

Abstract
Angiogenesis and osteogenesis are tightly coupled during bone development and regeneration. The vasculature supplies oxygen to developing and regenerating bone and also delivers critical signals to the stroma that stimulate mesenchymal cell specification to promote bone formation. Recent studies suggest that the hypoxiainducible factors (HIFs) are required for the initiation of the angiogenic–osteogenic cascade. Genetic manipulation of individual components of the HIF/vascular endothelial growth factor (VEGF) pathway in mice has provided clues to how coupling is achieved. In this article, we review the current understanding of the cellular and molecular mechanisms responsible for angiogenic–osteogenic coupling. We also briefly discuss the therapeutic manipulation of HIF and VEGF in skeletal repair. Such discoveries suggest promising approaches for the development of novel therapies to improve bone accretion and repair.
Keywords: Hypoxia-inducible factor, VEGF, Endochondral bone formation, Fracture repair
Vascularization of the skeleton
The skeleton is highly vascularized and receives 5–20% of resting cardiac output. [1–3]. In long bones, the nutrient artery serves as the primary vascular supply. Upon entering the diaphysis from the systemic circulation, this vessel branches into ascending and descending medullary arteries within the marrow cavity. Branching arterioles penetrate the endosteal surface to form the primary vascular supply for the diaphyseal cortex. In the skull, parietal bones are supplied by one major branch of the meningeal artery. From each of these vessels, separate branches supply the dura wherein a network of vessels covers the bone. Numerous fine vessels penetrate the periosteum and the dura and enter the cortical plates [4].
During development, hypoxia-driven diffusion of oxygen is a major stimulus for cellular differentiation and migration in the embryo. Hypoxic cells signal to vascular elements to ensure the proper differentiation and maintenance of the developing vasculature [5]. While the delivery of oxygen and nutrients to the bone is critical for the maintenance of healthy skeletal tissue, blood vessels also play a role in bone modeling and remodeling by mediating interactions between osteoblasts, osteocytes, osteoclasts, and vascular cells. These cells contain all of the cellular machinery necessary to perceive and respond to change in tissue oxygen tension and are responsible for coordinating bone formation both temporally and spatially during skeletal development and repair. Seminal work by Coolbaugh revealed the critical importance of these interactions, as surgical disruption of skeletal blood supply produced marked alterations in bone density and strength [6]. Subsequently, Trueta and Harrison demonstrated that interrupting the blood supply to the growth plate alters bone mineralization and the replacement of hypertrophic chondrocytes [7, 8]. Analogous anatomical relationships between the vasculature and bone compartments also exist in adult bone. For example, Hauge et al. [9] and Eriksen et al. [10] have suggested the existence of discrete vascular structures termed bone remodeling compartments that coordinate bone resorption and formation. In this setting, bone lining cells that express mediators of osteoclastogenesis such as receptor activator of nuclear factor kappa B ligand (RANKL) and osteoprotegerin seal off the compartment from the adjacent marrow, while an associated blood vessel serves as a conduit for circulating progenitor cells. Such anatomical compartmentalization would provide a mechanism to functionally link the vasculature to bone formation and resorption.
The purpose of this article is to highlight recent work on the cellular and molecular events responsible for the coupling of angiogenesis to osteogenesis during skeletal development and repair with a focus on results from genetically modified mice. These studies implicate the transcription factor hypoxia-inducible factor (HIF)-1α and its target gene vascular endothelial growth factor (VEGF) as critical regulators of angiogenic–osteogenic coupling.
Hypoxia-inducible factors and vascular endothelial growth factor
HIFs, which belong to the Per/Anrt/Sim subfamily of basic helix–loop–helix transcription factors [11, 12], are major regulators of the gene programs that orchestrate angiogenic–osteogenic coupling. The HIF family comprises three functionally nonredundant α subunits, HIF-1α, HIF-2α, and HIF-3α, which form a heterodimer with the HIF-1β subunit. The messenger RNAs (mRNAs) encoding HIF-1α and HIF-1β are widely expressed and have been detected in most human and rodent tissues [13]. However, while HIF-1β protein is constitutively expressed, HIF-1α protein expression is regulated by an oxygen-sensitive proteolytic mechanism. Under normoxic conditions, HIF-1α is rapidly degraded, but, when oxygen levels drop below 5%, expression is stabilized and its activity progressively increases with additional decreases in the oxygen tension [14].
HIFs do not directly perceive changes in oxygen tension. Rather, the expression and activity of the α-subunit is regulated by a class of 2-oxoglutarate-dependent and Fe2+-dependent dioxygenases [14]. Each α-subunit has an oxygen-dependent degradation domain that contains prolyl residues recognized by prolyl hydroxylase domain (PHD) proteins. It is important to note that these PHDs are distinct from the family of collagen prolyl hydroxylases. At oxygen levels above 5%, PHD proteins hydroxylate HIF-1α at P402 and/or P564. This modification allows the binding of the von Hippel-Lindau (VHL) tumor suppressor protein, a component of the E3 ubiquitin ligase complex that modifies HIF-1α with polyubiquitin chains and thereby targets it for proteasomal degradation. Under hypoxic conditions, prolyl hydroxylation is inhibited and HIF-1α protein accumulates [15, 16]. A second level of oxygen-dependent regulation is supplied by an asparaginyl hydroxylase referred to as factor-inhibiting Hif (FIH) [17]. FIH hydroxylates an asparagine residue (N803) in the carboxy-terminal transcriptional activation domain. This modification prevents the interaction of HIF-1 with transcriptional coactivators such as p300 and CBP.
HIFs arose early in evolution and control the activity of a broad array of genes. Indeed, binding sites, referred to as hypoxia-response elements, have been identified in the promoter regions of more than 100 putative target genes involved in a variety of cell processes [18]. Among these factors, VEGF is the best characterized proangiogenic factor that is activated by hypoxia and plays a critical role in angiogenesis during the development of most tissues including bone. VEGF family members, including VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placenta growth factor, are secreted, dimeric glycoproteins that activate one of three receptor tyrosine kinases [19]. Additional complexity in this family is manifested by the fact that alternative splicing of VEGF transcripts gives rise to multiple isoforms with different biological activity. Five VEGF-A isoforms have been identified in humans, while there are three major isoforms in the mouse (VEGF120, VEGF164, and VEGF188). Intracellular signaling events stimulated by VEGF are similar to other growth factors and induce responses such as cell proliferation, survival, and migration, but it is unique in its ability to transduce signals that direct the formation of vascular tubes and regulate vascular permeability. So important is VEGF signaling for angiogenesis that the deletion of even a single copy of the VEGF-A gene results in embryonic lethality owing to defective vascular development [20, 21].
Angiogenic–osteogenic coupling during endochondral bone formation
The endochondral skeleton is formed as a series of interconnected avascular cartilage models called anlagen. Chondrocytes arise from condensing regions of the mesenchyme, exit the cell cycle, differentiate, and become hypertrophic and hypoxic [22, 23]. As early as embryonic day (E)13.5–14.5 in the mouse tibia, vascular elements are recruited to the perichondrium. This is followed by vessel invasion into the hypertrophic cartilage at E14.5 to establish the primary ossification center. With continued development, vessels sprout within the metaphyseal region and continue to invade the hypertrophic cartilage [24, 25]. These events supply the osteoclastic precursors necessary to remove the cartilage anlage as well as the morphogenic signals required for osteoblast differentiation.
HIF-1α stimulates VEGF production in this microenvironment to initiate blood vessel invasion into cartilage. At E13.5, VEGF is expressed in the perichondrium, presumably to stimulate perichondrial angiogenesis [24]. This is followed by increased VEGF expression in the hypertrophic cartilage [26–28]. Genetic manipulation of VEGF expression has a profound impact on vascular invasion as well as subsequent endochondral bone development. Mice expressing only the soluble form of VEGF, VEGF120, but lacking VEGF188 and VEGF164 exhibit delayed blood vessel penetration into the perichondrium and decreased expression of osteoblastic differentiation markers [24, 29]. Vascular formation continues to be disturbed in neonatal VEGF120/120 mice and is accompanied by a reduction in bone growth with a 35% decrease in trabecular bone volume and a 34% increase in the size to the hypertrophic zone of the growth plate [29]. Similarly, administration of mFlt (1-3)-IgG, which completely blocked neoangiogenesis in the growth plates of 24-day-old mice, resulted in the expansion of the hypertrophic zone and decreased bone mass [30].
Hypoxia is a major driving force for angiogenesis during endochondral bone formation. The mesenchymal condensations of the presumptive endochondral bones as well as the fetal growth plate are hypoxic and express HIF-1α [31–33]. Tissue-specific deletion of this transcription factor via the expression of the Col2-Cre transgene induces a massive increase in epiphyseal chondrocyte death, indicating that HIF-1α is required for survival in this hypoxic tissue. Interestingly, HIF-1α deletion results only in a modest decrease in the expression of VEGF by hypertrophic chondrocytes [33], which suggests that VEGF production by hypertrophic chondrocytes may also be regulated by HIF-1α-independent mechanisms [34]. As an example of one such mechanism, VEGF is not upregulated in the hypertrophic chondrocytes of Runx2-deficient mice and as a result vascular invasion does not occur in these animals [28].
In addition to chondrocytes, osteoblasts also express components of the HIF pathway and promote skeletal vascularization during long-bone formation. Disrupting the expression of the E3 ligase VHL specifically in osteoblasts (ΔVHL) results in the overexpression of HIFs [34]. ΔVHL mice exhibit dramatic increases in VEGF expression associated with a striking and progressive increase in bone volume resulting from increased numbers and activity of osteoblasts. On the other hand, deletion of HIF-1α in osteoblasts decreases the diameter of bones in ΔHIF-1α mice and causes significant reductions in osteoid volume along with reduced numbers of osteoblasts. Interestingly, the amount of bone in the axial skeleton of these mutant mice is directly proportional to the amount of skeletal vascularity, which suggests that HIF-1α regulates bone formation via a cell nonautonomous mechanism. In support of this idea, manipulating the expression of HIFs in vitro by deleting VHL expression does not affect osteoblast function. Further, VEGF mRNA levels are upregulated by VHL deletion and endothelial sprouting from E17.5 metatarsals is entirely abolished by incubation with a VEGF-neutralizing antibody. It is likely that HIF-2α also functions in skeletal tissue, as the reduction in bone and blood vessel volume in ΔHIF-1α mice was less pronounced than would be expected from the elimination of all HIF signaling. The fact that mice lacking both VHL and HIF-1α have a phenotype intermediate to that of either single mutant further supports a role for HIF-2α, at least when HIF-1α is deleted.
Hypoxia is not the only stimulus that induces the expression of VEGF by osteoblasts. Prostaglandins E1 and E2 [35], transforming growth factor β [36], bone morphogenetic proteins [37], insulin-like growth factor 1 [38], endothelin 1 [39], and vitamin D3 [40] all increase the expression of VEGF in bone cells. Many of these same factors also increase the expression of HIFs [38, 41]. Likewise, other transcription factors that regulate VEGF expression may interact with HIFs. For example, β-catenin has been shown to regulate VEGF expression, has been implicated in skeletal vascularization [42], and may interact with HIF-1α at the VEGF promoter [43]. Taken together, these data emphasize the critical role of HIF-1α in regulating angiogenesis and suggest that this transcription factor acts as a proximate control point for many pathways that influence angiogenesis in the skeleton.
Angiogenic–osteogenic coupling during skeletal repair
Fracture healing recapitulates many of the stages of endochondral bone formation including the requirement for angiogenesis for bone regeneration. Following fracture, inflammatory signals stimulate the recruitment of mesenchymal progenitors that subsequently differentiate to chondrocytes and produce a cartilage matrix. Hypertrophic chondrocytes release signals that stimulate neoangiogenesis of the cartilaginous callus and then undergo apoptosis. The recruitment of osteoclasts initiates callus remodeling and its replacement with woven bone that is ultimately consolidated to fully support the mechanical demands of the skeleton [44, 45]. Just as the inhibition of angiogenesis during development impedes the replacement of chondrocytes with osteoblasts, delays in angiogenesis during fracture healing results in a tissue composed of chondrocytic cells rather than osteoblasts [46].
The abrupt interruption of oxygen and nutrient supply with consequential upregulation of HIF-1α has been proposed as a primary stimulus for new bone formation [47, 48]. VEGF is expressed at fracture sites [49, 50] and manipulation of VEGF activity alters fracture repair. VEGF expression is first evident during the cartilage formation phase, to stimulate vascular in-growth from the periosteum, but levels do not peak until the cartilage resorption–bone formation stages [45]. Application of exogenous VEGF enhances bone formation in mice and rabbits with segmental radius defects, while administration of a soluble chimeric VEGF receptor Flt-IgG dramatically reduces angiogenesis, bone formation, and callus mineralization [51]. Interestingly, the upregulation of both HIF-1α and VEGF have been shown to be influenced by age and diabetic status in wound-healing models [52, 53]. These observations may help to explain the impairment in fracture healing evident in diabetic humans [54, 55].
To suggest that VEGF is the only HIF-1α target involved in fracture healing would be an oversimplification. Rather, HIF-1α likely regulates a large number of genes involved in angiogenesis and the recruitment of progenitor cells. As an example, placental growth factor, a VEGF homolog that activates VEGFR-1, is regulated by HIF-1α [53] and is expressed at the fracture site. Mice lacking this growth factor do not exhibit a detectable bone phenotype, but fracture healing is impaired and accompanied by cartilage accumulation in the callus and poor vascularization [56]. Similarly, stromal-cell-derived factor 1 is regulated by HIF-1α [57], and manipulating the activity of this cytokines alters the recruitment of progenitor cells to sites of skeletal repair [58].
Distraction osteogenesis (DO) is a common surgical technique used to lengthen limbs and has been utilized as a model of bone regeneration to further examine the cellular mechanisms that couple angiogenesis and fracture healing. In this surgical procedure, intramembranous bone formation is locally induced by the application of an external fixation device that applies gradual mechanical distraction across the osteotomy [59]. During distraction, a close temporal and spatial relationship exists between formation of regenerated bone and vascular proliferation from the periosteum and medullary canal [47]. A migrating zone of proliferating fibroblast-like cells appears and aligns parallel to the vector of elongation and deposit collagen bundles. Capillaries then form between these bundles and osteoblasts emerge and arrange themselves along the collagen fibers. With continued elongation, woven bone is laid down and subsequently remodeled to lamellar bone.
At the center of the distraction gap, cells that exhibit morphological features consistent with osteoblasts are hypoxic and express both HIF-1α and VEGF [60]. Genetic and pharmacological manipulation of the HIF/VEGF pathway clearly demonstrates its role in bone healing following DO. Constitutive overexpression of HIFs, via the disruption of VHL in osteoblasts, markedly improved bone healing. The distraction gap rapidly fills with blood vessels and this is followed by increases in bone volume and callus strength. Inactivation of HIF-1α impairs both angiogenesis and bone regeneration as the distraction gap in these animals develop fewer blood vessels and accumulate less bone. Interestingly, administration of antibodies specific for VEGFR-1 and VEGFR-2 greatly diminishes the effect of HIF overexpression on bone regeneration. These data imply that VEGF signaling is necessary for bone healing and are supported by more recent studies using VEGF receptor blocking antibodies in wild-type mice [61].
The essential roles of HIF and VEGF during fracture repair suggest that this pathway could be targeted in therapeutic interventions to accelerate bone healing. Indeed, adenoviral-VEGF gene transfer and implantation of scaffolds containing VEGF encoding plasmids increase VEGF expression by local cell populations and enhance fracture repair by accelerating endochondral bone formation [62–64]. Implantation of modified cells that overexpress VEGF has also produced encouraging results [65].
The fact that HIF degradation is regulated by prolyl hydroxylation suggests that small molecule inhibitors of PHD can be designed to selectively activate the HIF/VEGF pathway to facilitate bone regeneration. Several molecules that sequester cofactors necessary for PHD activity have already been made available. For example, desferrioxamine (DFO) chelates iron, a cofactor necessary for PHD activity, and enhances HIF-1α expression, VEGF expression, and endothelial sprouting from E17.5 metatarsals in vitro [60]. Administration of this agent to the distraction gap strongly increased vascularity and significantly increased bone volume. Surprisingly, bone regeneration after DFO treatment was actually greater than in the genetic models, probably as a result of DFO's ability to activate signaling in osteoblasts as well as other cells in the distraction gap.
Cell-autonomous effects of HIFs and VEGF
In some experimental settings, HIF-1α and VEGF can directly regulate bone cell function without stimulating angiogenesis. The fetal growth plate has evolved an “out–in” gradient of oxygenation [33]. As indicated above, chondrocyte-specific disruption of HIF-1α induces massive apoptosis at the center of the growth plate (where oxygen tension is lowest), but viable chondrocytes at the periphery actually proliferate at a higher rate. These data suggest that HIF-1α is essential not only for survival in this hypoxic tissue but can also directly regulate chondrocyte proliferation. Since conditional knockout of VEGF results in a similar but considerably milder cell death phenotype at the center of the growth plate, it is likely that VEGF is at least a component of this chondrocyte survival pathway [66]. HIF-1α may also directly regulate chondrocyte differentiation and matrix accumulation since increased matrix accumulation is evident in the growth plates of mice lacking VHL in chondrocytes [67]. Indeed, hypoxia increases type II collagen accumulation via a HIF-1α-dependent mechanism in vitro [68], likely via the regulation of Sox-9 expression [69, 70].
The possibility that HIFs participate in osteoblast differentiation and function is still unclear. Overexpression of HIFs via the disruption of VHL did not directly affect osteoblast function in vitro [34]. On the other hand, a recent study suggests that HIF-2α may regulate the expression of the master osteoblastic regulator Runx2 [71]. Equally controversial is whether VEGF exerts direct effects on osteoblast function. Clearly, VEGF and its receptors (VEGFR-1/Flt-1 and VEGFR-2/Flk-1) are expressed by osteoblasts suggesting a possible autocrine and/or paracrine role for the growth factor [72]. Mice globally deficient for VEGFR-1 exhibit decreased bone volume, mineralizing surface, and mineral apposition rate, and this is accompanied by decreased mineralization of bone marrow stromal cells [73]. While these data are complicated by the fact that stromal cell populations may contain a considerable fraction of endothelial cells that may indirectly influence osteogenesis, a few studies do suggest that VEGF promotes osteoblast proliferation and differentiation and acts as a potent chemoattractant [72, 74–76]. Other studies have produced opposite effects. For example, Villars et al. [77] reported that VEGF does not affect the differentiation of human bone marrow stromal cells and that VEGFR-1 and VEGFR-2 are not detectable at the mRNA level in these cells [78]. Further, using a stem-cell-based gene therapy model, Peng et al. [79] demonstrated that VEGF treatment only improves bone regeneration when acting synergistically with BMP-4 to increase stromal cell recruitment and to enhance cell survival. Culture conditions may at least partially account for these discrepancies as hypoxia induces VEGFR-1 expression in bone marrow stromal cell populations via a HIF-1α-dependent mechanism [80]. Selective disruption of VEGF expression in cells of the osteoblast lineage should help to clarify these issues.
Finally, VEGF might also regulate the differentiation, migration, and activity of osteoclasts [81]. Cells of the monocytes/macrophage lineage, from which osteoclasts are derived, express VEGFR-1 and activation of this receptor stimulates migration [82]. The differentiation program of this cell lineage is initiated by the cytokine macrophage colony-stimulating factor (M-CSF) and the interaction of receptor activator of nuclear factor kappa B present in the cell surface of osteoclast precursors with its ligand RANKL expressed by cells of the osteoblast lineage. Mutations or loss of function in either gene results in osteopetrosis characterized by strikingly high bone mass and a lack of osteoclasts [83, 84]. Niida et al. [85, 86] demonstrated that, in the presence of RANKL, VEGF acting through VEGFR-1 can substitute for M-CSF. Juvenile op/op mice, which lack M-CSF, are osteopetrotic, but injection of VEGF stimulates osteoclastogenesis and bone resorption. Further, the administration of a VEGF-neutralizing antibody abolished the age-dependent resolution of the osteopetrotic phenotype.
Conclusion
Although most bone biologist readily accept the concept that angiogenesis is required for skeletal development, the mechanisms responsible for coupling these events have remained obscure. A growing body of evidence demonstrates that the HIF pathway is a central regulator that coordinates the initiation of angiogenesis and couples these events to bone formation. This requires precise temporal and spatial interactions of vascular and skeletal elements to ensure that bone is deposited at the right place and time. Based upon the experimental observations summarized in this review, we propose a model (Fig. 1) in which resident bone cell perceive reduced oxygen and nutrient availability and upregulate HIF-1α. HIF-1α target genes including VEGF are activated and function via one of two mechanisms. HIF-1α target genes may act directly on bone cells or they may stimulate new blood vessel formation and invasion into bone. Increasing numbers of blood vessels provide osteogenic cues and possibly deliver osteoblast progenitors that then mature and lay down new bone. The challenge for future work will be to identify the reciprocal signals emanating from vascular cells that promote osteogenesis. The observation that isolated endothelial cells augment osteoblast differentiation in vitro suggests that the mechanism involves more than the fulfillment of the osteoblast's metabolic demands. Further elucidation of these mechanisms may lead to a better understanding of the precise communication between angiogenesis and osteogenesis and aid in the design of new therapies to combat bone loss and accelerate bone healing.
Fig. 1.
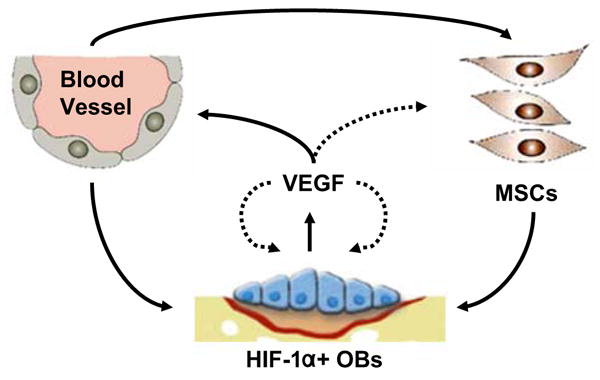
Model for angiogenic–osteogenic coupling in bone. The expression of HIF-1α by resident osteoblasts (OBs) during bone formation and repair leads to the upregulation of VEGF and other angiogenic signals. VEGF may stimulate bone formation via a cell nonautonomous mechanism (solid lines) that involves vascular invasion. Blood vessels direct osteoprogenitors (MSCs) to sites of bone formation or supply bone morphogenic signals that enhance osteoblast activity. Alternatively, VEGF may act as an autocrine/paracrine factor that acts directly on bone cells to increase their recruitment to sites of bone formation, differentiation, and activity (dotted lines)
Contributor Information
Ryan C. Riddle, Department of Pathology, University of Alabama at Birmingham, 1670 University Boulevard, G001, Birmingham, AL 35294, USA
Richa Khatri, Endocrine Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
Ernestina Schipani, Endocrine Unit, Massachusetts General Hospital and Harvard Medical School, Boston, MA, USA.
Thomas L. Clemens, Email: tclemens@uab.edu, Department of Pathology, University of Alabama at Birmingham, 1670 University Boulevard, G001, Birmingham, AL 35294, USA; Veterans Administration Medical Center, Birmingham, AL, USA.
References
- 1.Brooks M. The blood supply of bone. Buttersworths; London: 1971. [Google Scholar]
- 2.Tothill P, MacPherson JN. The distribution of blood flow to the whole skeleton in dogs, rabbits and rats measured with microspheres. Clin Phys Physiol Meas. 1986;7:117–123. doi: 10.1088/0143-0815/7/2/002. [DOI] [PubMed] [Google Scholar]
- 3.Gross PM, Heistad DD, Marcus ML. Neurohumoral regulation of blood flow to bones and marrow. Am J Physiol. 1979;237:H440–448. doi: 10.1152/ajpheart.1979.237.4.H440. [DOI] [PubMed] [Google Scholar]
- 4.Gross TS, Clemens TL. Vascular control of bone remodeling. In: Zaidi M, Bittar EE, Abedanjo OA, Huang CLH, editors. Advances in organ biology. JAI; Stamford: 1998. pp. 137–160. [Google Scholar]
- 5.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 6.Coolbaugh CC. Effects of reduced blood supply on bone. Am J Physiol. 1952;169:26–33. doi: 10.1152/ajplegacy.1952.169.1.26. [DOI] [PubMed] [Google Scholar]
- 7.Trueta J, Buhr AJ. The vascular contribution to osteogenesis V. The vasculature supplying the epiphysial cartilage in rachitic rats. J Bone Joint Surg Br. 1963;45:572–581. [PubMed] [Google Scholar]
- 8.Trueta J, Amato VP. The vascular contribution to osteogenesis. III. Changes in the growth cartilage caused by experimentally induced ischaemia. J Bone Joint Surg Br. 1960;42-B:571. doi: 10.1302/0301-620X.42B3.571. [DOI] [PubMed] [Google Scholar]
- 9.Hauge EM, Qvesel D, Eriksen EF, Mosekilde L, Melsen F. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J Bone Miner Res. 2001;16:1575–1582. doi: 10.1359/jbmr.2001.16.9.1575. [DOI] [PubMed] [Google Scholar]
- 10.Eriksen EF, Eghbali-Fatourechi GZ, Khosla S. Remodeling and vascular spaces in bone. J Bone Miner Res. 2007;22:1–6. doi: 10.1359/jbmr.060910. [DOI] [PubMed] [Google Scholar]
- 11.Semenza GL. HIF-1, O2, and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 12.Semenza GL. HIF-1 and human disease: one highly involved factor. Genes Dev. 2000;14:1983–1991. [PubMed] [Google Scholar]
- 13.Wenger RH, Rolfs A, Spielmann P, Zimmermann DR, Gassmann M. Mouse hypoxia-inducible factor-1α is encoded by two different mRNA isoforms: expression from a tissue-specific and a housekeeping-type promoter. Blood. 1998;91:3471–3480. [PubMed] [Google Scholar]
- 14.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 15.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. Embo J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Structure of an HIF-1α -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 17.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 19.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 20.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 21.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 22.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 23.Provot S, Schipani E. Molecular mechanisms of endochondral bone development. Biochem Biophys Res Commun. 2005;328:658–665. doi: 10.1016/j.bbrc.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 24.Zelzer E, McLean W, Ng YS, Fukai N, Reginato AM, Lovejoy S, D'Amore PA, Olsen BR. Skeletal defects in VEGF120/120 mice reveal multiple roles for VEGF in skeletogenesis. Development. 2002;129:1893–1904. doi: 10.1242/dev.129.8.1893. [DOI] [PubMed] [Google Scholar]
- 25.Colnot C, Lu C, Hu D, Helms JA. Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol. 2004;269:55–69. doi: 10.1016/j.ydbio.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Colnot CI, Helms JA. A molecular analysis of matrix remodeling and angiogenesis during long bone development. Mech Dev. 2001;100:245–250. doi: 10.1016/s0925-4773(00)00532-3. [DOI] [PubMed] [Google Scholar]
- 27.Carlevaro MF, Cermelli S, Cancedda R, Descalzi Cancedda F. Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: auto-paracrine role during endochondral bone formation. J Cell Sci. 2000;113(Pt 1):59–69. doi: 10.1242/jcs.113.1.59. [DOI] [PubMed] [Google Scholar]
- 28.Zelzer E, Glotzer DJ, Hartmann C, Thomas D, Fukai N, Soker S, Olsen BR. Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev. 2001;106:97–106. doi: 10.1016/s0925-4773(01)00428-2. [DOI] [PubMed] [Google Scholar]
- 29.Maes C, Carmeliet P, Moermans K, Stockmans I, Smets N, Collen D, Bouillon R, Carmeliet G. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 2002;111:61–73. doi: 10.1016/s0925-4773(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 30.Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5:623–628. doi: 10.1038/9467. [DOI] [PubMed] [Google Scholar]
- 31.Provot S, Schipani E. Fetal growth plate: a developmental model of cellular adaptation to hypoxia. Ann N Y Acad Sci. 2007;1117:26–39. doi: 10.1196/annals.1402.076. [DOI] [PubMed] [Google Scholar]
- 32.Provot S, Zinyk D, Gunes Y, Kathri R, Le Q, Kronenberg HM, Johnson RS, Longaker MT, Giaccia AJ, Schipani E. Hif-1α regulates differentiation of limb bud mesenchyme and joint development. J Cell Biol. 2007;177:451–464. doi: 10.1083/jcb.200612023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Wan C, Deng L, Liu X, Cao X, Gilbert SR, Bouxsein ML, Faugere MC, Guldberg RE, Gerstenfeld LC, Haase VH, Johnson RS, Schipani E, Clemens TL. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J Clin Invest. 2007;117:1616–1626. doi: 10.1172/JCI31581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harada S, Nagy JA, Sullivan KA, Thomas KA, Endo N, Rodan GA, Rodan SB. Induction of vascular endothelial growth factor expression by prostaglandin E2 and E1 in osteoblasts. J Clin Invest. 1994;93:2490–2496. doi: 10.1172/JCI117258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saadeh PB, Mehrara BJ, Steinbrech DS, Dudziak ME, Greenwald JA, Luchs JS, Spector JA, Ueno H, Gittes GK, Longaker MT. Transforming growth factor-β1 modulates the expression of vascular endothelial growth factor by osteoblasts. Am J Physiol. 1999;277:C628–637. doi: 10.1152/ajpcell.1999.277.4.C628. [DOI] [PubMed] [Google Scholar]
- 37.Deckers MM, van Bezooijen RL, van der Horst G, Hoogendam J, van Der Bent C, Papapoulos SE, Lowik CW. Bone morphogenetic proteins stimulate angiogenesis through osteoblast-derived vascular endothelial growth factor A. Endocrinology. 2002;143:1545–1553. doi: 10.1210/endo.143.4.8719. [DOI] [PubMed] [Google Scholar]
- 38.Akeno N, Robins J, Zhang M, Czyzyk-Krzeska MF, Clemens TL. Induction of vascular endothelial growth factor by IGF-I in osteoblast-like cells is mediated by the PI3K signaling pathway through the hypoxia-inducible factor-2α. Endocrinology. 2002;143:420–425. doi: 10.1210/endo.143.2.8639. [DOI] [PubMed] [Google Scholar]
- 39.Kozawa O, Kawamura H, Hatakeyama D, Matsuno H, Uematsu T. Endothelin-1 induces vascular endothelial growth factor synthesis in osteoblasts: involvement of p38 mitogen-activated protein kinase. Cell Signal. 2000;12:375–380. doi: 10.1016/s0898-6568(00)00061-9. [DOI] [PubMed] [Google Scholar]
- 40.Wang DS, Yamazaki K, Nohtomi K, Shizume K, Ohsumi K, Shibuya M, Demura H, Sato K. Increase of vascular endothelial growth factor mRNA expression by 1, 25-dihydroxyvitamin D3 in human osteoblast-like cells. J Bone Miner Res. 1996;11:472–479. doi: 10.1002/jbmr.5650110408. [DOI] [PubMed] [Google Scholar]
- 41.Liu XH, Kirschenbaum A, Lu M, Yao S, Dosoretz A, Holland JF, Levine AC. Prostaglandin E2 induces hypoxia-inducible factor-1α stabilization and nuclear localization in a human prostate cancer cell line. J Biol Chem. 2002;277:50081–50086. doi: 10.1074/jbc.M201095200. [DOI] [PubMed] [Google Scholar]
- 42.Chen M, Zhu M, Awad H, Li TF, Sheu TJ, Boyce BF, Chen D, O'Keefe RJ. Inhibition of β-catenin signaling causes defects in postnatal cartilage development. J Cell Sci. 2008;121:1455–1465. doi: 10.1242/jcs.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaidi A, Williams AC, Paraskeva C. Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9:210–217. doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 44.Gerstenfeld LC, Cullinane DM, Barnes GL, Graves DT, Einhorn TA. Fracture healing as a post-natal developmental process: molecular, spatial, and temporal aspects of its regulation. J Cell Biochem. 2003;88:873–884. doi: 10.1002/jcb.10435. [DOI] [PubMed] [Google Scholar]
- 45.Ai-Aql ZS, Alagl AS, Graves DT, Gerstenfeld LC, Einhorn TA. Molecular mechanisms controlling bone formation during fracture healing and distraction osteogenesis. J Dent Res. 2008;87:107–118. doi: 10.1177/154405910808700215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi IH, Ahn JH, Chung CY, Cho TJ. Vascular proliferation and blood supply during distraction osteogenesis: a scanning electron microscopic observation. J Orthop Res. 2000;18:698–705. doi: 10.1002/jor.1100180504. [DOI] [PubMed] [Google Scholar]
- 47.Danis A. Mechanism of bone lengthening by the Ilizarov technique. Bull Mem Acad R Med Belg. 2001;156:107–112. [PubMed] [Google Scholar]
- 48.Glowacki J. Angiogenesis in fracture repair. Clin Orthop Relat Res. 1998:S82–S89. doi: 10.1097/00003086-199810001-00010. [DOI] [PubMed] [Google Scholar]
- 49.Ferguson C, Alpern E, Miclau T, Helms JA. Does adult fracture repair recapitulate embryonic skeletal formation? Mech Dev. 1999;87:57–66. doi: 10.1016/s0925-4773(99)00142-2. [DOI] [PubMed] [Google Scholar]
- 50.Le AX, Miclau T, Hu D, Helms JA. Molecular aspects of healing in stabilized and non-stabilized fractures. J Orthop Res. 2001;19:78–84. doi: 10.1016/S0736-0266(00)00006-1. [DOI] [PubMed] [Google Scholar]
- 51.Street J, Bao M, deGuzman L, Bunting S, Peale FV, Jr, Ferrara N, Steinmetz H, Hoeffel J, Cleland JL, Daugherty A, van Bruggen N, Redmond HP, Carano RA, Filvaroff EH. Vascular endothelial growth factor stimulates bone repair by promoting angiogenesis and bone turnover. Proc Natl Acad Sci U S A. 2002;99:9656–9661. doi: 10.1073/pnas.152324099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bosch-Marce M, Okuyama H, Wesley JB, Sarkar K, Kimura H, Liu YV, Zhang H, Strazza M, Rey S, Savino L, Zhou YF, McDonald KR, Na Y, Vandiver S, Rabi A, Shaked Y, Kerbel R, Lavallee T, Semenza GL. Effects of aging and hypoxia-inducible factor-1 activity on angiogenic cell mobilization and recovery of perfusion after limb ischemia. Circ Res. 2007;101:1310–1318. doi: 10.1161/CIRCRESAHA.107.153346. [DOI] [PubMed] [Google Scholar]
- 53.Liu L, Marti GP, Wei X, Zhang X, Zhang H, Liu YV, Nastai M, Semenza GL, Harmon JW. Age-dependent impairment of HIF-1α expression in diabetic mice: correction with electroporation-facilitated gene therapy increases wound healing, angiogenesis, and circulating angiogenic cells. J Cell Physiol. 2008;217:319–327. doi: 10.1002/jcp.21503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cozen L. Does diabetes delay fracture healing? Clin Orthop Relat Res. 1972;82:134–140. [PubMed] [Google Scholar]
- 55.Loder RT. The influence of diabetes mellitus on the healing of closed fractures. Clin Orthop Relat Res. 1988:210–216. [PubMed] [Google Scholar]
- 56.Maes C, Coenegrachts L, Stockmans I, Daci E, Luttun A, Petryk A, Gopalakrishnan R, Moermans K, Smets N, Verfaillie CM, Carmeliet P, Bouillon R, Carmeliet G. Placental growth factor mediates mesenchymal cell development, cartilage turnover, and bone remodeling during fracture repair. J Clin Invest. 2006;116:1230–1242. doi: 10.1172/JCI26772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 58.Kitaori T, Ito H, Schwarz EM, Tsutsumi R, Yoshitomi H, Oishi S, Nakano M, Fujii N, Nagasawa T, Nakamura T. Stromal cell-derived factor 1/CXCR4 signaling is critical for the recruitment of mesenchymal stem cells to the fracture site during skeletal repair in a mouse model. Arthritis Rheum. 2009;60:813–823. doi: 10.1002/art.24330. [DOI] [PubMed] [Google Scholar]
- 59.Ilizarov GA. Clinical application of the tension–stress effect for limb lengthening. Clin Orthop Relat Res. 1990:8–26. [PubMed] [Google Scholar]
- 60.Wan C, Gilbert SR, Wang Y, Cao X, Shen X, Ramaswamy G, Jacobsen KA, Alaql ZS, Eberhardt AW, Gerstenfeld LC, Einhorn TA, Deng L, Clemens TL. Activation of the hypoxia-inducible factor-1α pathway accelerates bone regeneration. Proc Natl Acad Sci U S A. 2008;105:686–691. doi: 10.1073/pnas.0708474105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jacobsen KA, Al-Aql ZS, Wan C, Fitch JL, Stapleton SN, Mason ZD, Cole RM, Gilbert SR, Clemens TL, Morgan EF, Einhorn TA, Gerstenfeld LC. Bone formation during distraction osteogenesis is dependent on both VEGFR1 and VEGFR2 signaling. J Bone Miner Res. 2008;23:596–609. doi: 10.1359/JBMR.080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarkka T, Sipola A, Jamsa T, Soini Y, Yla-Herttuala S, Tuukkanen J, Hautala T. Adenoviral VEGF-A gene transfer induces angiogenesis and promotes bone formation in healing osseous tissues. J Gene Med. 2003;5:560–566. doi: 10.1002/jgm.392. [DOI] [PubMed] [Google Scholar]
- 63.Geiger F, Bertram H, Berger I, Lorenz H, Wall O, Eckhardt C, Simank HG, Richter W. Vascular endothelial growth factor gene-activated matrix (VEGF165-GAM) enhances osteogenesis and angiogenesis in large segmental bone defects. J Bone Miner Res. 2005;20:2028–2035. doi: 10.1359/JBMR.050701. [DOI] [PubMed] [Google Scholar]
- 64.Huang YC, Kaigler D, Rice KG, Krebsbach PH, Mooney DJ. Combined angiogenic and osteogenic factor delivery enhances bone marrow stromal cell-driven bone regeneration. J Bone Miner Res. 2005;20:848–857. doi: 10.1359/JBMR.041226. [DOI] [PubMed] [Google Scholar]
- 65.Geiger F, Lorenz H, Xu W, Szalay K, Kasten P, Claes L, Augat P, Richter W. VEGF producing bone marrow stromal cells (BMSC) enhance vascularization and resorption of a natural coral bone substitute. Bone. 2007;41:516–522. doi: 10.1016/j.bone.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 66.Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, Olsen BR. VEGFA is necessary for chondrocyte survival during bone development. Development. 2004;131:2161–2171. doi: 10.1242/dev.01053. [DOI] [PubMed] [Google Scholar]
- 67.Pfander D, Kobayashi T, Knight MC, Zelzer E, Chan DA, Olsen BR, Giaccia AJ, Johnson RS, Haase VH, Schipani E. Deletion of Vhlh in chondrocytes reduces cell proliferation and increases matrix deposition during growth plate development. Development. 2004;131:2497–2508. doi: 10.1242/dev.01138. [DOI] [PubMed] [Google Scholar]
- 68.Pfander D, Cramer T, Schipani E, Johnson RS. HIF-1α controls extracellular matrix synthesis by epiphyseal chondrocytes. J Cell Sci. 2003;116:1819–1826. doi: 10.1242/jcs.00385. [DOI] [PubMed] [Google Scholar]
- 69.Robins JC, Akeno N, Mukherjee A, Dalal RR, Aronow BJ, Koopman P, Clemens TL. Hypoxia induces chondrocyte-specific gene expression in mesenchymal cells in association with transcriptional activation of Sox9. Bone. 2005;37:313–322. doi: 10.1016/j.bone.2005.04.040. [DOI] [PubMed] [Google Scholar]
- 70.Amarilio R, Viukov SV, Sharir A, Eshkar-Oren I, Johnson RS, Zelzer E. HIF-1α regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development. 2007;134:3917–3928. doi: 10.1242/dev.008441. [DOI] [PubMed] [Google Scholar]
- 71.Tamiya H, Ikeda T, Jeong JH, Saito T, Yano F, Jung YK, Ohba S, Kawaguchi H, Chung UI, Choi JY. Analysis of the Runx2 promoter in osseous and non-osseous cells and identification of HIF2A as a potent transcription activator. Gene. 2008;416:53–60. doi: 10.1016/j.gene.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 72.Deckers MM, Karperien M, van der Bent C, Yamashita T, Papapoulos SE, Lowik CW. Expression of vascular endothelial growth factors and their receptors during osteoblast differentiation. Endocrinology. 2000;141:1667–1674. doi: 10.1210/endo.141.5.7458. [DOI] [PubMed] [Google Scholar]
- 73.Otomo H, Sakai A, Uchida S, Tanaka S, Watanuki M, Moriwaki S, Niida S, Nakamura T. Flt-1 tyrosine kinase-deficient homozygous mice result in decreased trabecular bone volume with reduced osteogenic potential. Bone. 2007;40:1494–1501. doi: 10.1016/j.bone.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 74.Midy V, Plouet J. Vasculotropin/vascular endothelial growth factor induces differentiation in cultured osteoblasts. Biochem Biophys Res Commun. 1994;199:380–386. doi: 10.1006/bbrc.1994.1240. [DOI] [PubMed] [Google Scholar]
- 75.Mayr-Wohlfart U, Waltenberger J, Hausser H, Kessler S, Gunther KP, Dehio C, Puhl W, Brenner RE. Vascular endothelial growth factor stimulates chemotactic migration of primary human osteoblasts. Bone. 2002;30:472–477. doi: 10.1016/s8756-3282(01)00690-1. [DOI] [PubMed] [Google Scholar]
- 76.Mayer H, Bertram H, Lindenmaier W, Korff T, Weber H, Weich H. Vascular endothelial growth factor (VEGF-A) expression in human mesenchymal stem cells: autocrine and paracrine role on osteoblastic and endothelial differentiation. J Cell Biochem. 2005;95:827–839. doi: 10.1002/jcb.20462. [DOI] [PubMed] [Google Scholar]
- 77.Villars F, Bordenave L, Bareille R, Amedee J. Effect of human endothelial cells on human bone marrow stromal cell phenotype: role of VEGF? J Cell Biochem. 2000;79:672–685. doi: 10.1002/1097-4644(20001215)79:4<672::aid-jcb150>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 78.Furumatsu T, Shen ZN, Kawai A, Nishida K, Manabe H, Oohashi T, Inoue H, Ninomiya Y. Vascular endothelial growth factor principally acts as the main angiogenic factor in the early stage of human osteoblastogenesis. J Biochem. 2003;133:633–639. doi: 10.1093/jb/mvg081. [DOI] [PubMed] [Google Scholar]
- 79.Peng H, Wright V, Usas A, Gearhart B, Shen HC, Cummins J, Huard J. Synergistic enhancement of bone formation and healing by stem cell-expressed VEGF and bone morphogenetic protein-4. J Clin Invest. 2002;110:751–759. doi: 10.1172/JCI15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Okuyama H, Krishnamachary B, Zhou YF, Nagasawa H, Bosch-Marce M, Semenza GL. Expression of vascular endothelial growth factor receptor 1 in bone marrow-derived mesenchymal cells is dependent on hypoxia-inducible factor 1. J Biol Chem. 2006;281:15554–15563. doi: 10.1074/jbc.M602003200. [DOI] [PubMed] [Google Scholar]
- 81.Nakagawa M, Kaneda T, Arakawa T, Morita S, Sato T, Yomada T, Hanada K, Kumegawa M, Hakeda Y. Vascular endothelial growth factor (VEGF) directly enhances osteoclastic bone resorption and survival of mature osteoclasts. FEBS Lett. 2000;473:161–164. doi: 10.1016/s0014-5793(00)01520-9. [DOI] [PubMed] [Google Scholar]
- 82.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood. 1996;87:3336–3343. [PubMed] [Google Scholar]
- 83.Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD, Nishikawa S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature. 1990;345:442–444. doi: 10.1038/345442a0. [DOI] [PubMed] [Google Scholar]
- 84.Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, Tan HL, Elliott G, Kelley MJ, Sarosi I, Wang L, Xia XZ, Elliott R, Chiu L, Black T, Scully S, Capparelli C, Morony S, Shimamoto G, Bass MB, Boyle WJ. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96:3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niida S, Kaku M, Amano H, Yoshida H, Kataoka H, Nishikawa S, Tanne K, Maeda N, Nishikawa S, Kodama H. Vascular endothelial growth factor can substitute for macrophage colony-stimulating factor in the support of osteoclastic bone resorption. J Exp Med. 1999;190:293–298. doi: 10.1084/jem.190.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Niida S, Kondo T, Hiratsuka S, Hayashi S, Amizuka N, Noda T, Ikeda K, Shibuya M. VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc Natl Acad Sci U S A. 2005;102:14016–14021. doi: 10.1073/pnas.0503544102. [DOI] [PMC free article] [PubMed] [Google Scholar]