

Abstract
N-type and P/Q-type Ca2+ channels are inhibited by neurotransmitters acting through G protein-coupled receptors in a membrane-delimited pathway involving Gβγ subunits. Inhibition is caused by a shift from an easily activated “willing” (W) state to a more-difficult-to-activate “reluctant” (R) state. This inhibition can be reversed by strong depolarization, resulting in prepulse facilitation, or by protein kinase C (PKC) phosphorylation. Comparison of regulation of N-type Ca2+ channels containing Cav2.2a α1 subunits and P/Q-type Ca2+ channels containing Cav2.1 α1 subunits revealed substantial differences. In the absence of G protein modulation, Cav2.1 channels containing Cavβ subunits were tonically in the W state, whereas Cav2.1 channels without β subunits and Cav2.2a channels with β subunits were tonically in the R state. Both Cav2.1 and Cav2.2a channels could be shifted back toward the W state by strong depolarization or PKC phosphorylation. Our results show that the R state and its modulation by prepulse facilitation, PKC phosphorylation, and Cavβ subunits are intrinsic properties of the Ca2+ channel itself in the absence of G protein modulation. A common allosteric model of G protein modulation of Ca2+-channel activity incorporating an intrinsic equilibrium between the W and R states of the α1 subunits and modulation of that equilibrium by G proteins, Cavβ subunits, membrane depolarization, and phosphorylation by PKC accommodates our findings. Such regulation will modulate transmission at synapses that use N-type and P/Q-type Ca2+ channels to initiate neurotransmitter release.
Keywords: neuromodulation, calcium channels, Gβγ subunits, protein phosphorylation
Neuronal voltage-gated Ca2+ channels are involved in multiple cellular functions including neurotranstransmitter release, Ca2+-mediated regulatory processes, and generation of dendritic action potentials. Electrophysiological and pharmacological studies distinguish at least six classes of Ca2+ currents designated L-, N-, P-, Q-, R-, and T-type (1, 2). Ca2+ channels consist of complexes of a pore-forming α1 subunit with α2δ, β, and γ subunits (2, 3). Ca2+ channels containing Cav2.2 α1 subunits (formerly α1B; ref. 4) are responsible for N-type currents, and Ca2+ channels containing Cav2.1 α1 subunits (formerly α1A; ref. 4) are responsible for both P- and Q-type Ca2+ currents (5–10). The Cav2.1 and Cav2.2 channels are located in presynaptic nerve terminals (10–13) and are responsible for the Ca2+ entry that triggers neurotransmitter release at most synapses (14, 15).
These two channel types are inhibited by neurotransmitter receptors acting through pertussis toxin-sensitive G proteins via membrane-delimited pathways that cause a positive shift in the voltage dependence of channel activation (16–18). This inhibition can be reversed by strong depolarization leading to prepulse facilitation of Ca2+ currents (19–22). The effect of G proteins has been modeled as a shift in channel state from a “willing” (W) state, in which activation occurs rapidly at relatively negative membrane potentials, to a “reluctant” (R) state, in which activation is slower and requires stronger depolarization (16). G-protein modulation is mediated by Gβγ subunits (23, 24) in part through direct binding to a site in the intracellular loop connecting domains I and II of the α1 subunit of the Ca2+ channel (25–28). N-type and P/Q-type Ca2+ channels are regulated also by protein kinase C (PKC). PKC can increase the activity of N-type and P/Q-type Ca2+ channels directly (29) and also can reverse their inhibition by G proteins (30, 31) by phosphorylating site(s) in the intracellular loop connecting domains I and II (28, 32). Such modulation of Ca2+ channels is thought to influence the efficiency of synaptic transmission strongly (17).
We have expressed specific isoforms of Cav2.1 and Cav2.2 channels in the human embryonic kidney cell line tsA-201 and analyzed their function, regulation by G proteins, facilitation by depolarizing prepulses, and modulation by PKC. Our results reveal surprising differences in modulation of these two channel types and suggest a common allosteric mechanism for Ca2+-channel modulation by these diverse regulatory pathways.
Experimental Procedures
cDNAs encoding α12.1 (rbA isoform; ref. 33), α12.2a (rbB-I isoform; ref. 5) and β1b were cloned in pMT2XS (34), α2δ (35) in pZEM228, and CD8 in EBO-pcD. tsA-201 cells were transfected with the α1, α2δ, and β cDNAs in 1:1:1 molar ratios plus CD8 by using either calcium phosphate or Lipofectamine (Stratagene) and incubated for at least 48 h. Positively transfected cells were identified by labeling with anti-CD8 antibody tagged with fluorophore and analyzed by whole-cell patch clamp as described (24, 36). Currents were recorded and filtered at 10 kHz with an eight-pole Bessel filter. Leak and capacitative currents were measured and subtracted using the P/-4 method. Cells were bathed in an external solution containing 100 mM Tris, 4 mM MgCl2, and 10 mM BaCl2 with pH adjusted to 7.3 with methanesulfonic acid. The internal pipette solution consisted of 120 mM aspartic acid, 5 mM CaCl2, 2 mM MgCl2, 10 mM Hepes, 10 mM EGTA, and 2 mM Mg-ATP with pH adjusted to 7.3 with CsOH. When indicated in the figure legends, GTPγS was added to the internal solution at a concentration of 0.6 mM. 1-oleoyl-2-acetyl glycerol (OAG) was prepared as a 2-mM stock in DMSO. Cells were preincubated with 20 μM OAG for 2–4 h before recording. Recordings were performed in the absence of added OAG. Experiments describing the voltage dependencies were fit with Boltzmann relationships of the form 100%/{1 + exp[(V − Vx)/k]}, in which Vx is the voltage of half activation (Va), inactivation (Vh), or prepulse facilitation (Vf), and k is a slope factor.
Results
Voltage-Dependent Gating of Expressed P/Q-Type and N-Type Ca2+ Channels.
P/Q-type Ca2+ channels composed of α12.1, α2δ, and β1b subunits and N-type Ca2+ channels composed of α12.2a, α2δ, and β1b subunits were expressed in the human embryonic kidney cell line tsA-201, and Ba2+ currents were measured in the whole-cell voltage-clamp configuration. Activation and inactivation kinetics during a test pulse to +30 mV are much slower for Cav2.2a than for Cav2.1 (Fig. 1A). Cav2.2a inactivates with a time constant of 313 ± 28 ms, whereas Cav2.1 exhibits a time constant of 127 ± 24 ms for inactivation (Fig. 1A). The voltage dependence of activation of Cav2.2a is more positive (Va = 42.2 mV) and more shallow than for Cav2.1 (Va = 23.6 mV; Fig. 1B, ○ and ●). Steady-state inactivation from closed states during a 4-s conditioning pulse was observed at more-negative potentials (Vh = −41.5 ± 0.42 mV) for Cav2.2a than for Cav2.1 (Vh = −25.4 ± 2.2 mV; Fig. 1B). Thus, these two channel types differ markedly in both activation and inactivation behavior.
Figure 1.

Voltage-dependent activation and inactivation of the Cav2.1 and Cav2.2a Ca2+ channels. (A) Rates of activation and inactivation. Ba2+ currents were recorded during 100-ms (Upper) or 1,000-ms (Lower) pulses to +30 mV from holding potentials of −60 mV (Cav2.1) or −70 mV (Cav2.2a). (B) Voltage dependence of activation and inactivation. To measure activation, tail currents were recorded at a holding potential of −60 mV (Cav2.1) or −70 mV (Cav2.2a) after a 4-ms (Cav2.1) or 50-ms (Cav2.2a) test pulse to the indicated potential. Means ± SEM of normalized tail currents were plotted as a function of the test voltage. ●, Cav2.1; ○, Cav2.2a. To measure inactivation, tail currents were recorded after a 4-s prepulse to the indicated potential followed by a 4-ms (Cav2.1) or 10-ms (Cav2.2a) test pulse to +30 mV. Means ± SEM of normalized tail currents were plotted as a function of prepulse voltage. ▪, Cav2.1; ▫, Cav2.2a. (C) Voltage dependence of activation in the presence of intracellular GTPγS. ▴, Cav2.1; ▵, Cav2.2a. Results for activation of Cav2.1 (●) and Cav2.2a (○) in the absence of GTPγS are replotted from B for comparison. (D) Voltage dependence of activation in the absence and presence of intracellular GDPβS for Cav2.1 (●, control; ▵, GDPβS) and Cav2.2a (○, control; ▿, GDPβS).
G Protein Modulation.
In the presence of GTPγS in the intracellular solution to activate G proteins, the voltage dependence of activation of Cav2.1 was shifted positively and was less steep than in control intracellular solution (Fig. 1C, compare ▴ with ●), reflecting inhibition of the Ca2+ channel by Gβγ caused by transition to the reluctant (R) state. In contrast, the voltage dependence of activation of Cav2.2a channels was unaffected by GTPγS (Fig. 1C, compare ○ with ▵). Under GTPγS-modulated conditions, the voltage dependence of activation of Cav2.1 was comparable to that of Cav2.2a channels without GTPγS (Fig. 1C, compare ▴ with ○). The positive value of Va and the shallow voltage dependence of activation of Cav2.2a suggest that it is tonically in the R state.
Because Cav2.2a in the absence of GTPγS resembles a G protein-modulated Ca2+ channel in the R state, we investigated whether the G protein inhibitor GDPβS could cause a negative shift in the voltage dependence of activation. No significant difference in activation of Cav2.1 or Cav2.2a was detected with 2 mM GDPβS in the intracellular solution (Fig. 1D), indicating that there was no tonic modulation of Cav2.2a by activated G proteins in tsA-201 cells. Similarly, we found that the sulfhydryl agent N-ethyl maleimide, a selective inhibitor of G proteins, had no effect on these channels (ref. 37 and data not shown). These data support the conclusion that Cav2.2a is tonically in the R state without G protein modulation, whereas Cav2.1 is tonically in the W state under basal conditions in tsA-201 cells.
Prepulse Facilitation.
In many cell types, G protein-mediated effects on the Ca2+ current can be reversed by brief depolarizing prepulses, leading to prepulse facilitation (19–21). This reversal of G protein modulation is thought to reflect a shift of Ca2+ channels from the R to the W state induced by strong depolarization (16). To determine the facilitation properties of Cav2.1 and Cav2.2a, we examined the voltage dependence of activation in the absence or presence of a depolarizing prepulse to +100 mV (Fig. 2). In the presence of GTPγS, the Ba2+ currents through Cav2.1 are facilitated over a wide voltage range by depolarizing prepulses (Fig. 2A; ref. 24). The midpoint of the activation curve (Va) of the facilitated current is shifted from 29.1 ± 3.5 mV to 18.6 ± 2.7 mV, and the facilitated activation curve is steeper than the control. Moreover, Cav2.1 activates more rapidly after a facilitating prepulse (Fig. 2A, Inset). These results are consistent with previous studies of G protein modulation and prepulse facilitation of P/Q-type Ca2+ channels in neurons (38, 39).
Figure 2.

Facilitation of Cav2.1 and Cav2.2a. (A) Prepulse facilitation of Cav2.1 channels. A 4-ms test pulse (test 1) to the indicated test potential was applied from the holding potential of −60 mV. After 1 s, a 10-ms conditioning prepulse to +100 mV was applied, the cell was repolarized to −60 mV for 10 ms, and a second 4-ms test pulse (test 2) identical to test-pulse 1 was applied. Means ± SEM of Ba2+ tail currents were plotted against test-pulse potentials. ○, test 1; ●, test 2. (B) Prepulse facilitation of Cav2.2a channels. As in A, except 10-ms test pulses were used, and the repolarization was for 1 ms to −70 mV. ○, test 1; ●, test 2. Current traces shown above A and B were recorded during and after test 1 and test 2 at +30 mV. (C) Voltage dependence of facilitation. A 4-ms (α1) or 10-ms (Cav2.2a) test pulse to +30 mV was applied from the holding potential of −60 mV (Cav2.1) or −70 mV (Cav2.2a). After 1 s, a 10-ms conditioning prepulse to the indicated potential was applied, the cell was repolarized to −60 mV (Cav2.1) for 10 ms or −70 mV (Cav2.2a) for 1 ms, and a second 4-ms (Cav2.1) or 10-ms (Cav2.2a) test pulse (test 2) to +30 mV was applied. Peak tail currents following each test pulse were measured. The facilitation ratio was calculated by dividing currents in test 2 by the currents in test 1, and means ± SEM of normalized facilitation ratios were plotted against the conditioning pulse potential. ●, Cav2.1; ○, Cav2.2a.
Surprisingly, although the expressed Cav2.2a channels were unaffected by GTPγS and GDPβS (Fig. 1 C and D), striking facilitation of Ba2+ currents through Cav2.2a was observed in the absence of G protein activation (Fig. 2B). After the prepulse to +100 mV, the voltage-dependent activation curve became steeper and was shifted from a Va value of 36.1 ± 5.3 to 24.8 ± 1.5 mV (Fig. 2B). In addition, the maximum level of Ba2+ current attained during the 10-ms test pulse increased at the most positive test-pulse potentials, and the rate of activation accelerated 25-fold from a time constant of 17.1 ± 2.0 to 0.69 ± 0.02 ms (Fig. 2B).
Facilitation increases with more-positive prepulses for both Cav2.1 and Cav2.2a (Fig. 2C). In fits with single Boltzmann equations, Cav2.2a shows a higher maximum facilitation ratio of 4.61 ± 0.71 compared with a ratio of 1.52 ± 0.10 for Cav2.1. Half-maximal facilitation of Cav2.1 is observed at 65.1 ± 4.1 vs. 97.6 ± 19 mV for Cav2.2a, and the voltage dependence of facilitation is much steeper for Cav2.1 (k = 14.7 ± 2.5) vs. Cav2.2a (k = 41.6 ± 2.8; Fig. 2C).
Rates of Onset and Reversal of Prepulse Facilitation.
The influence of prepulse duration on facilitation was examined by varying the length of the prepulse between 1 and 1,000 ms. For both channel types, the facilitation ratio increases, reaches a plateau, and then decreases with increasing duration of the prepulse (Fig. 3A). The form of these curves reflects the interaction of three processes: the rate of facilitation, the rate of reversal of facilitation, and the rate of inactivation during the prepulse. The rate of facilitation is 3-fold slower for Cav2.2a (τ = 15.4 ± 0.9 ms) than for Cav2.1 (τ = 4.8 ± 0.4 ms; Fig. 3B). Little facilitation of Cav2.1 is observed in the absence of GTPγS, and the Ba2+ current is depressed below the unfacilitated level for Cav2.1 and Cav2.2a during long prepulses (Fig. 3A) because of voltage-dependent inactivation. During single depolarizing pulses, the activity of Cav2.2a is inactivated with a time constant of 132 ± 26 ms at +100 mV vs. 462 ± 254 ms for Cav2.1 (Fig. 3C). Likewise, loss of facilitation after a prepulse is also much faster for Cav2.2a with a time constant of 4.5 ± 1.0 ms compared with Cav2.1 with a time constant of 51.3 ± 7.3 ms (Fig. 3D).
Figure 3.

Rates of onset and reversal of prepulse facilitation. (A) Time course of prepulse facilitation and inactivation. Facilitation was measured as in Fig. 2 A and C with prepulses of the indicated durations for Cav2.1, control (○), GTPγS (●). ▴, Cav2.2a. (B) Rate of onset of prepulse facilitation. Facilitation was measured as in Fig. 2 A and C with prepulses of the indicated durations. ●, Cav2.1 with GTPγS; ○, Cav2.2a, control. (C) Rates of inactivation. Inactivation was measured as in Fig. 1B by using prepulses of the indicated durations to +100 mV. ●, Cav2.1; ○, Cav2.2a. (D) Rates of reversal of prepulse facilitation. Facilitation was measured as in Fig. 2 A and B with a prepulse to +30 mV and repolarization for the indicated time periods before test-pulse 2. ●, Cav2.1; ○, Cav2.2a.
Regulation by PKC.
PKC has direct regulatory effects on N-type and P/Q-type Ca2+ channels in neurons (29) and also reverses their inhibition by neurotransmitters acting through G protein-coupled receptors (30, 31). Activation of PKC with OAG shifts the voltage dependence of activation of Cav2.1 in the presence of GTPγS from a midpoint of 33.3 ± 2.7 mV to a midpoint of 13.4 ± 1.8 mV (Fig. 4A), which completely reverses the effect of G protein activation and occludes voltage-dependent facilitation (Fig. 4A).
Figure 4.

Influence of PKC on Ca2+-channel gating and modulation. (A and B) Voltage dependence of activation and facilitation were measured as in Fig. 1 with and without treatment with OAG. (A) Cav2.1. Activation: ●, GTPγS; ○, GTPγS plus OAG; ▵, PKC-inhibitor peptide (IP). Facilitation: ▫, GTPγS plus OAG test 1; ▪, GTPγS plus OAG test 2. (B) Cav2.2a. ●, no addition; ○, OAG; ▵, OAG + PKC-IP. Facilitation: ▫, OAG test 1; ▪, OAG test 2. (C and D) Influence of PKC on inactivation. Voltage dependence of inactivation was measured as in Fig. 1 in the presence and absence of OAG. (C) Cav2.1: ○, OAG; ●, GTPγS; ▵, PKC-IP. (D) Cav2.2a: ○, OAG; ●, GTPγS; ▵, PKC-IP.
Because activation of Cav2.2a is not altered by GTPγS, we analyzed the effect of the PKC activator by using the control intracellular solution. Under these conditions, OAG shifted the voltage dependence of activation from a Va of 42.2 ± 1.1 to 37.3 ± 2.3 mV (Fig. 4B). Prepulse facilitation of Cav2.2a also was diminished significantly, although substantial facilitation remained. After activation of PKC with OAG, the activation curves for Cav2.2a channels before and after prepulse facilitation approached the same maximum (Fig. 4B) unlike the increase in maximum current observed with facilitation under control conditions (Fig. 2B). Thus, the intrinsic facilitation of α1 is inhibited by activation of PKC in a similar manner to the G protein-dependent facilitation of Cav2.1, but the inhibition is less complete.
Stimulation of PKC by OAG also accelerated inactivation for both channel types (Fig. 4 C and D). The time constant for inactivation at +30 mV was decreased from 127 ± 24 to 49 ± 16 ms for Cav2.1 and from 313 ± 28 to 115 ± 17 ms for Cav2.2a. A similar increase in inactivation rate was observed for test potentials from 15 to 35 mV. In addition, the midpoint for steady-state inactivation was shifted from −25.4 ± 2.2 to −47.5 ± 1.3 mV for Cav2.1 (Fig. 4C) and from −41.5 ± 0.42 to −55.2 ± 3.4 mV for Cav2.2a (Fig. 4D).
All the effects of OAG could be reversed by the PKC-IP in the pipette solution (Fig. 4). These results confirm the specificity of the effects of OAG and support the conclusion that phosphorylation by PKC alters activation, inactivation, voltage-dependent facilitation, and G protein modulation.
Effects of Ca2+-Channel β Subunits.
Caβ subunits influence Cav2 channel voltage dependence, prepulse facilitation, and modulation by G proteins (2, 40–43). Therefore, we examined key features of modulation of the Cav2.1 channel by G proteins in the absence of Caβ subunits, as well as in the presence of a different Caβ subunit, Caβ3 (44). Unfortunately, currents conducted by expressed α12.2a alone were too small to measure reliably.
In the presence of Caβ1b or Caβ3 subunits, whole-cell currents increased 6.5-fold from −101 ± 21 pA (n = 11) for α12.1 subunits alone to −660 ± 120 pA (Caβ1b; n = 28) or −667 ± 235 pA (Caβ3; n = 21), as for expression in Xenopus oocytes (8, 45). The voltage dependence of activation was similar for cells coexpressing either the Caβ1b or Caβ3 subunits (Fig. 5A) (Caβ1b, Va = 23.6 ± 1.4 mV; Caβ3, Va = 22.0 ± 0.3 mV). However, activation curves from cells expressing the α12.1 subunit alone had a more-positive voltage dependence (Fig. 5A; Va = 30.4 ± 1.6 mV).
Figure 5.

Effects of Cavβ subunits on properties of Cav2.1 Ca2+ channels. Cav2.1 and α2δ subunits were expressed without (▵) or with Ca2+-channel β1b (●) or β3 (○) subunits in tsA-201 cells. (A) Voltage dependence of activation and inactivation measured as in Fig. 1. (B) Time constants of inactivation determined during 1,000-ms long test pulses to the indicated potentials. (C) Cav2.1 and α2δ subunits were expressed with Ca2+-channel β3 subunits and studied in the presence (○) or absence (●) of intracellular GTPγS. (D) Cav2.1 and α2δ subunits were expressed and studied in the presence (▵) or absence (▴) of GTPγS. (E and F) Activation curves recorded with intracellular GTPγS in the absence (○) and presence (●) of a preceding conditioning prepulse as described for Fig. 1 from cells coexpressing α12.1, α2δ, and Caβ1b (E) or α12.1 and α2δ alone (F).
Coexpression of Caβ subunits also had substantial effects on inactivation. The time constant for inactivation of open channels during a depolarization to +15 mV decreased from 237 ± 29 ms (n = 3) to 110 ± 14 ms for Caβ1b and 88 ± 12 ms for Caβ3 (Fig. 5B), with less pronounced effects for stronger depolarizations. In contrast, the presence of Caβ subunits had little effect on the voltage dependence of steady-state inactivation (data not shown). Overall, expressing α12.1 subunits in the absence of Caβ subunits reduces current amplitude, shifts the voltage dependence of activation toward more positive potentials, and slows the rates of activation and inactivation. The resulting channels in their unmodulated state are similar to α12.1β1b channels modulated by G proteins or Cav2.2a channels under basal conditions.
Effects of Cavβ Subunits on G Protein Modulation and Prepulse Facilitation.
For channels containing α12.1 and Caβ3 subunits, Va shifted from 22.0 ± 0.3 mV to 33.8 ± 2.3 mV when the pipette solution contained GTPγS (Fig. 5C) as for channels with β1b subunits. In contrast, the positively shifted voltage dependence of channels containing α12.1 alone was affected little by GTPγS (Fig. 5D). This finding is consistent with the idea that channels having only α12.1 subunits cannot be modulated by G proteins because they are already in the R state. Coexpression of α12.1 subunits with β1b subunits (Fig. 5E) or β3 subunits (not shown) resulted in prepulse facilitation over a broad range of test-pulse voltages. However, when α12.1 was expressed without β subunits, there was little facilitation, even in the presence of intracellular GTPγS (Fig. 5F). Thus, although α12.1 expressed alone resembles Cav2.2a in its kinetic properties and its failure to be modulated by GTPγS, it differs in being affected little by prepulse facilitation protocols under any condition tested. These results are consistent with the idea that Caβ subunits are required for α12.1 to enter the W state.
Discussion
An Allosteric Model of G Protein Modulation.
G protein modulation of N-type Ca2+ channels is thought to shift them from a state in which activation is easier (W) to a state in which activation is slower and more difficult (R; ref. 16). The W and R states are thought to be intrinsic states of the Ca2+ channel, and G protein activation is thought to modulate the equilibrium between these states (16), but no direct evidence that the W and R states are intrinsic to the Ca2+ channel has been presented. The effect of G proteins is mediated by the Gβγ subunits (23, 24) through direct binding to site(s) in the α1 subunits (25–28). Extending the original model of Bean (16) to include binding of Gβγ subunits, G protein regulation can be considered as a cyclic allosteric regulation of Ca2+-channel functional state by binding of Gβγ as follows:
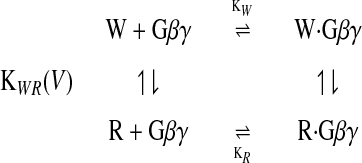 |
1 |
Activation from both the W and R states after depolarization involves voltage-dependent transitions through a series of closed states to an open state (46) accompanied by outward movement of gating charge at each step (47). For the R state, the initial voltage-dependent transitions to neighboring closed states or to the W state are slow (47), resulting in the reluctant gating mode.
This model is derived directly from the allosteric model of Monod et al. (48) and is similar in form to models used for neurotoxin activation of sodium channels (49) and block of sodium channels by local anesthetics (the “modulated receptor hypothesis”; ref. 50). The fraction of channels in the R state is controlled by the equilibrium constant (KWR) for the transition from R to W, the binding constants for Gβγ to the W and R states, and the concentration of activated Gβγ according to the following equation:
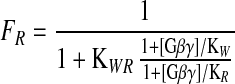 |
2 |
There are not enough quantitative data on intracellular G protein concentrations, G protein binding affinity, and voltage-dependent Ca2+-channel modulation to fit the parameters of this model objectively. However, our results for the different modulators studied here are qualitatively consistent with this model with two assumptions: (i) KWR is smaller for certain channel types such that they are tonically in the R state in the absence of G protein modulation; and (ii) each modulator allosterically alters KWR as a primary mechanism of action.
Different Values for KWR Account for Differences in Channel Modulation.
When expressed with Ca2+-channel β subunits, Cav2.1 activates at much more negative membrane potentials than Cav2.2a (Fig. 1), and its voltage dependence of activation is shifted to more positive membrane potentials by activation of G proteins. In contrast, Cav2.2a expressed with β subunits and Cav2.1 expressed without β subunits activate at more positive membrane potentials, and their voltage dependence of activation is unaffected by G protein activation or inhibition. These results are consistent with the conclusion that KWR is much smaller for these channels under our experimental conditions, causing most of these channels to be in the R state without G protein activation.
G Protein Activation Shifts α12.1β to the R State, Whereas α12.1 and α12.2aβ Are Intrinsically in the R State.
In this model, activation of G proteins with GTPγS shifts α12.1β to the R state and thereby shifts its voltage dependence of activation to more positive membrane potentials and slows its activation. In contrast, activation of G proteins with GTPγS has no effect on α12.1 alone or on α12.2aβ because all of the channels are in the R state, and no further shift can be caused by Gβγ. Inhibition of G proteins by GDPβS or N-ethyl maleimide also has no effect on facilitation of α12.2a, even though the heterotrimeric form of the G protein induced by GDPβS or N-ethyl maleimide is inactive in signal transduction. These results provide evidence that a Ca2+ channel can adopt the R state without G protein activation and therefore directly support the idea that the W and R states are intrinsic functional states of the Ca2+ channel.
Prepulse Facilitation Shifts α12.1β and α12.2aβ to the W State but Has a Smaller Effect on α12.1.
In the context of this model, the effect of strong depolarizing prepulses is viewed as a voltage-dependent increase in the value of KWR. This increase is large enough to shift α12.1β channels toward the W state even in the presence of activated G proteins. The voltage-dependent increase in KWR is also large enough to shift α12.2a to the W state, and thus prepulse facilitation is observed in the absence of G protein modulation. However, it is not large enough to cause substantial facilitation of channels containing α12.1 without Caβ subunits. The results with α12.2a provide the first evidence that prepulse facilitation is intrinsic to the Ca2+ channel itself and can occur without G protein modulation. The results with coexpression of Caβ subunits provide evidence that they can act by shifting channels from the R to the W state.
Phosphorylation by PKC Shifts Both α12.1β and α12.2aβ to the W State.
Previous work shows that activation of PKC in neurons can increase Ca2+-channel activity directly (29) and also can reverse G protein inhibition and thereby increase Ca2+-channel activity indirectly (30, 31). Our results show that these two seemingly independent effects of PKC can be considered as two manifestations of the same intrinsic regulatory mechanism. For α12.1β Ca2+ channels, G protein modulation is required to shift Ca2+ channels to the R state, and activation of PKC can shift them back toward the W state and thereby facilitate Ca2+-channel activation. For α12.2aβ channels, PKC can act directly because these channels are already in the R state without G protein activation and can be shifted back to W by phosphorylation. These results implicate the Ca2+ channel α1 subunits as the sites of phosphorylation that cause this regulatory effect of PKC, which is in agreement with results identifying required PKC phosphorylation sites in the intracellular loop connecting domains I and II (27).
Caβ Subunits Also Shift Ca2+ Channels to the W State.
Numerous studies have shown that coexpression of Caβ subunits shifts the voltage dependence of activation of Ca2+ channels containing α12.1 and α12.2 subunits to more negative potentials (2, 34, 40, 41, 45). Our results show that α12.1 expressed alone is tonically in the R state and that coexpression of Caβ subunits shifts it to the W state. Thus, one functional effect of Caβ subunits is to shift channels to the willing gating mode. This effect of Caβ subunits is expected to oppose G protein modulation, as observed in both neurons and Xenopus oocytes (41–43). Paradoxically, although Caβ subunits oppose G protein modulation, we find that a Caβ subunit is required for G protein modulation of α12.1 (see also ref. 51). This surprising result is expected from the allosteric model of channel modulation because, if α12.1 expressed alone is tonically in the R state, G proteins cannot inhibit its activity further and therefore seem inactive. Thus, modulation of the equilibrium between the W and R states is important for the effects of G proteins, PKC, and Caβ subunits.
Subtypes of N-type and P/Q-type Ca2+ Channels.
Multiple molecular isoforms of N-type and P/Q-type Ca2+-channel α1 subunits have been identified. Our experiments compare the properties of two specific molecular isoforms, the Cav2.2a isoform of N-type channels, and the Cav2.1 isoform of P/Q-type channels. The regulatory properties of Cav2.2a described here differ significantly from the properties of Cav2.2b described previously (32, 52). Cav2.2b channels activate more rapidly and at more negative membrane potentials than the Cav2.2a channels studied here, and G protein activation is required to observe voltage-dependent facilitation. Thus, it is likely that the Cav2.2b isoform is tonically primarily in the W state while the Cav2.2a isoform is tonically in the R state. The following paper (37) identifies a single amino acid residue that specifies this difference in channel gating properties.
Acknowledgments
We thank Drs. Charles Chavkin and Kenneth Mackie for comments on a draft of the manuscript. This work was supported by Research Grant NS22625 from the National Institutes of Health to W.A.C. and a postdoctoral fellowship from the Deutsche Forschungsgemeinschaft to S.H.
Abbreviations
- W
willing
- R
reluctant
- PKC
protein kinase C
- OAG
1-oleoyl-2-acetyl
- IP
inhibitor peptide
References
- 1.Zhang J-F, Randall A D, Ellinor P T, Horne W A, Sather W A, Tanabe T, Schwarz T L, Tsien R W. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]
- 2.Hofmann F, Biel M, Flockerzi V. Annu Rev Neurosci. 1994;17:399–418. doi: 10.1146/annurev.ne.17.030194.002151. [DOI] [PubMed] [Google Scholar]
- 3.Catterall W A. Annu Rev Biochem. 1995;65:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 4.Ertel E A, Campbell K P, Harpold M M, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch T P, Tanabe T, Birnbaumer L, et al. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 5.Dubel S J, Starr T V B, Hell J W, Ahlijanian M K, Enyeart J J, Catterall W A, Snutch T P. Proc Natl Acad Sci USA. 1992;89:5058–5062. doi: 10.1073/pnas.89.11.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams M E, Feldman D H, McCue A F, Brenner R, Velicelebi G, Ellis S B, Harpold M M. Neuron. 1992;8:71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- 7.Mori Y, Friedrich T, Kim M S, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, et al. Nature (London) 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 8.Sather W A, Tanabe T, Zhang J F, Mori Y, Adams M E, Tsien R W. Neuron. 1993;11:291–303. doi: 10.1016/0896-6273(93)90185-t. [DOI] [PubMed] [Google Scholar]
- 9.Bourinet E, Soong T W, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi G W, Nargeot J, Snutch T P. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 10.Westenbroek R E, Sakurai T, Elliott E M, Hell J W, Starr T V B, Snutch T P, Catterall W A. J Neurosci. 1995;15:6403–6418. doi: 10.1523/JNEUROSCI.15-10-06403.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen M W, Jones O T, Angelides K J. J Neurosci. 1991;11:1032–1039. doi: 10.1523/JNEUROSCI.11-04-01032.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robitaille R, Adler E M, Charlton M P. Neuron. 1990;5:773–779. doi: 10.1016/0896-6273(90)90336-e. [DOI] [PubMed] [Google Scholar]
- 13.Westenbroek R E, Hell J W, Warner C, Dubel S J, Snutch T P, Catterall W A. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- 14.Tsien R W, Lipscombe D, Madison D V, Bley K R, Fox A P. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 15.Dunlap K, Luebke J I, Turner T J. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 16.Bean BP. Nature (London) 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 17.Hille B. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 18.Dolphin A C. Exp Physiol. 1995;80:1–36. doi: 10.1113/expphysiol.1995.sp003825. [DOI] [PubMed] [Google Scholar]
- 19.Marchetti C, Carbone E, Lux H D. Pflügers Arch. 1986;406:104–111. doi: 10.1007/BF00586670. [DOI] [PubMed] [Google Scholar]
- 20.Tsunoo A, Yoshii M, Narahashi T. Proc Natl Acad Sci USA. 1986;83:9832–9836. doi: 10.1073/pnas.83.24.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elmslie K S, Zhou W, Jones S W. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. [DOI] [PubMed] [Google Scholar]
- 22.Ikeda S R. J Physiol (London) 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikeda S R. Nature (London) 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 24.Herlitze S, Garcia D E, Mackie K, Hille B, Scheuer T, Catterall W A. Nature (London) 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 25.Page K M, Stephens G J, Berrow N S, Dolphin A C. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herlitze S, Hockerman G H, Scheuer T, Catterall W A. Proc Natl Acad Sci USA. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Waard M, Liu H, Walker D, Scott V E S, Gurnett C A, Campbell K P. Nature (London) 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 28.Zamponi G W, Bourinet E, Nelson D, Nargeot J, Snutch T P. Nature (London) 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Tsien R W. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- 30.Swartz K J. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- 31.Swartz K J, Merritt A, Bean B P, Lovinger D M. Nature (London) 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- 32.Stea A, Soong TW, Snutch T P. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- 33.Starr T V B, Prystay W, Snutch T P. Proc Natl Acad Sci USA. 1991;88:5621–5625. doi: 10.1073/pnas.88.13.5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stea A, Dubel S J, Pragnell M, Leonard J P, Campbell K P, Snutch T P. Neuropharmacology. 1993;32:1103–1116. doi: 10.1016/0028-3908(93)90005-n. [DOI] [PubMed] [Google Scholar]
- 35.Ellis S B, Williams M E, Ways N R, Brenner R, Sharp A H, Leung A T, Campbell K P, McKenna E, Koch W J, Hui A, et al. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 36.Margolskee R F, McHendry-Rinde B, Horn R. BioTechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- 37.Zhong H, Li B, Scheuer T, Catterall W A. Proc Natl Acad Sci USA. 2001;98:4705–4709. doi: 10.1073/pnas.051629098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mintz I M, Bean B P. Neuron. 1993;10:889–898. doi: 10.1016/0896-6273(93)90204-5. [DOI] [PubMed] [Google Scholar]
- 39.Currie K P M, Fox A P. J Neurosci. 1997;17:4570–4579. doi: 10.1523/JNEUROSCI.17-12-04570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berrow N S, Campbell V, Fitzgerald E M, Brickley K, Dolphin A C. J Physiol (London) 1995;482:481–491. doi: 10.1113/jphysiol.1995.sp020534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell V, Berrow N S, Fitzgerald E M, Brickley K, Dolphin A C. J Physiol (London) 1995;485:365–372. doi: 10.1113/jphysiol.1995.sp020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourinet E, Soong T W, Stea A, Snutch T P. Proc Natl Acad Sci USA. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roche J P, Anantharam V, Treistman S N. FEBS Lett. 1995;371:43–46. doi: 10.1016/0014-5793(95)00860-c. [DOI] [PubMed] [Google Scholar]
- 44.Castellano A, Wei X, Birnbaumer L, Perez-Reyes E. J Biol Chem. 1993;268:3450–3455. [PubMed] [Google Scholar]
- 45.De Waard M, Campbell K P. J Physiol (London) 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boland L M, Bean B P. J Neurosci. 1993;13:516–533. doi: 10.1523/JNEUROSCI.13-02-00516.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones L P, Patil P G, Snutch T P, Yue D T. J Physiol (London) 1997;498:601–610. doi: 10.1113/jphysiol.1997.sp021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monod J, Wyman J, Changeux J-P. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 49.Catterall W A. J Biol Chem. 1977;252:8669–8676. [PubMed] [Google Scholar]
- 50.Hille B. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meir A, Bell D C, Stephens C J, Page K M, Dolphin A C. Biophys J. 2000;79:731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patil P G, deLeon M, Reed R R, Dubel S, Snutch T P, Yue D T. Biophys J. 1996;71:2509–2521. doi: 10.1016/S0006-3495(96)79444-4. [DOI] [PMC free article] [PubMed] [Google Scholar]