

Abstract
Tight regulation of both the NF-κB pathway and the autophagy process is necessary for maintenance of cellular homeostasis. Deregulation of both pathways is frequently observed in cancer cells and is associated with tumorigenesis and tumor cell resistance to cancer therapies. Autophagy is involved in several cellular functions regulated by NF-κB including cell survival, differentiation, senescence, inflammation, and immunity. On a molecular level, autophagy and NF-κB share common upstream signals and regulators and can control each other through positive or negative feedback loops, thus ensuring homeostatic responses. Here, we summarize and discuss the most recent discoveries that shed new light on the complex interplay between autophagy and NF-κB signaling pathways; this certainly has functional relevance in tumorigenesis and tumor responses to therapy.
Keywords: Autophagy, NF-κB, cancer, signaling pathways
1. Macroautophagy
In this section, we do not intend to cover the detailed signaling pathways involved in macroautophagy (referred to as autophagy hereafter) but to briefly summarize the regulation of autophagy in cancer cells. Readers interested in details of autophagy signaling pathways and regulation are referred to the several excellent and extensive reviews [1-7].
In eukaryotes, autophagy is a lysosomal process that induces the degradation of cytoplasmic constituents such as proteins, lipids, and organelles [8]. Autophagy is a pivotal regulator of several important physiological processes including development, cell survival, differentiation, and senescence (Figure 1) [2, 9]. This process is also involved in regulating inflammation and innate and adaptive immune responses [10-12]. Dysfunction of autophagy is linked to the development and the progression of human diseases (Figure 1) such as cancer, neurodegenerative disorders, liver diseases, inflammatory pathologies (such as Crohn's disease), and infectious diseases [13-15].
Figure 1.

Schematic representation of autophagy in mammals. The first step in the induction of autophagy is the formation of a multi-membrane structure, known as a phagophore which further elongates to form an autophagosome before enclosing cytoplasmic material. The autophagosome then fuses with the lysosome to form the autophagoly-sosome, in which the sequestered material is degraded by lysosomal hydrolases. The ULK1/2 complex and the class III phosphatidylinositol 3-kinase (Vps34) complex drive the first step of autophagosome formation (induction and nucleation). The elongation and the closure of the autophagosomal membranes (maturation) require the recruitment of two conjugation systems, Atg5-Atg12 and Atg8/LC3-phosphatidyl ethanolamine, to the phagophore. The lysosomal membrane protein Lamp 2 and the GTPase Rab7 together mediate the fusion of autophagosome with the lysosome. The major cellular functions of autophagy are shown. Depicted are the consequences of impaired autophagy in the development of certain human pathologies. For details, see text.
Autophagy contributes to the maintenance of cellular homeostasis by inducing the recycling of damaged organelles and toxic components such as aggregation-prone proteins [16-19]. Recent studies using tissue-specific deletion of autophagy-related genes have revealed the accumulation of ubiquitinated proteins and aggregates in autophagy-impaired tissues emphasizing the importance of basal autophagy in tissue homeostasis [20, 21]. By inducing degradation of cellular material, autophagy plays also an essential role in tissue remodeling during cell differentiation [2]. In response to starvation, autophagy is induced to provide nutrients and energy necessary to cope with changes in metabolism thus ensuring cell viability [21, 22]. In this sense, autophagy is essential for survival during the early neonatal starvation period [23]. Autophagy is also induced in response to stressful conditions including oxidative stress, hypoxia, endoplasmic reticulum stress, hormonal imbalance, pathogen-associated molecular patterns, DNA damage, and anti-cancer therapies [6, 24-26]. Under stress conditions, autophagy mostly plays a cytoprotective role; however, autophagy has been also shown to induce a cell death program [27-29].
Autophagy core machinery
Autophagy starts with the formation of a multi-membrane structure, the preautophagosomal membrane also called the phagophore (Figure 1). This membrane elongates to form the autophagic vesicle, known as autophagosome, before enclosing cytoplasmic components. When the autophagosome fuses with the lysosome, the autophagosomal contents are degraded and recycled to be used for the synthesis of macro-molecules and the production of energy [1, 3, 21, 30]. Genetic studies of autophagy in the yeast have led to the identification of autophagy -related genes (Atg genes) whose products orchestrate the steps of autophagosome formation, namely nucleation, expansion, uncoating, and completion. The core machinery of autophagy that drives autophagy in mammals involves three complexes formed by Atg proteins and lysosomal-associated membrane protein 2 (Lamp-2) and Rab7 proteins (Figure 1). The complex involved in the initiation of autophagy process is the Atg1/unc-51-like kinase (ULK) complex (ULK1/2, Atg13, Atg 101, Fip200 and Atg9). Next, the class III phosphatidylinositol 3-kinase (Vps34) complex (Vps34, Vps15, Atg6/Beclin1 and Barkor) generates phosphatidylinositol 3-phosphate (PI3P) by inducing the activity of PI3-kinase, a key event that subsequently leads to the elongation of autophagosomal membrane. Two conjugation systems comprised by Atg5-Atg12 and Atg8/microtubule-associated protein 1 light chain 3 (LC3) phosphatidyl-ethanolamine are required for the elongation and the closure of the auto-phagosomal membranes. Finally, the fusion of autophagosome with the lysosome requires the lysosomal membrane protein Lamp2 and the GTPase Rab7 [31-34]. The functions of the autophagy machinery core complexes are tightly regulated by different factors that favor or repress the induction of autophagy. For example, autophagy is positively regulated by the interaction of Atg6/Beclin 1 with Atg14L, ultraviolet radiation resistance associated gene (UVRAG), Bif-1, and Activating molecule in Beclin 1-regulated autophagy (Ambra1). Conversely, the binding of Beclin 1 with the proto-oncogene B-cell lymphoma 2 (Bcl-2) inhibits autophagy in several experimental settings [32].
Although autophagy has long been considered a non-selective process, a number of recent studies have revealed that autophagy can selectively degrade certain cytosolic cargos including protein aggregates, damaged organelles (such as mitochondria and peroxisomes), intracellular pathogens, and virus [17-19]. By regulating the turnover of damaged cellular constituents, autophagy prevents the accumulation of reactive oxygen species (ROS) and oxidative damage to DNA and may thus be a tumor-suppressive mechanism. In selective autophagy, cargos are recognized through interaction with specific receptor proteins. For example, p62 and NBR1 are cargo receptors for degradation of ubiquitinated proteins and Bnip3 (NIX) is required for the clearance of mitochondria (mitophagy) and cell death in response to hypoxia [19, 35].
Signaling pathways regulating autophagy
A variety of cellular stress signals converge to the mammalian target of rapamycin 1 (mTOR) complex 1 (mTORC1) to regulate autophagy upstream of the core machinery. During deprivation of nutrients and growth factors, autophagy is induced through inactivation of mTORC1, which drives the initial step of autophagy by triggering the activation the ULK1/2 complex [31]. Upstream of mTOR, several tumor suppressor genes, including LKB1, AMPK, TSC1/TSC2 and PTEN have been shown to inhibit mTORC1 and lead to the stimulation of autophagy. In contrast, tumor promoter pathways initiated by Class I PI3K, PKB/Akt, insulin and IGF receptors, Rheb and NF-κB activate mTORC1 and thereby repress autophagy [3, 25, 36-38]. Some other tumor regulatory molecules that control autophagy include oncoproteins Bcl-2 and BCl-xL (which inhibit Beclin 1-dependent autophagy), Ras (via class I PI3K), and hypoxia-inducible factor-1 (Hif-1) and tumor suppressors such as p53, DAPK, E2F-1, and FoxO proteins [6,39].
Autophagy and cancer
The relationship between autophagy and cancer is complex and depends on the tumor's nature, stage, and context. Autophagy can function as a tumor-suppressive mechanism or conversely as a tumor-promoting process [3, 38, 40-42].
Several genetic studies have established the link between the autophagy machinery and tumorigenesis [13]. For example, a monoallelic deletion of the essential gene Beclin 1 is frequently observed in ovarian, breast, and prostate cancers [43]. Defects in essential autophagy genes Atg5 and Beclin 1 promote tumorigenesis of certain cell lines. Allelic loss of Beclin 1 and deficiencies in expression of two other autophagy-regulating genes, Atg4C and Bif-1, render mice prone to tumor development [43-46]. Moreover, as mentioned above, the regulation of autophagy is tightly controlled by signaling pathways that regulate tumorigenesis. Indeed, inactivating mutations or allelic deletions in tumor suppressor genes whose products positively regulate autophagy (e.g., PTEN, TSC1/TSC2, and p53) are frequently observed in human tumors, emphasizing the tumor-suppressive function of autophagy. On the other hand, some tumors have aberrant activation of genes like Bcl-2, Akt, Class I PI3K, and NF-κB whose products have tumor promoter functions and repress autophagy [3, 38, 40-42]. However, in some instances oncogenes can promote autophagy stimulation as it has been shown for H-Ras (V12) and K-Ras (V12) [47].
Several mechanisms have been proposed to explain the tumor-suppressive role of autophagy. As stated above, one of them concerns the function of autophagy in the selective removal of toxic cellular components that produce ROS, a mechanism that would limit the incidence of DNA mutation and prevent chromosome instability and subsequent tumorigenesis [48] Autophagy may limit cell proliferation as demonstrated by results showing that enforced expression of Beclin 1 represses the growth of cancer cell xenografts [43, 49] and that Ras-induced downregulation of Beclin 1 expression enhances proliferation of malignant cells that remain viable upon their detachment from extracellular matrix [50]. In addition, through its ability to promote rapid protein turnover, autophagy facilitates the acquisition of the senescence phenotype, a tumor suppression mechanism that prevents tumor development through cell-cycle arrest, cellular remodeling, and secretion of specific cytokines [51]. The prevention of chronic tumor cell death mediated by autophagy may represent another tumor-suppressive function of this process. Indeed, in apoptosis-defective tumors exposed to metabolic stress, autophagy prevents chronic necrosis, limiting undesirable inflammatory responses that create a pro-tumorigenic environment [48]. The role of autophagy in the suppression of inflammation is also supported by the fact that defective autophagy enhances susceptibility to some inflammatory diseases such as Crohn's disease and pancreatitis [11].
On the other hand, autophagy contributes to tumor progression by maintaining the survival of established tumor cells in starvation conditions and in hypoxic microenvironments. This phenomenon might occur when cancer cells are located in the core of a solid tumor and are poorly vascularized; under these contexts, the cells have to survive under conditions of low oxygen (hypoxia) and reduced nutrients. Metabolic stress-induced autophagy may also render cancer cells more resistant to anti-cancer therapies [21, 52]. Moreover, the release pro-inflammatory factors such as the damage-associated molecular pattern molecule HMGB1 by stressed or dying tumor cells has been shown to favor the survival of stressed cells in tumor microenvironment by a mechanism that depends on the induction of autophagy [53, 54].
Autophagy, thus, appears to have a dual role in tumorigenesis: on the one hand, autophagy restricts tumor development by inhibiting inflammation, cell proliferation, and accumulation of cellular damages. On the other hand, autophagy sustains viability of established tumors exposed to environmental changes or subjected to anti-cancer treatments [3, 38, 40-42]. Depending on the context, autophagy may also promote or inhibit metastasis by limiting anoikis during extracellular matrix detachment or by promoting anti-tumor inflammatory responses and maintaining cancer cells dormancy, respectively [55].
Autophagy, cancer therapies and cell death
A variety of anticancer agents can induce autophagy; these include tamoxifen, rapamycin, temozolomide, histone deacetylase inhibitors, and a subset of DNA-damaging agents such as camptothecin, etoposide, and ionizing radiation [25, 26]. In most cases, the stimulation of autophagy by these agents results from a cytoprotective mechanism and confers tumor resistance to therapies. However, in some instances autophagy induced by anti-cancer therapy may facilitate the activation of cancer cell death programs [27, 29, 56]. Interestingly, there are molecular interconnections between autophagy and different cell death pathways. For example, the interplay between autophagy and apoptosis is demonstrated by extensive molecular crosstalk between these two pathways [27, 56,57]. Several mediators of death receptor-induced apoptosis, such as TRAIL, TNF, FADD, and DAPK, activate autophagy. Moreover, some pro-apoptotic regulators, such as RIP, JNK, ARF, E2F1, and Foxo, have recently been shown to positively regulate autophagy [27, 57]. p53, another apoptosis-regulatory protein, represses autophagy when localized in the cytoplasm; its nuclear localization leads to the activation of autophagy through transactivation of damage-regulated autophagy modulator (Dram) and Sestrin 2 genes [58]. Conversely, several anti-apoptotic proteins can repress autophagy by either forming a complex with Beclin 1 or Atg3 (BCl-2/BCl-XL and Flip, respectively) or by activating the mTOR pathway (Akt/PKB and NF-κB) [32, 36, 56, 59, 60]. Moreover, some of the autophagy core components, including Beclin 1, Atg5, and Atg4D, are cleaved during apoptosis, and the cleaved fragments induce cell death; this suggests that apoptosis events may change the function of autophagy-related proteins [56, 59, 61, 62].
2. NF-κB transcription factors
In this section, we briefly summarize the regulation of NF-κB transcription factor in cancer cells. Readers interested in details of NF-κB signaling pathways and regulation are referred to the following excellent reviews [63-69].
NF-κB family members
The NF-κB family of transcription factors regulates the expression of a broad range of genes involved in development, cell proliferation, survival, differentiation, and senescence. These transcription factors also play a pivotal role in regulating inflammation and the innate and adaptive immune responses [63, 70, 71]. The mammalian NF-κB family has five members: RelA (called also p65), c-Rel, Rel B, p50, and p52 [66, 71, 72]. These proteins contain the Rel homology domain that is essential for their homo- and hetero-dimerization and their binding to a specific DNA response sequences in the promoters of target genes called κB sites. Of note, each of the five members induces the activation of a selective set of genes that depend on the cell type and the stimulus. In most cells, the NF-κB dimers are sequestered in the cytoplasm as latent complexes through their binding to a member of the inhibitor of NF-κB (IκB) protein family including IκBα, IκBβ, IκBε and the IκB-like inhibitors p100and p105 [67].
NF-κB signaling pathways
Depending on the stimuli and the cell type, NF-κB activation can be promoted by two distinct pathways: the canonical and noncanonical pathways (Figure 2) [27, 63]. In both pathways, the activation of NF-κB is mediated by the activation of IκB kinase (IKK) complexes formed by either the catalytic kinases IKKα and IKKβ and the regulatory protein NEMO (IKKγ) or by IKKα homo-dimers [73, 74]. The canonical NF-κB pathway is activated primarily by stimulation of pro-inflammatory receptors such as TNF receptor family members, the Toll receptor family member, and antigen receptors [68, 69]. Certain anti-cancer therapies also activate this pathway [75-78]. These stimuli activate the IKK complex promoting the phosphorylation and subsequent proteasomal degradation of IκBα protein. This allows the translocation of the liberated NF-κB dimers into nucleus where they bind to the promoter regions of target genes that control cell survival, proliferation, inflammatory and innate immunity (Figure 2) [63-65, 76].
Figure 2.

Schematic model for the activation of NF-κB signaling pathways. In the canonical NF-κB pathway, stimulation of pro-inflammatory receptors activates the IKK complex (comprised of IKKα, IKKβ and IKKγ) leading to the phos-phorylation of IKBCX and its proteasomal degradation. This induces the translocation of p65/p50 dimers into the nucleus where the dimer binds to the promoter regions of a subset of target genes. In the noncanonical NF-κB pathway, stimulation of BAFF-R, CD40, and lymphotoxina-β receptor (LTβR) leads to the stabilization of NIK and results in the activation of the IKKα homo-dimers. Active IKKα, in turn, induces the phosphorylation of the NF-κB family member NF-κB2/p100 leading to its processing by the proteasome to generate p52. This allows the translocation of the p52/Rel B dimer into nucleus where it regulates the transcription of target genes. For details, see text.
The noncanonical NF-κB pathway is activated by stimulation of specific TNF receptors, including LTβR, CD40, and BAFFR, and plays a key role in the generation of lymphoid organs, in B-cell maturation, and in adaptive immunity [73, 79]. In the noncanonical pathway, the stabilization and the accumulation of NF-κB-inducing kinase (NIK) is an essential signal that triggers the activation of the IKKα homo-dimer [80, 81]. Conversely, stimulus that induces the proteasomal degradation of NIK promotes the negative regulation of the noncanonical NF-κB signaling events [80]. Once activated, IKKα induces the phosphorylation of NF-κB2/P100 (commonly associated with Rel B) leading to its processing by proteasome to generate p52. This allows the translocation of the p52/Rel B dimer into nucleus where it regulates the transcription of target gene (Figure 2) [79-81].
In context of normal cells, the stimulus-dependent activation of NF-κB is mostly transient due to establishment of feedback loops that terminate the activation. In both canonical and noncanonical pathways, one of the mechanisms that cells invoke to prevent the persistent activation of NF-κB is the inactivation of IKK. This can occur through IKK-mediated phosphorylation of its upstream activators (NIK in the noncanonical NF-κB pathway and RIP in canonical pathway), which subsequently induces their loss of function and promotes their proteasomal degradation. Of note, IKK can also inhibit itself by inducing its proper phosphorylation [73, 74]. Other repressors of NF-κB activation are A20 and CYLD, two proteins that mediate the K-63 deubiquitination and subsequent inactivation of signaling molecules like tumor necrosis factor receptor-associated factor (TRAF), RIP, and IKKγ that are required for NF-κB activation [82, 83]. Interestingly, these NF-κB repressors, as well as IKB family members (for example IκB-α), are target genes of NF-κB activation and are therefore function in negative feedback mechanisms [82, 83]. Moreover, post-translational modifications of the p65 subunitof NF-κB (e.g., phosphorylation, ubiquitination, and methylation) have been shown to affect IKK/NF-κB signaling events [76, 84]. In addition to the above mentioned regulators of NF-κB pathways, the activity of NF-κB in cancer cells is under the control of several tumor-regulatory pathways including Ras, EGF receptor, mTOR, Akt, STAT3, and p53 [67, 68, 85]. Therefore, different signaling pathways converge to NF-κB, and the process is tightly regulated by a counterbalance between activators and repressors to maintain appropriate cellular responses. The disruption of feedback inhibition mechanisms of NF-κB are linked to the development and the progression of several human diseases including inflammatory disorders and cancers [68, 81, 85, 86].
NF-κB and cancer
It is now well-established that NF-κB plays a critical role in the initiation, promotion, and progression of certain cancers due to the ability of this pathway to upregulate genes responsible for cell survival, invasion, angiogenesis, and metastasis [69, 71, 74, 81, 85, 87, 88]. NF-κB-mediated prevention of apoptosis is certainly one key event that favors the development of tumors and that plays a pivotal role in resistance to cancer therapies [75, 77, 78, 88-90]. Studies focused on identifying the mechanism involved in the anti-apoptotic role of NF-κB have shown that activation of this transcription factor results in an increase in expression of genes encoding for anti-apoptotic proteins like Bcl-2 family members, c-Flip, and IAPs (inhibitor of apoptosis) [87]. NF-κB activation also upregu-lates expression of antioxidant proteins that suppress ROS-dependent apoptosis and necrosis [88, 89].
Constitutive activation of NF-κB is observed in various types of malignancies. Activation may be due to alterations in expression of genes encoding NF-κB family members or their negative regulators or to persistence of NF-κB inducers such as pro-inflammatory cytokines (mostly, TNF and IL6) in the microenvironment of tumor cells [68, 69, 78, 81, 87, 88, 91, 92] The first evidence of the oncogenic role of NF-κB emerged after NF-κB subunits p50/p105 were cloned; this revealed homology of the cellular homologue c-Rel to the oncogene v-Rel encoded by the avian reticuloendotheliosis virus [85, 91]. Later, the over-activation of NF-κB members in a wide variety of types of tumors was demonstrated. For example, mutations in the gene encoding NF-κB2/P100 were detected in B and T cell lymphomas that resulted in loss of the inhibitory effects of the p100 protein and gain of transcriptional activity of the truncated p52 product [91, 92]. The transcriptional activity of p52 and p50 complexes is also enhanced by the Bcl-3 oncoprotein, a product of chromosomal translocation in B cell chronic lymphocytic leukemia. The overexpression of Bcl-3 has also been observed in various solid and lymphoid tumors, and patients with high levels of Bcl-3 have poor prognosis [93]. Amplification of c-Rel is observed in diffuse lymphoma with a large cell component, non-Hodgkin's B cell lymphoma, and in some solid tumors [92]. Mutations or deletions in IκBα and IκBε have been also detected in a subset of Hodgkin lymphomas, although their roles in the development of the disease is still not clear. Several experiments indicate that inhibition of IκBα degradation using a repressor form of this protein promotes apoptotic cell death and inhibits tumorigenesis in different animal models. In support to this tumor-suppressive role, constitutive degradation of IκB has been observed in various types of tumor cells [94, 95].
Although aberrant activation of IKKs has been observed in hematological and solid malignancies, no oncogenic mutations of the IKK genes have been yet discovered. Several lines of evidence support the idea that constitutive activation of IKK is due to the upregulation of IKK upstream activators or inactivation of its repressors. One example comes from recent studies in myeloma cell lines that led to the identification of mutations in genes encoding proteins that lead to the stabilization and accumulation of NIK, an upstream kinase of IKKα [96, 97]. The turnover of NIK is tightly regulated by IAP1 and c -IAP2, two ubiquitin ligases that target NIK for ubiquitination and thereby enhance its proteasomal degradation [98]. Mutational inactivation of c-IAP1 and c-IAP2 genes also occurs in myeloma cells and is correlated with the stabilization of NIK. Moreover, gain-of-function mutations in the NIK gene have been also observed in myeloma cells that may contribute to the accumulation of NIK protein. As stated above, A20 and CYLD, two proteins belonging to deubiquitinase family, can also attenuate the NF-κB responses by targeting signalling proteins that are required for IKK activation [82, 99]. Inactivating genetic alterations of A20 have been observed in various hematological malignancies and are linked with chronic NF-κB activation in these cancers [99]. Moreover, the overexpression of A20 in A20-deficient lymphoma cell lines inhibits both NF-κB activity and cell growth and triggers apoptotic cell death. Together, these data suggest that A20 has a tumor suppressor function [99]. Similarly, loss of CYLD1 function has been shown to promote tumorigenesis by enhancing NF-κB activation [82].
Although chronic activation of NF-κB is observed in the majority of cancer cells, genetic alterations in NF-κB members and regulatory signalling components are rare except in certain lymphoid malignancies [68, 81, 87]. This supports the idea that cancer cells have evolved other mechanisms to activate NF-κB. In fact, mutations in growth factor signalling pathways (e.g. k-Ras, EGF receptor, mTOR) or changes due to oncogenic viruses (T-cell leukemia virus type I, Epstein-Barr, Kaposi sarcoma-associated herpes virus) constitute other manners of persistent activation of NF-κB [68, 81, 100, 101]. Moreover, a link between NF-κB and inflammation-associated cancer progression has been demonstrated in several carcinogen/inflammation in vivo models by using conditional IKK knockout mice [72, 86]. These studies revealed that IKKβ-dependent NF-κB activation in premalignant-infiltrating immune cells leads to the production of cytokines that stimulate proliferation and enhance survival of malignant cells. The cytokines produced by immune cells, in turn, activate NF-κB in cancer cells to induce chemokines that attract additional inflammatory cells prolonging tumor-associated inflammation. IKKβ is therefore a key factor in inflammation-associated cancer progression and its inhibition may reduce tumor growth and enhance susceptibility to anti-cancer therapies. However, in some specific circumstances IKKβ and NF-κB act as tumor suppressors indicating that their inhibition would not always be beneficial to cancer patients [71].
3. The interplay between NF-κB and autophagy signalling pathways
3.1 Degradation of NF-κB signaling components through autophagy
Inhibition of Hsp90 induces autophagic degradation of IKK and NIK
IKKα, IKKβ, and IKKγ are targets for degradation by autophagy when heat shock protein 90 (Hsp90) function is inhibited (Figure 3a) by geldanamycin [102, 103]. Hsp90 is a chaperon protein, required for the correct folding and maturation of certain signaling proteins thus preventing their degradation under conditions of stress. Interestingly, geldanamycin-mediated degradation of IKK is prevented when autophagy is inhibited; but the clearance of Akt/PKB, another substrate of Hsp90 known to be degraded through the proteasomal pathway, is not affected by inhibition of autophagy. Thus, Hsp90 substrates are specifically degraded by either autophagy or proteasomal pathways. One example of such regulation has been recently reported in NB4 leukemia cells treated with se-lenite, a common dietary form of selenium with chemopreventive potential against various cancers [104]. Selenite treatment promotes down-regulation of Hsp90, which leads to inactivation of NF-κB pathway and inhibition of autophagy through downregulation of Beclin 1 expression. In NB4 cells, Hsp90 is a key regulator of the cell signaling pathway that switches cells treated with selenite from the autophagy program to apoptosis. Interestingly, in addition to induction of autophagy, specific inhibition of mitochondrial Hsp90 by gamitrinib elicits the organelle unfolded protein response (UPR), which also contributes to the repression of NF-κB activity [105].
Figure 3.

Autophagy mediates degradation of NF-κB signaling components. a) NIK and IKKs are targets for degradation by autophagy upon inhibition of Hsp90. This leads to inactivation of NF-κB pathway and, in turn, inhibition of autophagy through downregulation of an essential autophagy gene Beclin 1. The feedback regulatory loop between autophagy and NF-κB pathways is depicted. b) IKKβ degradation by autophagy through a mechanism that involves Keap1. In cells treated by TNF, Keap1 is responsible for the negative regulation of NF-κB through inhibition of the IKKβ phosphorylation and induction of IKKβ degradation by the autophagy pathway. c) IKKβ degradation by autophagy through a mechanism that involves Ro52. In cells infected with HTLV-1, Tax activates IKKβ-dependent NF-κB activation. Active IKKβ subsequently interacts with Ro52; this induces monoubiquitination of IKKβ and leads to its degradation by autophagy resulting in the termination of NF-κB activation. Ub represents ubiquitin. For details, see text.
NIK, an activator of IKKα in the non-canonical pathway, is another Hsp90 interacting protein. Disruption of the NIK/Hsp90 interaction by geldanamycin targets NIK for degradation by autophagy leading to the inhibition of both p100 processing and NF-κB activity (Figure 3a) [106]. Thus, both the proteasomal system and the autophagy pathway are involved in the degradation of NIK and the regulation of the NF-κB activity [98]. Taken together, these studies showed that Hsp90 is a novel regulatory bridge between autophagy and IKK/NF-κB pathways [37, 107]. Further studies are needed to determine the mechanisms involved in selective degradation of IKKs and NIK by autophagy. Interestingly, the activation of NF-κB by heat shock stress has been shown to activate, in turn, autophagy pathway [108] which underscores the notion that NF-κB and autophagy are mutually regulated under heat shock treatment. This will be further discussed in the chapter 3.2.
Keap1-dependent autophagy degradation of IKKβ
One clue in the mechanisms responsible for the control of autophagy-dependent degradation of IKK came from study that led to the identification of Kelch-like ECH-associated protein 1 (Keap1) [109] , as an interactor of IKK beta. Keap1 interacts with the kinase domain of IKKβ through its C-terminal domain. This domain is also required for the binding of Keap1 to the transcription factor NF-E2-related factor 2 (Nrf 2), which controls expression of certain antioxidant target genes. In response to TNF, Keap1 negatively regulates activation of NF-κB through inhibition of the IKKβ phosphorylation and induction of IKKβ degradation by autophagy pathway (Figure 3b). Moreover, Keap1 functions as an E3 ligase to mediate proteasomal degradation of IKKβ. Thus, Keap1 mediates degradation of IKKβ by both autophagy and proteasome pathways. However, Keap1 does not interact with IKKα, which is also degraded by autophagy implying another, not yet identified mechanism. Recently, the NF-κB family member p65 was shown to interact with Keap1 [110]. Moreover, current data suggest that a functional interplay between NF-κB components (IKKβ and p65) and Keap1 are necessary for the tight regulation of the transcription activity of both NF-κB and Nrf2 [111]. However, although various somatic mutations of Keap1 have been detected in some malignancies [112], a causal link between these mutations and the aberrant NF-κB activity observed in these tumors was not yet established. Interestingly, recent report has identified the inhibitor of NF-κB, IκBα as a new autophagic substrate. At the first phase following stimulation with TNF treatment, IKBOC is degraded through proteasome–dependent mechanism while at later phase it is cleared through the autophagic pathway [113]. Autophagy can thus lead to either the terminaison of NF-κB by inducing the clearance of IKK or the persistent activation of NF-κB though degradation of IκBα [113].
Ro52-dependent autophagy degradation of IKKβ
The E3 ubiquitin ligase Ro52 is another signaling molecule that targets IKKβ for degradation through the autophagy pathway [114]. In cells infected with HTLV-1, Tax oncoprotein is responsible for the persistent phosphorylation of IKKβ and constitutive activation of NF-κB activity (Figure 3b). Once activated, IKKβ interacts with Ro52, which mediates its monoubiquitination, a signal necessary for targeting IKKβ to auto-phagosomes. Inhibition of autophagy suppresses Tax/IKKβ-induced NF-κB activity suggesting that Ro52-mediated autophagy degradation of IKKβ represents a feedback mechanism for the termination of NF-κB activation by Tax. Interestingly, Ro52-mediated monoubiquitination of IKKβ occurs only under conditions of the persistent phosphorylation and activation of IKKβ (observed in response to Tax), which allows the formation of a stable interaction between Ro52 and IKKβ. Indeed, the monoubiquitination of IKKβ is not detected in response to TNF (another inducer of NF-κB activity) probably because the phosphorylation of IKKβ by this cytokine is transient and does not lead to the formation of a stable IKK/Ro52 complex. However, it is not yet clear whether the Ro-52-dependent monoubiquitination of IKKβ is a general response that occurs during persistent activation of IKKβ or a specific response triggered in cells expressing Tax.
Together, these results indicate that Hsp90, Keap1, and Ro52 are novel partners of IKKs, (especially IKKβ) which mediate the interplay between NF-κB and the autophagy pathway. In response to distinct stimuli, their specific interactions with IKKs regulate NF-κB activity through their ability to activate or repress degradation of IKKs through autophagy. It will be important to determine whether these interactions are mutually exclusive or whether some of these proteins cooperate to regulate IKKβ degradation. Determining this mechanism is essential to understanding how IKK/NF-κB activity can be switched on or off in response to different stimuli.
Regulation of NF-κB signaling components by autophagy-regulatory proteins
In addition to the direct effects of autophagy on NF-κB signaling components like IKKs and NIK, some proteins known to regulate autophagy have been reported to modulate NF-κB activity. For example, p62 (also known as sequesto-some-1 or SQSTM1), a cargo receptor for degradation of ubiquitinated proteins through the autophagy pathway, activates NF-κB in response to several stimuli including TNF, IL-1, receptor activator for NF-κB (RANK) ligand, and nerve growth factor [115]. Intriguingly, the impairment of both autophagy and apoptosis in immortal baby mouse kidney (iBMK) cells, an epithelial tumor cell line, results in the accumulation of p62, which is sufficient to inhibit NF-κB activity and to promote tumorigenesis [116]. The reason for seemingly opposite effects of p62 on the regulation of NF-κB is yet not clear.
Another protein involved in the formation of autophagosome with a link to the NF-κB pathway is Atg5. Atg5 interacts with Fas-associated protein with death domain (FADD), an upstream regulator of NF-κB [117-119]. This interaction is required for IFNγ-induced autophagy-dependent death in HeLa cells and contributes to the regulation of autophagy and activation of caspase 8 during mitogenic stimulation of T lymphocytes. However, whether NF-κB regulation was affected in HeLa cells treated by IFNγ was not explored. Beclin 1 is another essential autophagy protein that has been shown to regulate NF-κB activity in context of cells exposed to cigarette smoke extract. The partial loss of Beclin 1 in fibroblasts from Beclin 1 heterozygous knockout mice increases p65 phosphorylation and prevents apoptosis mediated by cigarette smoke extract. However, the mechanism by which Beclin 1 regulates NF-κB, has not been elucidated [120].
Several recent studies have revealed molecular crosstalk between tuberous sclerosis proteins TSC1 and TSC2 (upstream inhibitors of mTOR activation and activators of autophagy) and NF-κB pathway [121-124]. NF-κB signaling and cell survival are attenuated in TSC1- and-TSC2-deficient cells stimulated by DNA damage or TNF [121]. The addition of rapamycin, an inhibitor of mTOR activity, restores NF-κB activation and cell survival in TSC2-deficient cells suggesting that TSC2-mediated mTOR inhibition contributes to NF-κB activation [121]. Interestingly, IKKβ in turn, can activate the mTOR pathway in TNF-treated cells through a mechanism that involves its interaction with phosphorylated TSC1 [122]. Moreover, the IKKα subunit is required for activation of mTOR in PTEN-deficient cancer cells and in cells exposed to insulin [123, 124].
3.2 Regulation of autophagy by NF-κB
NF-κB activation occurs in response to various signals such as ligation of death receptors or pathogen recognition receptors (PRRs), oncogenes, virus, and DNA damage [68, 101, 125]. Recently, several studies have highlighted the regulation of autophagy in response to inducers of NF-κB-mediated signaling.
Regulation of autophagy by NF-κB activity
In response to TNF, NF-κB is activated in most cells through the canonical NF-κB pathway and leads to the upregulation of several anti-apoptotic and anti-oxidants genes [90, 126]. TNF-dependent activation of NF-κB represses autophagy in Ewing's sarcoma, breast, and leukemia cancer cell lines, whereas loss of NF-κB activation in these cells triggers the reactivation of autophagy [36, 127] (Figure 4a). The repression of autophagy in TNF-treated cells is associated with the activation of mTOR pathway, a negative regulator of autophagy. This is consistent with studies showing that IKK activation is required for mTOR activation in certain cell types [122, 123, 124]. The negative regulation of autophagy by NF-κB is also supported by a study showing that inhibition of NF-κB activity results in an enhancement of starvation-induced autophagy in cells derived from myelodysplastic patients with poor prognosis [128]. Similarly, the suppression of prolonged NF-κB activity in macrophages exposed to Escherichia coli promotes autophagy and increased cell survival whereas NF-κB-proficient macrophages undergo to cell death under the same condition [129] (Figure 4a). It should be mentioned that autophagy can be an effector of the innate immune response by targeting some pathogens for degradation by lysosomes [10-12]. These results support the idea that, in response to certain pathogens, cells initiate a regulatory circuit to override the repression of autophagy, and this protects them against the microbe.
Figure 4.

Regulation of autophagy by NF-κB signaling pathways. a) NF-κB activity regulates the extent of autophagy in cells through NF-κB inducers that regulate autophagy either in a positive or a negative manner. TNF-mediated NF-κB activation represses autophagy through both ROS inhibition and mTOR activation. In macrophages exposed to E. coli, NF-κB activation inhibits autophagy and triggers cell death program. TLR4 stimulation activates autophagy by inducing the ubiquitination (Ub represents ubiquitin) of Beclin 1 by TRAF6, an upstream activator of NF-κB. In turn, NF-κB activation upregulates the expression of A20, a factor that inhibits the ubiquitination of Beclin 1 and thereby limits autophagy. Upon hypoxia, ROS-mediated the activation of both NF-κB and Hif-1α is responsible for the induction of autophagy. In response to heat shock stress, autophagy is induced through a mechanism that involves NF-κB activation, b) NF-κB regulates the transcription of autophagy regulatory genes. In some experimental settings, p65, a member of NF-κB family, upregulates Beclin 1 mRNA level and induces autophagy. In response to DNA damage, the p52 subunit of NF-κB subunit controls autophagy by modulating the expression of both Skp2 and p27KIP1. The p65 sub-unit of NF-κB blocks E2F1-dependent Bnip 3 promoter binding and Bnip 3 transcription, thus inhibiting mitochondrial turnover via autophagy. c) NF-κB signaling components regulate autophagy. TAK1 enhances autophagy through AMP-mediated inhibition of mTORC1. X represents an as-yet-unidentified factor that is required for autophagy induction by TAK1. Under starvation condition, active IKKs induce autophagy through an AMPK inhibition-dependent of mTOR. In starved cells, IKK activity also promotes the upregulation of several essential autophagy genes and a subset of anti-apoptotic genes through an NF-κB independent or dependent mechanism, respectively.
The induction of autophagy by TNF in the absence of functional NF-κB activity requires the accumulation of ROS (mainly superoxide), as activation of antioxidants responses do not occur when NF-κB is inhibited [36, 127]. The link between autophagy and NF-κB inactivation and ROS accumulation is supported by a study showing that sex-related differences in autophagy in the Syrian hamster are controlled by ROS production through a mechanism that relies on the activities of NF-κB and p53 [130]. The key role of ROS in the regulation of autophagy has been also demonstrated under starvation conditions and in response to several anti-cancer therapies [24, 131]. Although the superoxide is assumed to be the major ROS species involved in modulation of autophagy [131], hydrogen peroxide also acts as a sensor for the regulation of autophagy. For example, in senescence cells, the overproduction of hydrogen peroxide due the NF-κB-dependent upregulation of manganese superoxide dismutase (MnSOD) antioxidant results in the induction of both autophagy and cell death [132]. Thus, depending on the cellular context, the duration and the intensity of NF-κB activity, the redox-regulating function of NF-κB can act either as an inhibitor or an activator of autophagy. Interestingly, ROS, in turn, can activate NF-κB activity and promotes autophagy. For example, upon hypoxia, MCF7 epithelial cancer cells activate the oxidative stress pathway in adjacent cancer-associated fibroblasts resulting in the activation of both NF-κB and Hif-1α [133]. This activation is sufficient for the induction of autophagy and leads to the degradation of Caveolin-1 (one component of caveolae membranes involved in receptor-independent endocytosis). The clearance of Caveolin 1 mediated by NF-κB/Hif-1α prevent the death of adjacent cancer cells suggesting that caveolin-1 may be a relevant biomarker for certain types of cancer cells (Figure 4a).
Additional evidence for the positive regulation of autophagy by NF-κB came from study that investigated the consequences of NF-κB activation during the recovery period after heat shock treatment. Upon heat shock, NF-κB is activated through a mechanism that is independent of IκB kinase activity and IκBα degradation but dependent on the thermolability of the IκB/NF-κB complex (Figure 4a). NF-κB activation enhances autophagy and increases the likelihood of cell survival, probably by inducing the degradation of misfolded and aggregated proteins that accumulate during heat shock stress [108]. In addition to this pro-survival function of NF-κB-dependent induction of autophagy, in the context of glanglioside-induced astrocyte activation, NF-κB activation is involved in the induction of cell death through an autophagy-dependent mechanism [134].
Upon recognition of pathogen infections or tissue damage, PRRs (e.g., Toll-like receptors, TLRs) and NOD family proteins activate multiple signaling pathways, including NF-κB-mediated signaling, which trigger the induction of the innate immune responses to defend against pathogen infections [10-12]. The link between Toll-like receptor-mediated autophagy and NF-κB signaling has been recently investigated in a macrophage cell line subjected to the TLR4 agonist lipopolysaccharide (LPS) [135]. TLR4 stimulation relies on the recruitment of Beclin 1 into the TLR4-signaling complex and requires the K63-linked ubiquitination of Beclin 1 by TRAF6, an upstream activator of NF-κB signaling pathway. The deubiquitination enzyme A20 (a target gene product of NF-κB) inhibits the ubiquitination of Beclin 1 and limits autophagy in response to TLR4 signaling (Figure 4a). Thus, TRAF6 and A20 can stimulate or inhibit autophagy by regulating the ubiquitination and deubiquitination of Beclin 1, respectively. Since the duration and intensity of TRAF6 activation and the A20 levels vary depending on the cell type and the stimulus, this can explain, at least in part, why NF-κB can act as either an inhibitor or an activator of autophagy. Of note, several primary cell lines fail to activate autophagy upon PRR engagement but, as of yet, TRAF6 and A20 activities have not been determined in these cells. The signaling pathways that lock the cascade linking NF-κB stimulation and autophagy induction is a matter for investigation in further studies.
Transcription regulation of autophagy-regulatory genes by NF-κB
The analysis of promoters of murine and human Beclin 1 genes led to the identification of several NF-κB consensus sequences suggesting that NF-κB regulates Beclin 1 expression. p65, a member of NF-κB family, specifically interacts with the κB site of Beclin 1 as revealed by in vitro and in vivo studies [136]. As a consequence, p65 induces the upregulation of Beclin 1 mRNA that is associated to the activation of autophagy in several cellular settings including T cell receptor-dependent activation of Jurkat cells (Figure 4b). Of note, p65-dependent Beclin 1 upregulation is dependent on the cellular context and the stimulus; p65 overexpression does not upregulate Beclin 1 expression in some cell lines and Beclin 1 mRNA levels are not increased in response to TNFα (unpublished data of our laboratory) or heat shock, which activate the p65/NF-κB signaling pathway [108].
Interestingly, NF-κB not only induces the upregulation of Beclin 1 but promotes deubiquitination of Beclin 1 by inducing the expression of A20 [135]. As stated above, induction of A20 expression, at least in response to TLR engagement, is thought to be the mechanism for the termination of NF-κB-dependent activation autophagy. However, it is unclear whether this kind of negative feedback loop occurs specifically in response to TLR activation or represents a general mechanism for interrupting the initial induction of autophagy.
In addition to the direct effect on the transcription of the pro-autophagic gene Beclin 1, NF-κB family members function cooperatively with other transcription factors to control the expression of genes involved in the regulation of autophagy. For example, S-phase kinase-associated protein 2 (Skp2) is an NF-κB target gene whose product functions as the receptor component of the Skp1/Cul1/F-box ubiquitin ligase complex that induces the degradation of p27 Kip1, an activator of the autophagy pathway [137]. In fact, silencing of the p52 NF-κB subunit inhibits Skp2 expression and results in increased levels of p27Kip1 and an induction of autophagy. Following DNA damage, Skp2 is also a direct target of the p53 tumor suppressor, which in concert with NF-κB regulates Skp2 expression. This regulation is dependent upon the activity of the Akt and the glycogen synthase kinase 3β (GSK3β pathways. When Akt is active, the p52 NF-κB subunit and p53 cooperatively induce Skp2 expression whereas the inactivation of Akt leads to the phosphorylation of p52 NF-κB sub-unit by GSK3β and thereby the repression of Skp2 expression (Figure 4b). Thus, the GSK3β-dependent phosphorylation of p52 plays a critical role in the regulation of autophagy and apoptosis in response to DNA damage. Another example is the NF-κB-dependent transcriptional regulation of Bnip 3, an important regulator of mitochondrial turnover via autophagy and cell death [35]. Under basal condition, p65 blocks binding of E2F-1 to the Bnip3 promoter thus inhibiting Bnip3 transcription (Figure 4b). Conversely, loss of NF-κB activity (which occurs, for example, during hypoxia) de-represses the Bnip3 promoter, Bnip3 is transcribed and cell death occurs [138]. Moreover, E2F-1-dependent Bnip3 transcription is epigenetically regulated by the histone deacetylase HDAC1 [139]. Thus, NF-κB and HDAC1 serve as molecular switches that regulated E2F-1-dependent Bnip3 expression, thereby regulating cell death mediated the E2F-1 tumor suppressor.
Regulation of autophagy by NF-κB signaling components
Molecules known to signal both up- and downstream of NF-κB are also sensors in the regulation of autophagy. Transforming growth factor-β-activating kinase 1 (TAK1), an upstream kinase of the IKK complex, has been shown to activate autophagy in breast epithelial cells subjected to Trail treatment [140]. TAK1-induced autophagy is dependent on AMP-mediated inhibition of mTORC1, a potent repressor of autophagy, and leads to cell protection against Trail-induced cytotoxicity (Figure 4c). Similar results were obtained with IL-1, which activates autophagy through a TAK1/AMPK-dependent mechanism. Interestingly, despite its ability to activate TAK1, TNF fails to activate AMPK, suggesting that TAK1 is essential but not sufficient for the effective activation of both AMPK and autophagy. Thus, TAK1 acts in conjunction with not-yet-identified regulatory factors to induce autophagy. It is not yet known whether NF-κB activation is required for TAK1-mediated autophagy.
The IKK subunits, critical sensors of the NF-κB signaling pathway, have been shown to regulate autophagy. Of note, IKKs can also control signaling pathways that are independent of the NF-κB [64, 141]. IKK subunits are necessary for the optimal induction of autophagy in response to starvation and rapamycin, the best known autophagy-inducing stimuli [142, 143] (Figure 4c). Moreover, the BH3-mimetic ABT737, a molecule that disrupts the interaction between Beclin 1 and Bcl-2/Bcl-XL, has been also shown to stimulate autophagy through a mechanism that involves IKK [144]. In response to starvation, rapamycin and ABT737, constitutively active IKK subunits signal autophagy through a canonical pathway that involves AMPK activation, mTOR inhibition, c-Jun N-terminal kinase (JNK) phosphorylation, p53 depletion, and dissociation of Beclin 1 from Bcl-2 [142-144]. Disruption of NF-κB activity, either due to a mutant form of IκBα that represses IKK-induced nuclear translocation of p65 or in p65-null cells, fails to prevent IKK-induced autophagy, indicating that IKK can operate through a mechanism that does not involve NF-κB. The investigation of autophagy responses in mice displaying a liver-specific conditional knockout of IKKβ revealed a defect in autophagy in liver but not in the other organs in response to starvation or rapamycin treatment. This in vivo data support the idea that IKKβ can function as a positive regulator of autophagy.
Under periods of prolonged nutrient deprivation, IKK activity promotes the upregulation of three essential autophagic genes LC3, Atg5, Beclin1, through an NF-κB independent mechanism (Figure 4c). This constitutes a positive feedback loop mechanism for the regulation of autophagy under starvation [145]. In starved cells, IKK activity also controls another pathway leading to the activation of both canonical and non-canonical pathways of NF-κB, thus resulting in the upregulation of NF-κB-dependent anti-apoptotic genes (e.g., Bnip 3, Bcl-xl, and cIAP-2) [145]. The identification of additional IKK-dependent substrates will lead to a better understanding of how IKK activates autophagy in both a transcriptionally-dependent and -independent manner.
Conclusion
The studies mentioned in this review shed light on the mutual control that exists between the autophagy and the NF-κB signaling pathways. Autophagy may constitute a pathway through which NF-κB signaling components are specifically degraded which results either in the termination of the initial NF-κB activation or conversely in the persistent activation of this process (Figure 3). In turn, NF-κB signaling can regulate autophagy through different routes that depend on the cellular context and the stimulus: NF-κB members can either activate or inhibit signaling pathways that lead to the induction of autophagy and regulates the transcription of a subset of pro-autophagic-regulating genes as well. Moreover, NF-κB signaling components can promote autophagy independently of NF-κB through stimulation of signaling pathways that activate autophagy or by inducing the transactivation of a subset of autophagy-related genes (Figure 4).
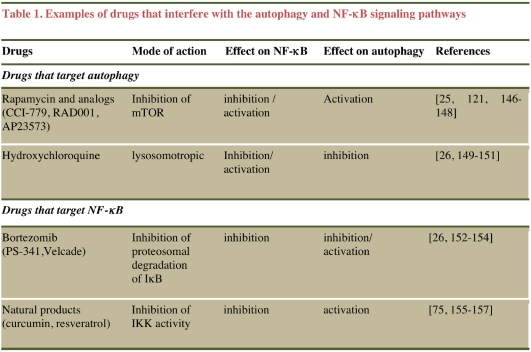
Since the regulation of both autophagy and NF-κB pathways overlap with that of multiple pathways that control tumorigenesis, a more detailed molecular dissection of the cross-regulatory circuits that exist between autophagy and NF-κB is required to better understand how these pathways interact to regulate tumorrelated responses. It is worth noting that several anti-cancer therapies that have been used in cancer clinical trials have the ability to influence both the autophagy and the NF-κB signaling pathways (Table 1). Fundamental insights into the functional interplay between autophagy and NF-κB pathways may provide clues for developing new strategies for combining drugs that target both pathways to improve effectiveness of cancer therapies. One example of such a strategy is the use of hydroxychloroquine (an potent inhibitor of autophagy) in combination with bortezomib (a molecule that targets the NF-κB pathway) in patients that manifest relapsed-refractory myeloma; this combination is being evaluated in an ongoing Phase I-II clinical trial [26].
Table 1.
Examples of drugs that interfere with the autophagy and NF-κB signaling pathways
 |
Acknowledgments
This work was supported by funds from the In-stitut National de la Santé et de la Recherche Médicale (INSERM), the University of Bordeaux, IFR 66 and by a grant from Ligue contre le Cancer comité de la Gironde and a regional administration grant from Région Aquitaine. A.T. is supported by a PhD fellowship from the Conseil Régional Aquitaine/INSERM. We regret being unable to reference all of the important published papers on autophagy and NF-κB.
Glossary
Abbreviations
- AMP kinase
adenosine monophos-phate-activated protein kinase
- Atg
autophagy-related genes
- Bcl-2
B-cell lymphoma 2
- CYLD1
cylin-dromatosis
- DAPK
Death associated Protein Kinase
- Dram
damage-regulated autophagy modulator
- FADD
Fas-Associated protein with Death Domain
- Fox
forkhead box
- GSK3β
glycogen synthase kinase 3β
- Hif-1
hypoxia-inducible factor
- HMGB1
high-mobility group box 1
- Hsp
heat shock protein
- IKK
IkB kinase
- iBMK
immortal baby mouse kidney
- IL-1R
interleukin receptor
- JNK
c-Jun N-terminal kinase
- Keap1
kelch-like ECH-associate protein 1
- LC3
microtubule-associated protein 1 light chain 3; Lamp; lysosomal-associated membrane protein
- LKB 1
liver kinase B1
- LPS
lipopolysaccharide
- LTβR
lymphotoxin-β receptor
- TNF-R
tumor necrosis factor receptor
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- NF-κB
nuclear factor κ-light-chain-enhancer of activated B cells
- Nfr-2
NFE2-related factor 2
- NIK
NF-κB inducing kinase
- MnSOD
manganese superoxide dismutase
- PRR
pathogen recognition receptor
- PI3P
phosphatidylinositol 3-phosphate
- PI3K
phosphatidylinositol 3-kinase
- PKB
protein kinase B
- PTEN
phosphatase and tensin homolog
- RANK
receptor activator for NF-κB
- RIP
receptor-interacting proteins
- ROS
reactive oxygen species
- TAK1
transforming growth factor- β-activating kinase 1
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- TSC
tuberous sclerosis complex
- TRAF
tumor necrosis factor receptor-associated factor
- TRAIL
TNF-related apoptosis-inducing ligand
- Vps 34
vacuolar protein sorting 34, ULK, Unc-51 like kinase 1
- UVRAG
ultraviolet radiation resistance associated gene
Declaration of conflicts of interest
None
References
- 1.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Botti J, Djavaheri-Mergny M, Pilatte Y, Codogno P. Autophagy signaling and the cogwheels of cancer. Autophagy. 2006;2:67–73. doi: 10.4161/auto.2.2.2458. [DOI] [PubMed] [Google Scholar]
- 4.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 5.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22:212–217. doi: 10.1016/j.ceb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salminen A, Kaarniranta K. Regulation of the aging process by autophagy. Trends Mol Med. 2009;15:217–224. doi: 10.1016/j.molmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delgado MA, Deretic V. Toll-like receptors in control of immunological autophagy. Cell Death Differ. 2009;16:976–983. doi: 10.1038/cdd.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kroemer G, White E. Autophagy for the avoidance of degenerative, inflammatory, infectious, and neoplastic disease. Curr Opin Cell Biol. 2010;22:121–123. doi: 10.1016/j.ceb.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 17.Komatsu M, Ichimura Y. Selective autophagy regulates various cellular functions. Genes Cells. 2010;15:923–933. doi: 10.1111/j.1365-2443.2010.01433.x. [DOI] [PubMed] [Google Scholar]
- 18.Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregateprone proteins. FEBS Lett. 2010;584:2635–2645. doi: 10.1016/j.febslet.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 19.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011:7. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. 2008;15:344–357. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010;21:683–690. doi: 10.1016/j.semcdb.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–1348. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 24.Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy. 2010;6:838–854. doi: 10.4161/auto.6.7.12113. [DOI] [PubMed] [Google Scholar]
- 25.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 26.Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–666. doi: 10.1158/1078-0432.CCR-10-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 28.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011 doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fengsrud M, Roos N, Berg T, Liou W, Slot JW, Seglen PO. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: effects of vinblastine and asparagine on vacuole distributions. Exp Cell Res. 1995;221:504–519. doi: 10.1006/excr.1995.1402. [DOI] [PubMed] [Google Scholar]
- 31.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 34.Tanida I. Autophagy basics. Microbiol Immunol. 2011;55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- 35.Mazure NM, Pouyssegur J. Hypoxiainduced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22:177–180. doi: 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Codogno P. Regulation of autophagy by NFkappaB transcription factor and reactives oxygen species. Autophagy. 2007;3:390–392. doi: 10.4161/auto.4248. [DOI] [PubMed] [Google Scholar]
- 37.Xiao G. Autophagy and NF-kappaB: fight for fate. Cytokine Growth Factor Rev. 2007;18:233–243. doi: 10.1016/j.cytogfr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, Kroemer G. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta. 2009;1793:1524–1532. doi: 10.1016/j.bbamcr.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 39.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 40.Hippert MM, O'Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer Res. 2006;66:9349–9351. doi: 10.1158/0008-5472.CAN-06-1597. [DOI] [PubMed] [Google Scholar]
- 41.Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- 42.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 44.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7:961–967. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koneri K, Goi T, Hirono Y, Katayama K, Yamaguchi A. Beclin 1 gene inhibits tumor growth in colon cancer cell lines. Anticancer Res. 2007;27:1453–1457. [PubMed] [Google Scholar]
- 50.Yoo BH, Wu X, Li Y, Haniff M, Sasazuki T, Shirasawa S, Eskelinen EL, Rosen KV. Oncogenic ras-induced down-regulation of autophagy mediator Beclin-1 is required for malignant transformation of intestinal epithelial cells. J Biol Chem. 2010;285:5438–5449. doi: 10.1074/jbc.M109.046789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011;21:113–119. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tang D, Loze MT, Zeh HJ, Kang R. The redox protein HMGB1 regulates cell death and survival in cancer treatment. Autophagy. 2010;6:1181–1183. doi: 10.4161/auto.6.8.13367. [DOI] [PubMed] [Google Scholar]
- 54.Kang R, Tang D, Schapiro NE, Livesey KM, Farkas A, Loughran P, Bierhaus A, Lotze MT, Zeh HJ. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol. 2010;22:241–245. doi: 10.1016/j.ceb.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leber B, Andrews DW. Closing in on the link between apoptosis and autophagy. F1000 Biol Rep. 2010;2:88. doi: 10.3410/B2-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 58.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 59.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 60.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–277. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R, Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S, Vandenabeele P. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010;1:e18. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 64.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 65.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–186. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 67.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 68.Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-kappaB addiction and its role in cancer: ‘one size does not fit all’. Oncogene. 2010 doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiao G, Fu J. NF-kappaB and cancer: a paradigm of Yin-Yang. Am J Cancer Res. 2011;1:192–221. [PMC free article] [PubMed] [Google Scholar]
- 70.Hayden MS, Ghosh S. NF-kappaB in immunobiology. Cell Res. 2011;21:223–244. doi: 10.1038/cr.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 2006;13:759–772. doi: 10.1038/sj.cdd.4401838. [DOI] [PubMed] [Google Scholar]
- 72.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 73.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arkan MC, Greten FR. IKK- and NFkappaB-Mediated Functions in Carcinogenesis. Curr Top Microbiol Immunol. 2010 doi: 10.1007/82_2010_97. [DOI] [PubMed] [Google Scholar]
- 75.Nakanishi C, Toi M. Nuclear factorkappaB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5:297–309. doi: 10.1038/nrc1588. [DOI] [PubMed] [Google Scholar]
- 76.Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-kappaB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17:281–293. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 77.Ralhan R, Pandey MK, Aggarwal BB. Nuclear factor-kappa B links carcinogenic and chemopreventive agents. Front Biosci (Schol Ed) 2009;1:45–60. doi: 10.2741/S6. [DOI] [PubMed] [Google Scholar]
- 78.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Espinosa L, Bigas A, Mulero MC. Alternative nuclear functions for NF-kappaB family members. Am J Cancer Res. 2011;1:446–459. [PMC free article] [PubMed] [Google Scholar]
- 80.Thu YM, Richmond A. NF-kappaB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev. 2010;21:213–226. doi: 10.1016/j.cytogfr.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harhaj EW, Dixit VM. Deubiquitinases in the regulation of NF-kappaB signaling. Cell Res. 2011;21:22–39. doi: 10.1038/cr.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wertz IE, Dixit VM. Signaling to NFkappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010;2:a003350. doi: 10.1101/cshperspect.a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 85.Basseres DS, Baldwin AS. Nuclear factorkappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- 86.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 88.Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev. 2008;18:19–26. doi: 10.1016/j.gde.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luo JL, Kamata H, Karin M. IKK/NFkappaB signaling: balancing life and death–a new approach to cancer therapy. J Clin Invest. 2005;115:2625–2632. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dutta J, Fan Y, Gupta N, Fan G, Gelinas C. Current insights into the regulation of programmed cell death by NF-kappaB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 91.Courtois G, Gilmore TD. Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843. doi: 10.1038/sj.onc.1209939. [DOI] [PubMed] [Google Scholar]
- 92.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–6947. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 93.Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS., Jr Selective activation of NFkappa B subunits in human breast cancer: potential roles for NF-kappa B2/p52 and for Bcl-3. Oncogene. 2000;19:1123–1131. doi: 10.1038/sj.onc.1203412. [DOI] [PubMed] [Google Scholar]
- 94.Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Mutations in the IkBa gene in Hodgkin's disease suggest a tumour suppressor role for IkappaBalpha. Oncogene. 1999;18:3063–3070. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- 95.Jungnickel B, Staratschek-Jox A, Brauninger A, Spieker T, Wolf J, Diehl V, Hansmann ML, Rajewsky K, Kuppers R. Clonal deleterious mutations in the IkappaBalpha gene in the malignant cells in Hodgkin's lymphoma. J Exp Med. 2000;191:395–402. doi: 10.1084/jem.191.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, Zhao H, Stephens O, Santra M, Williams DR, Dang L, Barlogie B, Shaughnessy JD, Jr, Kuehl WM, Staudt LM. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, Braggio E, Henry T, Zhu YX, Fogle H, Price-Troska T, Ahmann G, Mancini C, Brents LA, Kumar S, Greipp P, Dispenzieri A, Bryant B, Mulligan G, Bruhn L, Barrett M, Valdez R, Trent J, Stewart AK, Carpten J, Bergsagel PL. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh WC, Mak TW, Korneluk RG, Cheng G. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol. 2008;9:1371–1378. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nat Rev Cancer. 2010;10:332–341. doi: 10.1038/nrc2775. [DOI] [PubMed] [Google Scholar]
- 100.Baker RG, Hayden MS, Ghosh S. NFkappaB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baritaki S, Bonavida B. Viral infection and cancer: the NF-kappaB/Snail/RKIP loop regulates target cell sensitivity to apoptosis by cytotoxic lymphocytes. Crit Rev Immunol. 2010;30:31–46. doi: 10.1615/critrevimmunol.v30.i1.20. [DOI] [PubMed] [Google Scholar]
- 102.Qing G, Yan P, Xiao G. Hsp90 inhibition results in autophagy-mediated proteasomeindependent degradation of IkappaB kinase (IKK) Cell Res. 2006;16:895–901. doi: 10.1038/sj.cr.7310109. [DOI] [PubMed] [Google Scholar]
- 103.Yan P, Qing G, Qu Z, Wu CC, Rabson A, Xiao G. Targeting autophagic regulation of NFkappaB in HTLV-I transformed cells by geldanamycin: implications for therapeutic interventions. Autophagy. 2007;3:600–603. doi: 10.4161/auto.4761. [DOI] [PubMed] [Google Scholar]
- 104.Jiang Q, Wang Y, Li T, Shi K, Li Z, Ma Y, Li F, Luo H, Yang Y, Xu C. Hsp90-mediated inactivation of NF{kappa}B switches autophagy to apoptosis through becn1 transcriptional inhibition in selenite-induced NB4 cells. Mol Biol Cell. 2011 doi: 10.1091/mbc.E10-10-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Siegelin MD, Dohi T, Raskett CM, Orlowski GM, Powers CM, Gilbert CA, Ross AH, Plescia J, Altieri DC. Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J Clin Invest. 2011 doi: 10.1172/JCI44855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Qing G, Yan P, Qu Z, Liu H, Xiao G. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 2007;17:520–530. doi: 10.1038/cr.2007.47. [DOI] [PubMed] [Google Scholar]
- 107.Djavaheri-Mergny M, Codogno P. Autophagy joins the game to regulate NF-kappaB signaling pathways. Cell Res. 2007;17:576–577. doi: 10.1038/cr.2007.58. [DOI] [PubMed] [Google Scholar]
- 108.Nivon M, Richet E, Codogno P, Arrigo AP, Kretz-Remy C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy. 2009;5:766–783. doi: 10.4161/auto.8788. [DOI] [PubMed] [Google Scholar]
- 109.Kim JE, You DJ, Lee C, Ahn C, Seong JY, Hwang JI. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell Signal. 2010;22:1645–1654. doi: 10.1016/j.cellsig.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 110.Yu M, Li H, Liu Q, Liu F, Tang L, Li C, Yuan Y, Zhan Y, Xu W, Li W, Chen H, Ge C, Wang J, Yang X. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell Signal. 2011;23:883–892. doi: 10.1016/j.cellsig.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 111.Stepkowski TM, Kruszewski MK. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med. 2011 doi: 10.1016/j.freeradbiomed.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 112.Li QK, Singh A, Biswal S, Askin F, Gabrielson E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. J Hum Genet. 2011;56:230–234. doi: 10.1038/jhg.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Colleran A, Ryan A, O'Gorman A, Mureau C, Liptrot C, Dockery P, Fearnhead H, Egan LJ. Autophagosomal I{kappa}B{alpha} degradation plays a role in the long term control of tumor necrosis factor-{alpha}-induced NF-{kappa}B activity. J Biol Chem. 2011 doi: 10.1074/jbc.M110.199950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Niida M, Tanaka M, Kamitani T. Downregulation of active IKK beta by Ro52-mediated autophagy. Mol Immunol. 2010;47:2378–2387. doi: 10.1016/j.molimm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–269. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 118.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 119.Bell BD, Walsh CM. Coordinate regulation of autophagy and apoptosis in T cells by death effectors: FADD or foundation. Autophagy. 2009;5:238–240. doi: 10.4161/auto.5.2.7512. [DOI] [PubMed] [Google Scholar]
- 120.Kim HP, Wang X, Chen ZH, Lee SJ, Huang MH, Wang Y, Ryter SW, Choi AM. Autophagic proteins regulate cigarette smoke-induced apoptosis: protective role of heme oxygenase-1. Autophagy. 2008;4:887–895. doi: 10.4161/auto.6767. [DOI] [PubMed] [Google Scholar]
- 121.Ghosh S, Tergaonkar V, Rothlin CV, Correa RG, Bottero V, Bist P, Verma IM, Hunter T. Essential role of tuberous sclerosis genes TSC1 and TSC2 in NF-kappaB activation and cell survival. Cancer Cell. 2006;10:215–226. doi: 10.1016/j.ccr.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 122.Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, Sun HL, Li LY, Ping B, Huang WC, He X, Hung JY, Lai CC, Ding Q, Su JL, Yang JY, Sahin AA, Hortobagyi GN, Tsai FJ, Tsai CH, Hung MC. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–455. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 123.Dan HC, Baldwin AS. Differential involvement of IkappaB kinases alpha and beta in cytokine- and insulin-induced mammalian target of rapamycin activation determined by Akt. J Immunol. 2008;180:7582–7589. doi: 10.4049/jimmunol.180.11.7582. [DOI] [PubMed] [Google Scholar]
- 124.Dan HC, Adli M, Baldwin AS. Regulation of mammalian target of rapamycin activity in PTEN-inactive prostate cancer cells by I kappa B kinase alpha. Cancer Res. 2007;67:6263–6269. doi: 10.1158/0008-5472.CAN-07-1232. [DOI] [PubMed] [Google Scholar]
- 125.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 126.Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228:225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- 127.Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G, Codogno P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–30382. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 128.Fabre C, Carvalho G, Tasdemir E, Braun T, Ades L, Grosjean J, Boehrer S, Metivier D, Souquere S, Pierron G, Fenaux P, Kroemer G. NF-kappaB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2007;26:4071–4083. doi: 10.1038/sj.onc.1210187. [DOI] [PubMed] [Google Scholar]
- 129.Schlottmann S, Buback F, Stahl B, Meierhenrich R, Walter P, Georgieff M, Senftleben U. Prolonged classical NF-kappaB activation prevents autophagy upon E. coli stimulation in vitro: a potential resolving mechanism of inflammation. Mediators Inflamm. 2008;2008:725854. doi: 10.1155/2008/725854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Vega-Naredo I, Caballero B, Sierra V, Huidobro-Fernandez C, de Gonzalo-Calvo D, Garcia-Macia M, Tolivia D, Rodriguez-Colunga MJ, Coto-Montes A. Sexual dimorphism of autophagy in Syrian hamster Harderian gland culminates in a holocrine secretion in female glands. Autophagy. 2009;5:1004–1017. doi: 10.4161/auto.5.7.9610. [DOI] [PubMed] [Google Scholar]
- 131.Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–790. doi: 10.1089/ars.2008.2270. [DOI] [PubMed] [Google Scholar]
- 132.Deruy E, Gosselin K, Vercamer C, Martien S, Bouali F, Slomianny C, Bertout J, Bernard D, Pourtier A, Abbadie C. MnSOD upregulation induces autophagic programmed cell death in senescent keratinocytes. PLoS One. 2010;5:e12712. doi: 10.1371/journal.pone.0012712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, Tsirigos A, Migneco G, Witkiewicz A, Balliet R, Mercier I, Wang C, Flomenberg N, Howell A, Lin Z, Caro J, Pestell RG, Sotgia F, Lisanti MP. The autophagic tumor stroma model of cancer or “battery-operated tumor growth": A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–4306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hwang J, Lee HJ, Lee WH, Suk K. NFkappaB as a common signaling pathway in ganglioside-induced autophagic cell death and activation of astrocytes. J Neuroimmunol. 2010;226:66–72. doi: 10.1016/j.jneuroim.2010.05.037. [DOI] [PubMed] [Google Scholar]
- 135.Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Copetti T, Demarchi F, Schneider C. p65/RelA binds and activates the beclin 1 promoter. Autophagy. 2009;5:858–859. doi: 10.4161/auto.8822. [DOI] [PubMed] [Google Scholar]
- 137.Barre B, Perkins ND. The Skp2 promoter integrates signaling through the NF-kappaB, p53, and Akt/GSK3beta pathways to regulate autophagy and apoptosis. Mol Cell. 2010;38:524–538. doi: 10.1016/j.molcel.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 138.Shaw J, Yurkova N, Zhang T, Gang H, Aguilar F, Weidman D, Scramstad C, Weisman H, Kirshenbaum LA. Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc Natl Acad Sci U S A. 2008;105:20734–20739. doi: 10.1073/pnas.0807735105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gang H, Dhingra R, Wang Y, Mughal W, Gordon JW, Kirshenbaum LA. Epigenetic Regulation of E2F-1-Dependent Bnip3 Transcription and Cell Death by Nuclear Factor-kappaB and Histone Deacetylase-1. Pediatr Cardiol. 2011;32:263–266. doi: 10.1007/s00246-011-9893-z. [DOI] [PubMed] [Google Scholar]
- 140.Herrero-Martin G, Hoyer-Hansen M, Garcia-Garcia C, Fumarola C, Farkas T, Lopez-Rivas A, Jaattela M. TAK1 activates AMPKdependent cytoprotective autophagy in TRAILtreated epithelial cells. EMBO J. 2009;28:677–685. doi: 10.1038/emboj.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chariot A. The NF-kappaB-independent functions of IKK subunits in immunity and cancer. Trends Cell Biol. 2009;19:404–413. doi: 10.1016/j.tcb.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 142.Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S, Tailler M, Delahaye N, Tesniere A, De Stefano D, Younes AB, Harper F, Pierron G, Lavandero S, Zitvogel L, Israel A, Baud V, Kroemer G. IKK connects autophagy to major stress pathways. Autophagy. 2010;6:189–191. doi: 10.4161/auto.6.1.10818. [DOI] [PubMed] [Google Scholar]
- 143.Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S, Tailler M, Delahaye N, Tesniere A, De Stefano D, Younes AB, Harper F, Pierron G, Lavandero S, Zitvogel L, Israel A, Baud V, Kroemer G. The IKK complex contributes to the induction of autophagy. EMBO J. 2010;29:619–631. doi: 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Malik SA, Orhon I, Morselli E, Criollo A, Shen S, Marino G, Benyounes A, Benit P, Rustin P, Maiuri MC, Kroemer G. BH3 mimetics activate multiple pro-autophagic pathways. Oncogene. 2011 doi: 10.1038/onc.2011.104. [DOI] [PubMed] [Google Scholar]
- 145.Comb WC, Cogswell P, Sitcheran R, Baldwin AS. IKK-dependent, NF-kappaBindependent control of autophagic gene expression. Oncogene. 2010 doi: 10.1038/onc.2010.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Levati L, Ruffini F, Muzi A, Umezawa K, Graziani G, D'Atri S, Lacal PM. Placenta growth factor induces melanoma resistance to temozolomide through a mechanism that involves the activation of the transcription factor NF-kappaB. Int J Oncol. 2011;38:241–247. [PubMed] [Google Scholar]
- 147.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev. 2008;22:1490–1500. doi: 10.1101/gad.1662308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yamini B, Yu X, Dolan ME, Wu MH, Darga TE, Kufe DW, Weichselbaum RR. Inhibition of nuclear factor-kappaB activity by temozolomide involves O6-methylguanine induced inhibition of p65 DNA binding. Cancer Res. 2007;67:6889–6898. doi: 10.1158/0008-5472.CAN-06-4496. [DOI] [PubMed] [Google Scholar]
- 149.Park J, Kwon D, Choi C, Oh JW, Benveniste EN. Chloroquine induces activation of nuclear factor-kappaB and subsequent expression of pro-inflammatory cytokines by human astroglial cells. J Neurochem. 2003;84:1266–1274. doi: 10.1046/j.1471-4159.2003.01623.x. [DOI] [PubMed] [Google Scholar]
- 150.Yi AK, Peckham DW, Ashman RF, Krieg AM. CpG DNA rescues B cells from apoptosis by activating NFkappaB and preventing mitochondrial membrane potential disruption via a chloroquine-sensitive pathway. Int Immunol. 1999;11:2015–2024. doi: 10.1093/intimm/11.12.2015. [DOI] [PubMed] [Google Scholar]
- 151.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Mycinduced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Belloni D, Veschini L, Foglieni C, Dell'Antonio G, Caligaris-Cappio F, Ferrarini M, Ferrero E. Bortezomib induces autophagic death in proliferating human endothelial cells. Exp Cell Res. 2010;316:1010–1018. doi: 10.1016/j.yexcr.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 153.Periyasamy-Thandavan S, Jackson WH, Samaddar JS, Erickson B, Barrett JR, Raney L, Gopal E, Ganapathy V, Hill WD, Bhalla KN, Schoenlein PV. Bortezomib blocks the catabolic process of autophagy via a cathepsindependent mechanism, affects endoplasmic reticulum stress and induces caspasedependent cell death in antiestrogen-sensitive and resistant ER+ breast cancer cells. Autophagy. 2010;6:19–35. doi: 10.4161/auto.6.1.10323. [DOI] [PubMed] [Google Scholar]
- 154.Ocio EM, Mateos MV, Maiso P, Pandiella A, San-Miguel JF. New drugs in multiple myeloma: mechanisms of action and phase I/II clinical findings. Lancet Oncol. 2008;9:1157–1165. doi: 10.1016/S1470-2045(08)70304-8. [DOI] [PubMed] [Google Scholar]
- 155.Jia YL, Li J, Qin ZH, Liang ZQ. Autophagic and apoptotic mechanisms of curcumininduced death in K562 cells. J Asian Nat Prod Res. 2009;11:918–928. doi: 10.1080/10286020903264077. [DOI] [PubMed] [Google Scholar]
- 156.Singletary K, Milner J. Diet, autophagy, and cancer: a review. Cancer Epidemiol Biomarkers Prev. 2008;17:1596–1610. doi: 10.1158/1055-9965.EPI-07-2917. [DOI] [PubMed] [Google Scholar]
- 157.Delmas D, Solary E, Latruffe N. Resveratrol, a phytochemical inducer of multiple cell death pathways: apoptosis, autophagy and mitotic catastrophe. Curr Med Chem. 2011;18:1100–1121. doi: 10.2174/092986711795029708. [DOI] [PubMed] [Google Scholar]