. A unique kinetic phenotype was observed for residues 779–782 (LAIA) located in the lower third of segment IIS6; a shift in the voltage dependence of activation was accompanied by a deceleration of activation at hyperpolarized potentials, a deceleration of deactivation at all potentials (I781P and I781T), and decreased inactivation. These findings indicate that Ile-781 substitutions both destabilize the closed conformation and stabilize the open conformation of CaV1.2. Moreover there may be a flexible center of helix bending at positions 779–782 of CaV1.2. These four residues are completely conserved in high voltage-activated calcium channels suggesting that these channels may share a common mechanism of gating.
The entry of Ca2+ through voltage-gated Ca2+ channels has direct effects on muscle contraction, release of hormones and neurotransmitters, hearing, vision, gene expression, and other important physiological functions (2). The pore-forming α1-subunits of voltage-gated Ca2+ channels are composed of four homologous domains formed by six transmembrane segments (S1–S6) that are linked together on a single polypeptide (3). A membrane depolarization initiates channel openings (activation) and closures (inactivation). These events can be considered a multistep process consisting of a conformational change in the voltage sensor, a transmission of the signal to the pore region, the opening of the pore, and channel closure due to inactivation. The voltage-sensing machinery is formed by multiple charged amino acids located in segment S4 and adjacent structures of each domain (4). A large number of amino acids involved in Ca2+ channel inactivation have been identified and several molecular mechanisms for this process have been proposed (for reviews see Refs. 5-7).
The molecular mechanism of the voltage-dependent pore opening of Ca2+ channels, however, is less studied and largely unknown. The first attempt to localize the structural elements in Ca2+ channel α1-subunits that are involved in channel activation was made by Tanabe et al. (8) who constructed chimeric channels in which sequence stretches of a slow activating (“skeletal muscle-like”) CaV1.1 α1-subunit were replaced by sequences from a fast activating (“cardiac-like”) CaV1.2 α1-subunit. The chimeras activated slowly if repeat I of the CaV1.2 α1-subunit was replaced by the CaV1.1 α1-sequence. In a later study, replacement of domains I, II, and III of the low voltage and fast activating CaV3.1 α1-subunit with the corresponding domains of the high voltage-activated CaV1.2 α1-subunit resulted in a high voltage-activated channel (9). An important role of domains I and III but not II and IV on midpoint voltage and time constants of activation was reported by Garcia et al. (10) who mutated the arginines in the S4 segments of all four domains of a chimeric channel to neutral or negative amino acids. The removal of prolines that are conserved in segments IS4 and IIIS4 of voltage-gated Ca2+ channels resulted in shortening of channel open time, whereas introduction of extra prolines to corresponding positions of IIS4 and IVS4 lengthened the channel open time (11).
Our present study was initiated by the recent finding that a novel retinal disorder is caused by a point mutation (I745T) in segment IIS6 of the CaV1.4 α1-subunit that shifts the voltage dependence of CaV1.4 channel activation by approximately −30 mV (1, 12). As CaV1.4 channels express only at low density in mammalian cell lines (13) we have decided to study the functional roles of this residue and neighboring residues in segment IIS6 by introducing and characterizing mutations in the homologous CaV1.2 channel. Our findings demonstrate that residue Ile-781 and three neighboring residues (Leu-779, Ala-780, and Ala-782) play a key role in gating of the CaV1.2 channel.
EXPERIMENTAL PROCEDURES
Mutagenesis
The CaV1.2 α1-subunit coding sequence (GenBank™ X15539) in-frame 3′ to the coding region of a modified green fluorescent protein (GFP)4 was kindly donated by Dr. M. Grabner (14). For electrophysiological studies we used the plasmid lacking the GFP tag. Substitutions in segment IIS6 of the CaV1.2 α1-subunit were introduced by the “gene SOEing” technique (15). In particular, an isoleucine to threonine mutation was introduced in position 781, and further substitutions were made at this position to proline, leucine, alanine, asparagine, glutamine, and arginine. The mutated fragments were cloned into a BamHI-AflII-cassette (nucleotides 1265 and 2689, numbering according to the CaV1.2 α1-subunit coding sequence). This cassette was also used in creation of C769P, G770P, N771P, Y772P, I773P, L774P, L775P, N776P, V777P, F778P, L779P, A780P, A782P, and V783P. Mutations that did not lead to functional channels were recloned into the GFP-tagged vector (14) to study membrane targeting. All constructs were checked by restriction site mapping and sequencing.
Cell Culture and Transient Transfection
Human embryonic kidney tsA-201 cells were grown at 5% CO2 and 37 °C to 80% confluence in Dulbecco’s modified Eagle’s/F-12 medium supplemented with 10% (v/v) fetal calf serum and 100 units/ml penicillin/streptomycin. Cells were split via trypsin EDTA and plated on 35-mm Petri dishes (Falcon) at 30–50% confluence ~16 h before transfection. Subsequently tsA-201 cells were co-transfected with cDNAs encoding wild-type or mutant CaV1.2 α1-subunits with auxiliary β1a (16) or β2a (17) as well as α2-δ1-subunits (18). The transfection of tsA-201 cells was performed using the FuGENE 6 transfection reagent (Roche Applied Science) following standard protocols.
Ionic Current Recordings and Data Acquisition
Barium currents (IBa) through voltage-gated Ca2+ channels were recorded at 22–25 °C using the patch clamp technique (19) by means of an Axopatch 200A patch clamp amplifier (Axon Instruments) 36–48 h after transfection. The extracellular bath solution contained BaCl2 5 mm, MgCl2 1 mm, HEPES 10 mm, and choline-Cl 140 mm, titrated to pH 7.4 with methanesulfonic acid. Patch pipettes with resistances of 1–4 megohms were made from borosilicate glass (Clark Electromedical Instruments) and filled with pipette solution containing CsCl 145 mm, MgCl2 3 mm, HEPES 10 mm, and EGTA 10 mm, titrated to pH 7.25 with CsOH. All data were digitized using a DIGIDATA 1200 interface (Axon Instruments), smoothed by means of a four-pole Bessel filter and stored on computer hard disc. 300- and 100-ms current traces were sampled at 10 kHz and filtered at 5 kHz, for the steady state inactivation protocol, currents were sampled at 1 kHz and filtered at 0.5 kHz, and tail currents were sampled at 50 kHz and filtered at 10 kHz. Leak currents were subtracted digitally using average values of scaled leakage currents elicited by a 10 mV hyperpolarizing pulse or electronically by means of an Axopatch 200 amplifier. Series resistance and offset voltage were routinely compensated. The pClamp software package (Version 7.0 Axon Instruments, Inc.) was used for data acquisition and preliminary analysis. Microcal Origin 5.0 was employed for analysis and curve fitting.
The voltage dependence of activation was determined from current-voltage (I-V) curves that were fitted according to the following modified Boltzmann term: I = Gmax (V – Vrev)/(1 + exp[(V0.5,act – V)/kact]) where VreV is extrapolated reversal potential, V is membrane potential, I is peak current, Gmax is maximum membrane conductance, V0.5,act is voltage for half-maximal activation, and kact is slope factor. The time course of current activation was fitted to a mono-exponential function: I(t) = A[exp(−t/τ)] + C; where I(t) is current at time t, A is the amplitude coefficient, τ is a time constant, and C is steady state current. The voltage dependence of IBa inactivation (inactivation curve) was measured using a multistep protocol to account for run-down (see Ref. 1). The pulse sequence was applied every 40 s from a holding potential of −100 mV. Inactivation curves were drawn according to a Boltzmann equation: IBa,inact = Iss + (1 – Iss)/(1 + exp[(V – V 0.5,inact.)/k)] where V is membrane potential, V0.5,inact is midpoint voltage, k is the slope factor, and Iss is the fraction of non-inactivating current. Data are given as mean ± S.E. Statistical significance was assessed with the Student’s unpaired t test.
Confocal Imaging
The confocal images were obtained ~30 h after transfection. Data illustrated are representative for 15–20 tsA-201 cells from three independent experiments. Confocal images were acquired with a Zeiss LSM-510 confocal laser scanning microscope, using a 63× (1.4 NA) oil immersion objective. The plasma membrane was stained with 1 μm FM4-64 (amphiphilic styryl dye, Molecular Probes). Images were acquired using an argon laser (excitation, 488 nm; emission BP505–530 nm emission filter) for the GFP-tagged CaV1.2 α1-subunits and a He-Ne laser (excitation, 543 nm; emission filter, LP650 nm) for FM4-64.
RESULTS
Mutation I781T Shifts CaV1.2 Channel Activation to More Hyperpolarized Voltages
Residue Ile-745 in segment IIS6 of the CaV1.4 α1-subunit corresponds to residue Ile-781 in the CaV1.2 α1-subunit (Fig. 1A). The effects of I781T substitution on CaV1.2 channel activation were investigated after expressing wild-type or mutant CaV1.2 α1-subunits together with auxiliary β1a and α2-δ1-subunits in tsA-201 cells. Families of inward Ba2+ currents (Fig. 1B) and the corresponding current-voltage (I-V) curves (Fig. 1C) and voltage dependences of channel activation (Fig. 2A) and inactivation (Fig. 2B) are shown. For both wild-type and I781T channels, the current reversed at ~50 mV indicating that the I781T mutation does not affect ion selectivity (Fig. 1C). The midpoint voltage of activation changed from −9.9 ± 1.1 mV in the wild-type channel to −37.7 ± 1.2 mV in the I781T channel (Fig. 2A). I781T also shifted the voltage dependence of inactivation. The midpoint voltage of the inactivation curve changed from −38.7 ± 1.0 mV in wild-type channels to −57.8 ± 0.7 mV in I781T channels (Fig. 2, B and C). Wild-type channels were inactivated by 65 ± 4%, whereas I781T channels were inactivated by 47 ± 3% during a 300-ms pulse at the peak of their respective I-V curves (0 mV, −30 mV; Fig. 1B, TABLE ONE).
FIGURE 1. Functional analysis of I781T, the CaV1.2 mutant corresponding to I745T CaV1.4.
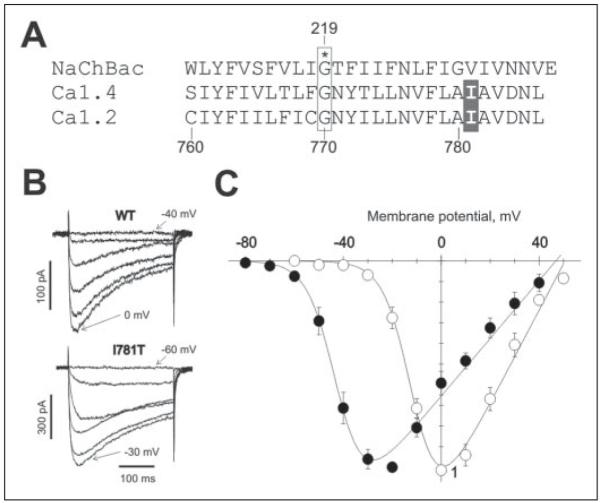
A, aligned pore-lining sequences of NaChBac, a bacterial sodium channel from Bacillus halodurance, and IIS6 segments of the L-type channels CaV1.2 and CaV1.4. The residue analogous to a conserved glycine in the M2 helix of the potassium channel MthK (31) is boxed, while the isoleucine residue of interest to the present study is shaded. B, representative families of IBa through wild-type (top) and I781T mutant (bottom) channels during depolarizing test pulses from −100 mV (threshold and maximal voltages are indicated, 10-mV increments). Wild-type or I781T mutant CaV1.2 α1-subunits were coexpressed together with β1a and α2-δ1-subunits. C, averaged current-voltage relationships of the wild-type (n = 8, open circles) and I781T mutant (n = 7, filled circles) channels.
FIGURE 2. Ile-781 substitutions shift the voltage dependences of CaV1.2 channel activation and inactivation.

A, averaged voltage dependences of activation of wild-type (n = 8), I781P (n = 8), I781T (n = 7), I781A (n = 7), I781L (n = 5), and I781N (n = 4) channels. B, averaged voltage dependences of steady state inactivation for wild-type (n = 3), I781P (n = 5), and I781T (n = 3). Solid lines represent fits to Boltzmann functions. C, correlation between potentials of half-maximal activation (V0.5,act) and half-maximal inactivation (V0.5,inact) of the Ile-781 mutants. Linear regression yielded a correlation coefficient of 0.97.
TABLE ONE.
The influence of IIS6 pore mutations on voltage-dependent gating of CaV1.2
| Half-activation | Slope | r 300 | |
|---|---|---|---|
| mV | mV | % | |
| Co-expression of β1a | |||
| Wild-type | −9.9 ± 1.1 | 6.3 ± 0.7 | 65 ± 4 |
| I781T | −37.7 ± 1.2 | 7.2 ± 1.0 | 47 ± 3 |
| I781P | −47.2 ± 1.1 | 6.3 ± 0.6 | 53 ± 6 |
| I781L | −20.6 ± 1.2 | 6.1 ± 0.2 | 63 ± 7 |
| I781A | −29.5 ± 0.6 | 7.9 ± 0.5 | 51 ± 4 |
| I781N | −34.6 ±1.6 | 9.1 ± 0.4 | 37 ± 6 |
| I781Q | No currents | ||
| I781R | No currents | ||
| C769P | 2.8 ± 0.8 | 4.9 ± 1.0 | 67 ± 7 |
| G770P | −11.4 ± 1.3 | 5.8 ± 0.3 | 62 ± 4 |
| N771P | −26.9 ± 1.1 | 4.9 ± 0.2 | 43 ± 4 |
| Y772P | No currents | ||
| I773P | −14.2 ± 1.8 | 2.9 ± 0.6 | 65 ± 6 |
| L774P | No currents | ||
| L775P | No currents | ||
| N776P | No currents | ||
| V777P | No currents | ||
| F778P | −9.3 ± 0.8 | 7.7 ± 0.3 | 23 ± 3 |
| L779P | −29.78 ± 1.90 | 6.0 ± 0.4 | 3 ± 1 |
| A780P | −37.1 ± 0.7 | 5.7 ± 0.8 | 2 ± 1 |
| A782P | −35.7 ± 1.4 | 9.8 ± 0.3 | 30 ± 5 |
| V783P | No currents | ||
| Co-expression of β2a | |||
| Wild-type | −13.8 ± 0.9 | 6.0 ± 0.5 | 16 ± 5 |
| I781T | −44.9 ± 1.4 | 8.8 ± 1.0 | 25 ± 4 |
| I781P | −52.3 ± 1.3 | 7.3 ± 0.8 | 27 ± 6 |
Midpoints and slope factors of the activation curves and amount of channel inactivation during a 300-ms pulse (r300) to the peak potentials of the I–V curve.
The Voltage Dependence of CaV1.2 Gating Was Also Shifted by Other Ile-781 Substitutions
The data presented in Figs. 1 and 2 demonstrate that Ile-781 plays a pivotal role in CaV1.2 gating. To gain insight into the structural requirements at this position, we subsequently replaced Ile-781 by residues of different size, polarity and charge. Replacement of Ile-781 by leucine, alanine, asparagine, and proline caused a shift in channel activation ranging from approximately −10 mV for I781L to approximately −37 mV for I781P (TABLE ONE). The same mutations also shifted the channel inactivation curves by values ranging from approximately −6 mV (I781L) to approximately −30 mV (I781P) (Fig. 2, B and C). Interestingly, the changes in the voltage dependences of channel activation and inactivation occurred in parallel. This is evident from Fig. 2C where the midpoint voltages of the activation curves are plotted versus the midpoint voltages of the inactivation curves. Fitting the data with a regression function revealed a correlation coefficient of 0.97.
Ile-781 Substitutions Showed Slow Activation Gating at Hyperpolarized Voltages and Slow Deactivation
The voltage dependences of the activation time constants for the Ile-781 substitution mutants were also determined (Fig. 3). Between −30 mV and +30 mV, activation of the wild-type and mutant channels occurred with a similar time course (wild-type, τm ranged between 3.2 ms (−30 mV) and 2.5 ms (30 mV); I781P, τm ranged between 3.9 ms (−30 mV) and 2.1 ms (10 mV) (Fig. 3B)). At voltages negative to the threshold potential of wild-type CaV1.2 activation (−40 mV, Fig. 2A), mutant channels displayed decelerated activation kinetics (Fig. 3). At −50 mV, no channel activation was observed in wild-type and I781L (Fig. 3A), whereas the four remaining mutant channels activated at comparatively slow rates (see Fig. 3B for time constants). Moreover, activation of I781P at −60 mV was even slower (τm ~25 ms). In other words, the more the voltage dependence of channel activation was shifted (TABLE ONE), the more slowly the channels activated at hyperpolarized voltages (Fig. 3B).
FIGURE 3. Ile-781 substitutions affect the activation kinetics of CaV1.2.
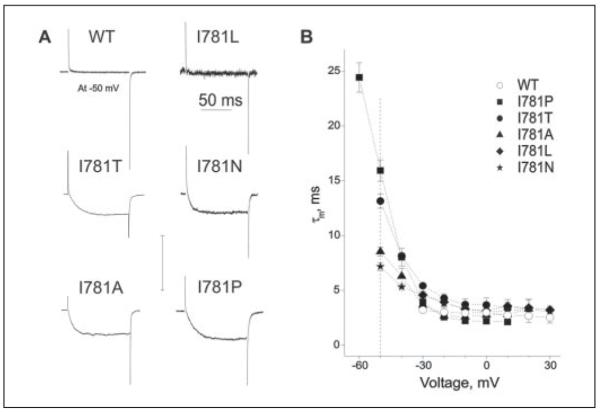
A, representative traces of IBa through wild-type and Ile-781 mutant channels. IBa were evoked during depolarizations from the holding potential of −100 mV to −50 mV (see dashed line in B). The activation time constants (τm) were obtained by fitting the activation phase of IBa to a mono-exponential function (I781T, 14.0 ms; I781N, 6.9 ms; I781A, 8.6 ms; and I781P, 14.8 ms). B, voltage dependence of mean time constants of activation for wild-type channels (n = 8, open circles) in comparison to I781L (n = 5, filled diamonds), I781T (n = 7, filled circles), I781N (n = 4, stars), I781A (n = 6, filled triangles) and I781P (n = 8, filled squares) mutant channels.
To gain insight into the stability of the open channel conformation, we also analyzed the voltage dependence of deactivation for I781P and I781T, the mutants that induced the strongest effects on channel activation. The two mutant channels deactivated more slowly than the wild-type channel at all potentials with deactivation of I781P being slower than that of I781T (Fig. 4).
FIGURE 4. Mutations I781P and I781T slowed the deactivation kinetics of CaV1.2.
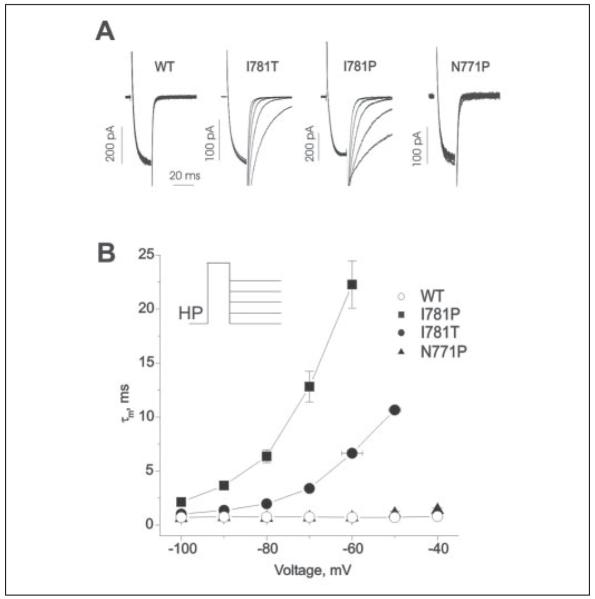
A, representative tail currents for wild-type, I781T, I781P, and N771P channels. Currents were activated during a 20-ms conditioning depolarization to 0 mV for wild-type, −40 mV for I781P, −30 mV for I781T, and −20 mV for N771P. Deactivation was recorded during subsequent repolarizations with 10-mV increments starting from −100 mV (inset). B, mean time constants of channel deactivation for wild-type, I781P, I781T, and N771P mutant CaV1.2 channels are plotted versus test potential. Time constants were estimated by fitting current deactivation to a mono-exponential function (see “Experimental Procedures”).
Proline Substitution of Gly-770
A similar kinetic phenotype to that observed for the I781T substitution (slow deactivation, slow activation at hyperpolarized voltages, and decelerated inactivation) was previously reported for a bacterial sodium channel (NaChBac) when a highly conserved glycine (Gly-219) in the central third of S6 was substituted by proline (20). To evaluate an analogous role of Gly-770 in the corresponding position of segment IIS6 of CaV1.2 (Fig. 1A), we mutated this amino acid to proline. G770P had, however, neither significant effects on the current kinetics (Fig. 5A, second current family from top; Fig. 5B) nor on the voltage dependence of channel activation and inactivation (Fig. 6). N771P adjacent to G770P was found to shift the voltage dependence of channel activation and inactivation (Fig. 6B, TABLE ONE) and to substantially slow the time course of inactivation (Fig. 5A, TABLE ONE). N771P did not, however, show slow channel activation at hyperpolarized voltages (τm ranged between 1.3 ± 0.2 and 2.9 ± 0.2 ms at all voltages, Fig. 5), and channel deactivation was not decelerated (Fig. 4).
FIGURE 5. Positional specificity of the effects of proline substitution on the activation kinetics of CaV1.2.

A, left panel shows α-helical representation of the amino acid sequence of segment IIS6 of the α1-1.2 subunit. Functional proline mutants are shown in gray, non-functional mutants are shown in black. Right panel shows representative families of IBa through wild-type and mutant channels. Barium currents were evoked during depolarizing test pulses from a holding potential of −100 mV (increments, 10-mV). Currents at threshold potentials are indicated. B, voltage dependence of channel activation time constants (τm). Data were obtained by fitting the activation phase of currents to a mono-exponential function. The dashed line represents the voltage dependence of I781P activation from Fig. 3. The inset illustrates the voltage dependence of channel activation time constants of the slowly activating A780P mutant.
FIGURE 6. Positional specificity of the effects of proline substitution on the voltage dependence of CaV1.2 gating.

A, voltage dependence of activation of wild-type and mutant channels. Solid lines represent fits to Boltzmann functions. B, correlation between potentials of half-maximal activation (V0.5,act, TABLE ONE) and half-maximal inactivation (V0.5,inact). Linear regression analysis (resulting line is shown) yielded a correlation coefficient of 0.98. Because of its unusual kinetic phenotype (see inset in Fig. 5) A780P was not included in the correlation analysis.
Positional Specificity of I781P
Proline substitution of Ile-781 caused the most dramatic changes in channel activation (Figs. 2-4). To examine the positional specificity of I781P on CaV1.2 channel gating we substituted all amino acids between Cys-769 and Val-783 by prolines (Figs. 5 and 6, TABLE ONE). Substitution of the two alanine residues flanking Ile-781 (A780P and A782P) and of L779P substantially shifted the voltage dependence of activation (Fig. 6, TABLE ONE). A782P and L779P also showed decelerated channel activation at hyperpolarized voltages (Fig. 5B). Additionally channel inactivation was decelerated for A782P and not detectable for A780P and L779P during a 300-ms pulse (Fig. 5A, TABLE ONE). Two peculiarities were observed for A780P. This mutation i) shifted the voltage dependence of channel activation but not inactivation (Fig. 6B), and ii) this mutation slowed the kinetics of channel activation at all voltages (Fig. 5B, inset). Proline substitutions C769P, I773P, and F778P caused smaller or non-significant shifts in the voltage dependence of activation (Fig. 6A, TABLE ONE).
Evidence for Membrane Targeting of Non-functional Mutants
Six constructs (Y772P, L774P, L775P, N776, V777P, and V783P) did not conduct Ba2+ currents. To investigate whether the lack of current observed for these mutants could be because of an impairment of plasma membrane targeting, we examined the subcellular distribution of GFP-tagged mutants (14) by confocal microscopy. Wild-type and mutant GFP-tagged CaV1.2 α1-subunits were co-transfected with β1a and α2-δ1-subunits in tsA-201 cells, and the plasma membrane was visualized by staining with FM4-64 (Fig. 7). Consistent with previous reports (21-23), the wild-type CaV1.2 α1-subunit localized predominantly at the plasma membrane with some intracellular labeling also evident. Similarly, images taken from cells expressing the non-functional channels (shown are two representative mutants) demonstrated that all mutant subunits were targeted to the plasma membrane, with some intracellular staining also detected. These findings demonstrate that the lack of current observed for these mutants cannot be attributed to a failure of the mutant GFP-tagged CaV1.2 α1-subunits to reach the plasma membrane.
FIGURE 7. Localization of wild-type and mutant CaV1.2 α1-subunits within tsA-201cells.
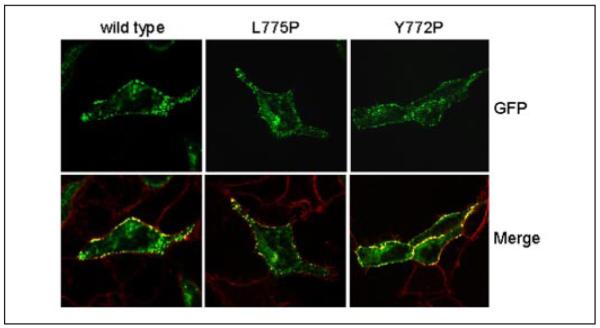
Transiently transfected tsA-201 cells expressing wild-type and two representative non-functional CaV1.2 channels, L755P and Y772P, are shown. The wild-type and mutant GFP-tagged (green) CaV1.2 α1-subunits were co-expressed with β1a and α2-δ1-subunits, and the cells were stained with FM4-64 (red), a plasma membrane marker. In these merged images, GFP-tagged α1-subunits located in the plasma membrane are seen as punctate yellow clusters.
Bay K8644 and β-Subunit Modulation of Ile-781 Mutants
A shift of voltage dependence of activation toward more negative voltages is a hallmark of Bay K8644 action. It was therefore interesting to analyze if the shift in activation gating caused by the naturally occurring mutation I781T would affect BAY K8644 action. At potentials corresponding to the maximum of the current voltage relationships (0 mV for WT and −30 mV for I781T, Fig. 1C) 100 nm of BAY K8644 induced similar stimulation (2.3 ± 0.3-fold, n = 3 in WT and 2.6 ± 0.3, n = 3 in the I781T mutant) (Fig. 8, A and B). The voltage dependences of channel activation were shifted to comparable extents, −5.9 ± 1.3 mV (n = 4) in I781T and −6.7 ± 1.4 mV in WT (n = 3) (Fig. 8C).
FIGURE 8. Mutation I781T does not significantly affect Bay K8644 action on L-type Ca2+ channels.

A, representative traces of IBa through WT and mutant I781T CaV1.2 channels before (control) and in the presence of 100 nm Bay K8644. Currents were evoked by depolarizations from −100 mV to voltages corresponding to the maximum of the current voltage relationship (i.e. 0 mV for WT and −30 mV for mutant I781T). B, stimulation of IBa through WT (2.3 ± 0.3, n = 3) and mutant I781T channels (2.6 ± 0.3, n = 3) by 100 nm Bay K8644 (protocol as in A) was not significantly different. C, 100 nm Bay K8644 shifted the midpoints of the voltage dependences of activation in WT and mutant I781T to similar extents (by −6.7 ± 1.4 mV in WT, n = 3, open circles, and by −5.9 ± 1.3 mV, n = 4, filled circles in I781T, p < 0.05). The dashed lines illustrate the corresponding Boltzmann curves in the absence of the drug (taken from Fig. 2).
It is well established that β-subunits modulate the gating of high voltage-activated Ca2+ channels (for reviews see Refs. 24-26). To elucidate whether the observed changes in the voltage dependence of channel activation interfere with β-subunit modulation, we co-expressed wild-type, I781T, and I781P CaV1.2 α1-subunits with β1a- or β2a-subunits as well as the α2-δ1-subunit and analyzed the resulting activation curves. For each of these channels, co-expression of the β2a-subunit gave a small hyperpolarizing shift in the voltage dependence of channel activation and slower channel inactivation when compared with coexpression with the β1a-subunit (Fig. 9, TABLE ONE). These findings suggest that these Ile-781 mutants do not substantially affect β-subunit modulation.
FIGURE 9. I781T and I781P substitutions did not interfere with modulation of CaV1.2 channel gating by β-subunits.

Comparison of voltage dependence of activation for wild-type, I781T and I781P CaV1.2 channels, containing either β1a- (open circles) or β2a-(filled circles) subunits.
DISCUSSION
In this study we demonstrate a crucial role of a cluster of amino acids (Leu-779, Ala-780, Ile-781, Ala-782) in the pore-forming segment IIS6 in activation and inactivation of the CaV1.2 channel. This finding is related to a recently described channelopathy, where a mutation of the corresponding isoleucine to threonine in CaV1.4 (I745T) causes a severe X-linked retinal disorder (12). As previously described for the I745T CaV1.4 mutant (1), I781T resulted in an almost identical shift in the voltage dependence of activation (Fig. 1C) and slowed inactivation of CaV1.2 (TABLE ONE) suggesting a common mechanism of gating disturbances in both channel types. Additional analyses revealed that the I781T mutation also caused a negative shift in the voltage dependence of inactivation (Fig. 2B), slowed activation at hyperpolarized voltages (Fig. 3), and slowed deactivation at all potentials (Fig. 4). These changes in gating of CaV1.2 favor transitions to the open state and can be explained by a destabilization of the closed state (i.e. an acceleration of the forward transition to the open state, see 4), a stabilization of the open state (i.e. a deceleration of the backward transition to the closed state, see Ref. 20), or both.
Ile-781 Substitutions May Destabilize the Closed State
The five Ile-781 substitutions that produced functional channels caused similar alterations in channel gating (Fig. 2). It seems more feasible to explain this finding by a destabilization, rather than by a stabilization, of channel conformations. A possible mechanism for destabilization of the closed state could be a reduction in hydrophobic interactions in position 781 with neighboring residues in the S6-bundle crossing region. This explanation is in line with the lower hydrophobicity and larger shifts in the activation curve of threonine when compared with alanine and leucine (Fig. 2A). Thus, hydrophobic interactions of Ile-781 with neighboring residues in the bundle crossing region might contribute to stabilization of the closed conformation in wild-type channels. Changes in hydrophobicity alone can, however, not explain the full picture. Asparagine is less hydrophobic than threonine (27), although it causes a similar shift (Fig. 2C). As leucine differs from isoleucine by the position of only one methyl group and showed the smallest shift, we speculated that Ile-781 forms part of a tightly fitted region in the closed channel state. Proline caused the largest effect on channel activation. Its hydrophobicity is, however, closer to the wild-type isoleucine than to threonine and asparagine. Proline might disturb the interaction with neighboring amino acids in the closed state by favoring bending of the α-helix (28) and may thereby favor an open pore conformation.
Evidence for Stabilization of the Open State
The deceleration of deactivation observed for I781P and I781T (Fig. 4) is a strong indication of a stabilization of the open channel conformation. A stabilization of the open state is also supported by the reduced rate of channel inactivation that was observed for the majority of Ile-781 mutants (TABLE ONE); a prolonged sojourn in the open channel state would result in a slower net transition rate from the open to the inactivated state. These findings indicate that a point mutation in one S6 segment can be sufficient to slow channel closure.
We speculate that closure of voltage-gated Ca2+ channels requires the return of all four pore-forming elements into the resting state. If one element of the gate structure does not precisely fit the resting (closed) state, then the conducting pore may remain open for a longer period.
Positional Specificity of Ile-781
To examine the positional specificity of I781P we substituted the residues between Cys-769 and Val-783 by prolines (see Fig. 5A). Amino acids causing prominent shifts in the voltage dependence of channel activation and inactivation were localized in a cluster near the intracellular channel mouth (I781P > L779P, A782P, A780P). The more the voltage dependence of channel activation was shifted, the slower these channels activated at hyperpolarized potentials (Figs. 5 and 6A). A782P showed moderately decreased inactivation, whereas inactivation was severely reduced for L779P and A780P suggesting that all three residues contribute to both activation and inactivation gating to different extents.
In the upper part of IIS6, N771P caused a shift (−17 mV) of the activation curve and moderately slowed inactivation, but did not shift the inactivation curve markedly and did not affect the rate of channel activation at hyperpolarized voltages, (Fig. 5B, Fig. 6B, TABLE ONE). Moreover, unlike I781P and I781T, the voltage dependence of channel deactivation was unaltered in this mutant (Fig. 4B). Together these findings suggest that mutation N771P does not stabilize the open state but is more likely to destabilize the closed state.
The correlation between shifts in the activation and inactivation curves observed for all mutants with the exception of A780P represents an interesting finding per se Figs. 2B and 6B. A model describing channel activation in terms of a voltage-sensing machinery, a discrete voltage-independent mechanism of the pore opening and inactivation events might reproduce this correlation.
Although correctly targeted to the plasma membrane, some proline mutants did not conduct barium currents (Fig. 7). These data suggest that proline substitutions in Ca2+ channel S6 segments may disrupt the functionality of the conducting pore. Similar observations were recently made for proline substitutions in the central part of NaChBac segment S6 (29). We cannot exclude, however, that other substitutions in these positions would produce functional Ca2+ channels with altered gating properties.
Bay K8644 and β-Subunit Modulation
Slowing of deactivation and a shift of steady state activation as observed for the naturally occurring mutation I781T and other effective mutations is reminiscent of BAY K8644 action. Interestingly, mutation I781T neither prevented a further shift of channel activation nor affected the stimulation of the current (Fig. 8). These data suggest that Bay K8644 and mutation I781T affect the channel via independent mechanisms. Functional and structural studies suggest that the modulation of channel activation and inactivation by β-subunits occurs by a direct modulation of the movement of the pore-forming segment IS6 (26, 30). The I781P and I781T mutants showed the same shift in the voltage dependence of channel activation and slowed inactivation that occurs when wild-type α1 subunits are coexpressed with β2a rather than β1a (Fig. 9, TABLE ONE). These findings suggest that β-subunit modulation of segment IS6 and the effects of residue 781 on pore stability are determined by independent mechanisms.
Significance of Ile-781 and Adjacent Residues for Activation Gating of Ca2+ Channels
The crystal structure of the 2-TM helix calcium-gated potassium channel MthK revealed that a conserved glycine at position 83 is responsible for bending the pore-lining M2 helix leading to channel opening (31). Previous studies have shown that the corresponding glycine (Gly-219) in the bacterial voltage-gated sodium channel NaChBac represents a “gating hinge” (20). This conclusion is based on two principle findings, i) G219P shifts the activation curve to hyperpolarized voltages, and ii) this shift is accompanied by a deceleration of deactivation kinetics. Mutating the corresponding Gly-770 in CaV1.2 to proline did not cause significant effects on channel gating (Fig. 6, TABLE ONE). Localization of a gating hinge at residue Gly-770 is therefore not likely. The kinetic phenotype of Ile-781 and adjacent residues (Leu-779, Ala-780, and Ala-782) observed in the present study is, however, similar to the one described for NaChBac construct G219P (20). Our data therefore suggest that helix bending is more likely to occur close to the inner channel mouth in CaV1.2. Residues 779–782 are conserved in high voltage-activated Ca2+ channels, and it is tempting to speculate that this sequence (LAIA) might participate in a common mechanism of gating.
Acknowledgments
We thank Dr. Thomas Peterbauer for his support in confocal imaging. The excellent technical assistance of Hannelore Kadlec was greatly appreciated.
Footnotes
This work was supported in part by a grant from Fonds zur Förderung der wissen-schaftlichen Forschung (FWF) 15914 (to S. H.).
The abbreviations used are: GFP, green fluorescent protein; WT, wild-type; NaChBac, bacterial sodium channel; Bay K8644, 1,4-dihydro-2,6-dimethyl-5-nitro-4-(2-(trifluoromethyl)phenyl)pyridine-3-carboxylic acid methyl ester.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Hemara-Wahanui A, Berjukow S, Hope CI, Dearden PK, Wu SB, Wilson-Wheeler J, Sharp DM, Lundon-Treweek P, Clover GM, Hoda JC, Striessnig J, Marksteiner R, Hering S, Maw MA. Proc. Natl. Acad. Sci. U. S. A. 2005;102:7553–7558. doi: 10.1073/pnas.0501907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E. Pharmacol. Rev. 2003;55:579–581. doi: 10.1124/pr.55.4.8. [DOI] [PubMed] [Google Scholar]
- 3.Catterall WA. Annu. Rev. Cell Dev. Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 4.Yifrach O, MacKinnon R. Cell. 2002;111:231–239. doi: 10.1016/s0092-8674(02)01013-9. [DOI] [PubMed] [Google Scholar]
- 5.Hering S, Berjukow S, Sokolov S, Marksteiner R, Weiss RG, Kraus R, Timin EN. J. Physiol. 2000;528:237–249. doi: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stotz SC, Zamponi GW. Trends Neurosci. 2001;24:176–181. doi: 10.1016/s0166-2236(00)01738-0. [DOI] [PubMed] [Google Scholar]
- 7.Shi C, Soldatov NM. J. Biol. Chem. 2002;277:6813–6821. doi: 10.1074/jbc.M110524200. [DOI] [PubMed] [Google Scholar]
- 8.Tanabe T, Adams BA, Numa S, Beam KG. Nature. 1991;352:800–803. doi: 10.1038/352800a0. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Stevens L, Klugbauer N, Wray D. J. Biol. Chem. 2004;279:26858–26867. doi: 10.1074/jbc.M313981200. [DOI] [PubMed] [Google Scholar]
- 10.Garcia J, Nakai J, Imoto K, Beam KG. Biophys. J. 1997;72:2515–2523. doi: 10.1016/S0006-3495(97)78896-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaguchi H, Muth JN, Varadi M, Schwartz A, Varadi G. Proc. Natl. Acad. Sci. U. S. A. 1999;96:1357–1362. doi: 10.1073/pnas.96.4.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hope CI, Sharp DM, Hemara-Wahanui A, Sissingh JI, Lundon P, Mitchell EA, Maw MA, Clover GM. Clin. Exp. Ophthalmol. 2005;33:129–136. doi: 10.1111/j.1442-9071.2005.00987.x. [DOI] [PubMed] [Google Scholar]
- 13.Koschak A, Reimer D, Walter D, Hoda JC, Heinzle T, Grabner M, Striessnig J. J. Neurosci. 2003;23:6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grabner M, Dirksen RT, Beam KG. Proc. Natl. Acad. Sci. U. S. A. 1998;95:1903–1908. doi: 10.1073/pnas.95.4.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Gene (Amst.) 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 16.Ruth P, Rohrkasten A, Biel M, Bosse E, Regulla S, Meyer HE, Flockerzi V, Hofmann F. Science. 1989;245:1115–1118. doi: 10.1126/science.2549640. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei XY, Birnbaumer L. J. Biol. Chem. 1992;267:1792–1797. [PubMed] [Google Scholar]
- 18.Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A, et al. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 19.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Pflugers Arch. Eur. J. Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y, Yarov-Yarovoy V, Scheuer T, Catterall WA. Neuron. 2004;41:859–865. doi: 10.1016/s0896-6273(04)00116-3. [DOI] [PubMed] [Google Scholar]
- 21.Gerster U, Neuhuber B, Groschner K, Striessnig J, Flucher BE. J. Physiol. 1999;517:353–368. doi: 10.1111/j.1469-7793.1999.0353t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao T, Bunemann M, Gerhardstein BL, Ma H, Hosey MM. J. Biol. Chem. 2000;275:25436–25444. doi: 10.1074/jbc.M003465200. [DOI] [PubMed] [Google Scholar]
- 23.Gao T, Chien AJ, Hosey MM. J. Biol. Chem. 1999;274:2137–2144. doi: 10.1074/jbc.274.4.2137. [DOI] [PubMed] [Google Scholar]
- 24.Birnbaumer L, Qin N, Olcese R, Tareilus E, Platano D, Costantin J, Stefani E. J. Bioenerg. Biomembr. 1998;30:357–375. doi: 10.1023/a:1021989622656. [DOI] [PubMed] [Google Scholar]
- 25.Arikkath J, Campbell KP. Curr. Opin. Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 26.Hering S. Trends Pharmacol. Sci. 2002;23:509–513. doi: 10.1016/s0165-6147(02)02104-1. [DOI] [PubMed] [Google Scholar]
- 27.Kyte J, Doolittle RF. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 28.Cordes FS, Bright JN, Sansom MS. J. Mol. Biol. 2002;323:951–960. doi: 10.1016/s0022-2836(02)01006-9. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Scheuer T, Catterall WA. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17873–17878. doi: 10.1073/pnas.0408270101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]