

Abstract
Intra-Vital Microscopy (IVM) is used to visualize tumors in animals and analyze various aspects of cancer physiology such as tumor vascularization, cell migration and metastasis. The main advantages of IVM include the real -time analysis of dynamic processes with single-cell resolution. The application of IVM, however, is limited by the availability of animal models that carry visually accessible tumors. These models have evolved over time to become more and more relevant to human tumors. The latest step is the development of a pseudo-orthotopic, syngeneic model for tumor growth and metastasis. In this model, tissue from a variety of mouse organs are grafted in a dorsal skinfold chamber and allowed to revascularize, whereupon tumor cell spheroids are implanted. These spheroids develop into tumors that bear a much closer resemblance to human tumors than xenografts. Unlike xenografts, the vasculature is well-ordered and, because the model is syngeneic, there are no cross-species host immune reactions. The use of fluorescence-tagged pseudo-organs and tumor cells allows IVM analysis and provides real-time access to the development of tumors that closely resemble the real disease. This model can be used to test therapeutics and to image tumor development and stroma-tumor interactions.
Keywords: Intra-Vital Microscopy (IVM), mouse dorsal chamber model, solid tumor, cancer, vascularization, cell migration and metastasis
Introduction
Over 1,5 million new cancer cases and 569,490 deaths from cancer were projected to occur in the United States in 2010 [1]. Cancer incidence rates have decreased in recent times, but despite progress in detection and treatment the death toll remains high and new therapeutic approaches are needed.
There are many physiological aspects to cancer progression that must be understood if we are to fight cancer more effectively [2]. A tumor is not just a mass of cancer cells that are proliferating without control. There are many stresses associated with the development of a tumor that cancer cells need to overcome such as, for example, the lack of oxygen when the tumor mass increases (hypoxia) and the resulting decrease in extracellular pH (acidification), or even the stresses caused by chemo or radiotherapy. As a result, a fraction of cancer cells develop resistance to various forms of cell death. In addition, myriad changes take place in the surrounding tissue that apparently promote tumor survival.
This concept was proposed by Paget in his famous ‘seed and soil’ hypothesis as early as 1889 [3] and has since been supported by large amounts of data. While a normal tissue environment inhibits the proliferation of cancer cells and slows down tumor formation, disruption of this environment caused by chronic injury, inflammation or hereditary alterations in key genes regulating tissue remodeling may help initiate cancer. In any case, the environment found in the vicinity of tumors is hardly normal, as the stroma reacts to the presence of cancer cells [4, 5]. Stroma changes include the recruitment of cancer-associated fibroblasts, smooth muscle cells and endothelial cells, and of immune cells such as tumor associated macro-phages, tumor-infiltrating lymphocytes and leukocytes [6, 7]. Conversely, stroma cells alter the behavior of epithelial cancer cells by secreting extracellular proteins, cytokines, growth factors, proteases, etc. Dramatic changes in gene expression can be measured in all cell types during cancer progression, including tumor epithelial, stroma endothelial, myofibroblasts, and immune cells. In addition, it now appears that genetic alterations can also arise in the micro-environment of tumors (reviewed in [8]). Thus, reciprocal interactions take place between stromal and epithelial cancer cells that promote cancer progression by increasing cell proliferation, causing the formation of new blood vessels through angiogenesis, remodeling the extracellular matrix, and supporting the metastatic spread of the tumor cells [5, 9, 10].
Metastasis is a complex process in which cancer cells leave a primary organ, migrate through basement membranes and connective tissue into lymphatic or blood vessels, then extravasate into a distant organ to establish a new tumor [11, 12]. Complications resulting from the development of metastases are responsible for 90% of cancer-related death [13]. Designing therapeutic strategies to kill metastatic cells or to prevent their colonization of recipient tissues is, therefore, a major goal of cancer research. Despite its clinical relevance however, metastasis is still poorly understood. There are many unanswered questions regarding how metastatic cells leave the tumor and establish themselves in a new tissue, where they may stay dormant for a long time or immediately grow into a new tumor [14, 15]. It is thought that accumulated genetic and epigenetic changes in a sub-population of tumor cells eventually allow these cells to undergo the metastatic voyage. In this hypothesis, metastasis is a late result of tumor progression. Another hypothesis, supported by evidence from epidemiological studies, contends that pre-malignant tumor cells can disseminate early and evolve independently from the primary tumor (discussed in [12, 16-18]).
The inability to directly observe dynamic processes in vivo has been a major obstacle to the study of cancer. At present the most relevant animal models of human cancer, such as transgenic mouse models, do not allow for the visualization of the molecular and microenvironmental events that influence tumor formation, growth, vascularization and metastasis. Indeed, solid tumors are often buried inside the body, which precludes direct observation, and metastatic lesions can be detected only once they are established and after the animal is sacrificed. Histological studies (epidemiologic studies in human, or studies in animal models of cancer) and measurements of tumor size or number of metastatic foci have provided important clues, but with a static view. Thus, extensive autopsies must be performed to reconstruct a time-resolved model of progression. Revascularization, cell migration or adaptation to a new environment during colonization cannot be observed directly, and reconstruction of these parameters using comparative histology is problematic because each tumor is different. New whole-body imaging technologies represent a real progress in cancer research as they now measure tumor growth in real-time and detect metastasis at relatively early stages [19-22]. However, it is still very difficult to detect small metastases either in patients or in animal models, and events that occur at intermediate phases of cancer progression cannot be monitored. Intravital Microscopy (IVM) is a powerful tool for the real-time visualization of previously unobserved mechanisms, thereby providing crucial information regarding several aspects of cancer progression and metastasis. In this review, we discuss the design of animal models of cancer that are compatible with the use of IVM.
Advantages of IVM
Interest in IVM has been renewed by the recent availability of very sophisticated imaging technologies and the constant improvements in molecular probes and analysis tools.
The history of IVM and new advances in imaging techniques are thoroughly reviewed in [23], whereas detailed descriptions of imaging technologies including fluorescent light microscopy, laser-scanning confocal microscopy, laser-scanning multiphoton microscopy, are described in [24]. Earlier studies commonly used in vivo dyes such as rhodamine-based CMTMR to visualize cancer cells or animal tissue. Later on, cells were rendered permanently fluorescent by stable expression of GFP-fusion proteins. There are now many more options with the availability of multiple fluorescence colors, FRET, the possibility to distinguish tissues in the recipient animals using fluorescent transgenics [25, 26], and the possibility to stain tumor cells cytoplasm and nucleus with different colors [27].
The main advantage of IVM is the real-time visualization of cellular events at a very high resolution. IVM permits the direct observation of dynamic cellular processes while they are taking place and the high magnification of images shows transitional mechanisms at the cellular level, or even at the molecular level. For example Tada et al. successfully tracked single-particle quantum dots conjugated with HER-antibodies to analyze the molecular mechanism behind the transport of drug carriers in vivo [28]. Time scales of observation vary from seconds to several weeks. In a study that strikingly illustrates the real-time possibilities of dynamic IVM, the binding of fluorophore-conjugated antibody to endothelial cells could be visualized in vivo, demonstrating that caveolae operate as pumps and move the antibody within seconds from the blood across the endothelium into lung tissue [29]. Events that take place within hours are extremely easy to follow, whereas observations can be made over days or weeks if precautions are taken to “label” the area of observation and ensure that the same region is being observed. For example, in their study in mouse mammary chambers, Kedrin et al. “optically marked individual tumor cells expressing photoswitchable proteins” to monitor the migration of defined groups of cells over time [30].
Importantly, IVM offers the possibility to follow tumor growth in a non-invasive, non-destructive manner. Tumor-related parameters can be measured in living animals and in real-time, including tumor growth or regression, angiogenesis, infiltration by immune cells, tumor cell migration, all in the context of the host. Since repeated measurements of physiological parameters can be made from the same animal, the technique considerably reduces the number of animals needed to obtain a time-resolved picture of cancer progression compared to methods that require euthanizing animals at each time-point.
Unique observations have been made in several areas of cancer, particularly angiogenesis and cancer cells migration. For example, IVM was applied to the study of tumor microcirculation and lymphatic systems and showed that the tumor vasculature in xenograft tumors is characterized by a variety of structural and functional abnormalities that impair blood flow, resulting in the appearance of zones of necrosis and/or hypoxia (reviewed in [31]). The high resolution of IVM also allows the visualization of microvessels and capillaries. Therefore, very detailed studies can be performed about blood vessel formation, blood flow, leakage, migration of cancer cells through vessel walls during intravasation or extravasation (migration of cells from the tumor into a blood vessel, or migration of tumor cells from the blood vessel into the tissue, respectively). The reader will find further review of the use of IVM applied to angiogenesis research in [32] and [33].
Another remarkable application of IVM is the observation of cell migration patterns within primary tumors, which is impossible to track in real-time using other approaches (See [23, 34] for reviews of IVM works on metastasis). Indeed, high resolution IVM allows one to visualize how cancer cells migrate within a tumor, then break away from the tumor and intravasate into blood vessels [35, 36]. Tracking cells inside tumors using IVM revealed heterogeneity within a tumor, with differences in how cells migrate in different areas of the tumor. Sahai's laboratory suggested that cancer cells chose between several migratory mechanisms depending on the microenvironment. Cancer cells undergo an epithelial-to-mesenchymal transition and acquire fibroblast-like migratory properties, or they use amoeboid moves similar to leukocytes in order to invade surrounding tissue [37, 38].
The mouse dorsal skinfold chamber model
The application of IVM to the study of cancer is limited by the availability of animal models that bear visually accessible tumors. Therefore, the implantation of transparent windows (“chambers”) on rodents was invented to enable microscopic observations in cancer research. The dorsal skinfold chamber model is used extensively with hamsters for the study of microcirculation and angiogenesis [39]. Chambers in rats have also been used to investigate cancer, but rat models lack the versatility of mouse models. The elegant dorsal skinfold model was described by Algire in 1943. Recent reviews provide an excellent technical description of the model, including apparatus and surgical procedures, examples of applications and model variations [24, 40]. Briefly, platinum chambers with a viewing transparent window are placed by surgery into the dorsal skin of mice as shown in Figure 1, an adaptation described in [41]. Plastic frames, described recently, are lighter than metal ones while maintaining the characteristics of the windows [42].
Figure 1.
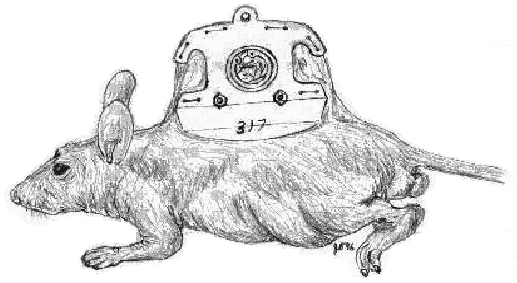
Cartoon illustrating the dorsal skinfold chamber in a nude mouse
An implanted tumor, blood vessels, and host tissue are observed directly through the transparent window. Thus, tumor growth can be visualized and quantified over time along with the development of the vasculature, at high resolution and without tissue damage. This permits “chronic” studies in which repeated analysis are made over a prolonged period. In our laboratory for example, it is now possible to observe tumors for up to 90 days.
Fluorescent tumor-derived cells are implanted under the glass of the chambers. Virtually any strain of mouse can be used. Nude or SCID immunodeficient mice are used to implant human cells, whereas syngeneic mice may be used for the implantation of mouse cancer cells, such as tumor cell lines derived from transgenic models of cancer. Alternatively, small pieces of a growing tumor, originating from xenografts or from transgenic mice, can be implanted either dissociated or as small fragments. This provides less control over cell composition, since tumors are more heterogeneous than cell lines (although cells dissociated from tumors can be sorted by flow cytometry). However, tumor tissues usually do not stably express fluorescent proteins and vital dyes need to be used instead, limiting the total period of observation. Alternatively, tumor tissue can be obtained from human patients.
A few laboratories, including ours, use tumor cells transfected with histone H2B-GFP fusion protein, which was initially developed by Kanda et al. for the observation of chromosome dynamics in living cells [43]. Histone H2B-GFP is incorporated into the chromatin without affecting cell cycle progression. Following implantation of H2B-GFP-transfected cells in mouse chambers, the development of the resultant tumors is followed by IVM in living animals and as a function of time. Tumor cell fluorescence allows one to measure angiogenic activity, infiltration by immune cells, tumor cell migration, and parameters pertaining to tumor growth or regression such as mitosis, apoptosis and cell cycle arrest [44-47]. The number of cancer cells in a growing tumor can be determined accurately from the fluorescence intensity by using a calibration curve [47, 48]. H2B-GFP makes it very simple to visualize metaphase-telophase DNA and apoptotic/pyknotic nuclei using high magnification images. Therefore the number of cells undergoing mitosis or apoptosis can be calculated. Finally, vascular parameters (vascular area, vascular length; average tumor vessel diameter and vascular density) are analyzed and calculated from video recordings using a photodensitometric computer software [49, 50]. Dual color analysis is achieved by implanting red-fluorescent cells in GFP-mice, or green-fluorescent tumor cells into red fluorescent mice [26, 51]. This strategy provides high-resolution images in which the vasculature, the host tissue, and immune cells can easily be distinguished from the implanted tumor cells.
A major drawback of the dorsal skinfold chamber model, however, lies in its limited relevance to tumor physiology. This model is, in fact, quite similar to the traditional sub-cutaneous xenograft model and similarly fails to represent many aspects of clinical cancer, especially with regard to metastasis, drug sensitivity and angiogenesis (xenograft tumors are notoriously poorly vascularized). Mouse cancer cells can be implanted in syngeneic animals, whereas human tumors can be studied using nude or SCID mice [52], but large numbers of human cells, usually between 106 and 107 cells, have to be injected for the tumor to survive and grow. While many human tumor cells successfully form tumors in the chamber model in these conditions, some cell lines fail to grow and undergo massive apoptosis [46]. We have found that the human tumors that do grow in the chamber often become encapsulated, thereby excluding macrophages and other immune cells that normally infiltrate the tumor mass. Tumor-associated macrophages (TAMs) have a dual influence. On the one hand, they may facilitate angiogenesis, tumor cell proliferation, and metastasis during tumor progression. On the other hand, TAMs also participate in the immune anti-tumor defense through cytotoxic activities [53]. Figure 2 (A, B) shows micro-photographs obtained after implantation of HT1080 human fibrosarcoma cells, which became encapsulated. We were able to demonstrate that human tumors that failed to encapsulate were massively infiltrated by macrophages, and that these tumors were, in fact, eliminated by the mouse inate immune system [46]. In contrast, syngeneic tumors formed from mouse LLC cells (Lewis Lung Carcinoma) were not encapsulated and bone marrow cells, including macrophages, were recruited as shown in Figure 2C, D. Thus, the inability to effectively exclude the innate immune system by encapsulation was a main impediments to the survival of small human micro-tumors in the dorsal skinfold chamber in nude mice.
Figure 2.

Growth of human tumor cells HT1080 (A, B) and growth of mouse tumor cells (C, D) in the dorsal skinfold chamber. Non-fluorescent tumor cells were implanted in the dorsal chambers of nude mice that had previously been irradiated and transplanted with GFP+ bone marrow. A succesfully growing human tumor becomes encapsulated (arrow in panel A), effectively excluding bone marrow-derived cells from the tumor, and is poorly vascularized. Mouse tumors do not become encapsulated, but vascularize (V=blood vessel in panel C). Note the denser bone marrow-derived microenvironment (green cells) in mouse tumors (panel D) compared to human tumors (panel B). Yellow bar = 25 μm in panels B and D; 100 μm in panels A and C. Reproduced with permission from Frost et al., In Vivo (2003) 17(5):377-88.
Stroma in the chamber model: the syngeneic pseudo-orthotopic dorsal chamber model
There is now a large body of literature indicating that the tumor microenvironment is crucial for the progression of almost every type of cancer and that orthotopic implantation of cancer cells recapitulate human disease much more closely than subcutaneous implantation [54]. Tumors grow faster and develop better vasculature when the cancer cells are implanted into the relevant organ. In addition, many tumors do not form metastases unless they are implanted orthotopically. Orthotopic implantation also enhances the tumorigenicity with respect to tumor growth and penetrance. This explains, in part, why the various types of cancer that have been modeled in transgenic mice mimic human cancer much more faithfully than xenograft models. However, many solid epithelial tumors that develop in transgenic animals or following orthotopic implantation are difficult to observe because they arise deep in the body.
The very distinctive benefit of the chamber model is to make possible the visualization of tumors that are normally buried and inaccessible, such as pancreatic, prostate or lung tumors. Still, the dorsal skinfold represents a non-orthotopic environment for most tumors except melanomas. In an insightful study, Shan et al. describe a method in which a microscope window was placed over the mammary gland of female rats to examine angiogenesis and the proliferation of breast cancer cells in an orthotopic environment [55]. Through the use of fluorescent tags and “photoswitching” in mouse mammary windows, Kedrin et al. stained tumor cells differently in various tumor areas to demonstrate that the microenvironment around tumor blood vessels was more supportive of metastatic behaviors than areas away from blood vessels [30]. Thus, mammary windows will allow one to directly observe cancer cell growth and division, migration, invasion and intravasation within mammary gland microenvironments. While this approach does, indeed, elegantly solve the problem of monitoring tumor development in the orthotopic environment for mammary tumors, it does not apply to other tumors of interest such as prostate or lung cancer.
To resolve this issue, a novel “pseudo-orthotopic” model has been developed in our laboratory. This method emerged from early experiments showing that various syngeneic tissues grafted in rodent dorsal chambers could re-vascularize and survive over long periods of time [56]. Successfully grafted tissues include spleen, myocardium, spongious bone [56] and adipose tissue [57]. Endometrial tissue was also successfully implanted in dorsal chambers to study angiogenesis and test the efficacy of drugs against endometriosis [58, 59]. In our “pseudo-orthotopic” model, small pieces of orthotopic tissue from donor mice were co-implanted with the tumor cells into the dorsal skinfold chamber of syngeneic C57BL/6 mice to mimic the microenvironment of the tumor. We observed that various types of tissue implanted in the chambers survived and revascularized, and that tumor-derived cell lines thrived upon co-implantation with their respective stroma. For example, we compared the growth characteristics of mouse prostate cancer cells implanted in dorsal chambers with or without prostate tissue. The proliferation of tumor cells was 5 times higher when prostate tissue was present. The vascular area, average vessel diameter, and vascular density were also consistently higher, whereas tumors implanted without prostate tissue were poorly angiogenic [47]. Thus, co-implanting mouse prostate cancer cells with prostate stroma provides the tumor cells with an environment that better reflects the clinical disease compared to purely subcutaneous models. Importantly, re-vascularized stromal tissue remains viable for long periods of time (up to 2-3 months in our laboratory).
Using this model we demonstrated that androgen ablation through surgical castration induces a profound regression of the vasculature surrounding prostate tumors [47, 60]. Androgen ablation is the standard therapeutic approach to prostate cancer that progresses after surgery and/or radiation therapy. Although control of the disease is always achieved at first, almost all patients then progress to androgen-independent disease. It is generally believed that the initial population of androgen-dependent cells undergoes rapid apoptosis upon hormone withdrawal. However, our data indicate that castration decreases cell proliferation rather than induce apoptosis, and also has anti-angiogenic effects.
The source of the vasculature surrounding the tumor was determined by implanting minced prostate tissue derived from GFP transgenic mice into mouse dorsal chambers. Thus, the implanted green-fluorescent stromal cells can be distinguished from host tissues. We observed that the GFP-positive endothelium in the prostate stroma revascularized with GFP-positive vessels rather than with host vasculature. Thereafter the GFP-positive endothelial cells reattached to the pre-existing vasculature underneath the tumor spheroid and flowing red blood cells could be seen [47]. In another set of experiments, breast cancer cells stably transfected with H2B-mCherry (red) were co-implanted with mammary fat pad tissue from a lactating GFP+ mouse and allowed to grow for 2 weeks. Intense GFP fluorescence was detected in the vasculature of the growing tumor, indicating that the engrafted tissue provided the blood vessels to the tumor (unpublished). Thus, tumor -associated vascularization occurs through recruitment of vessels from the co-implanted stromal tissue.
A variety of tissues were successfully implanted including lymph node, prostate, liver, lung, bone marrow, and mammary fat pad. In addition, several cancer cell lines have been successfully grown in the corresponding tissue such as prostate TRAMP-C2 [47, 60], breast N202 [61, 62], ovarian MOVCAR, and renal carcinoma RenCa, most of them generated from relevant transgenic models. To improve our prostate cancer model, we now use PTEN-P2 and PTEN-CaP2 mouse carcinoma cell lines. These cells, generated in Dr. H. Wu's laboratory, were derived from PTEN+/-and PTEN-/-transgenic mice, respectively, [63]. The prostate-specific PTEN-deficient mouse model is considered one the most relevant to human prostate cancer. H2B-GFP was transfected into these cells and green-fluorescent PTEN-P2 and PTEN-CaP2 cell spheroids were implanted on top of prostate tissue from donor mice in the dorsal chamber of recipient mice. Figure 3 shows pictures of the PTEN-P2 and PTEN-CaP2 micro-tumors growing as a function of time. Tumor growth was reflected by an increase in overall size as well as an increase in the brightness of the tumor area due to a higher density of fluorescent cells within the tumor. These tumors were highly angiogenic. Importantly, the growth characteristics and hormone-dependency of hormone-sensitive prostate tumors (P2 cells) and hormone-insensitive (CaP2 cells) in the pseudo-orthotopic chamber model corresponded to previously published data [63]. These tumor cells will be co-implanted with prostate tissue from a tomato mouse [64] for dual-color imaging studies.
Figure 3.

H2B-GFP-positive PTEN-P2 and PTEN-CaP2 cell sheroids were co-implanted with prostate tissue into mouse dorsal chambers. Tumors were imaged at the indicated times using IVM. One can see the increase in tumor size and in tissue vascularization. Yellow bar = 1mm in the tumor panels 1 and 3; 100 mm in panels 2 and 4.
Study of metastasis in the chamber model
After cancer cells break away from the primary tumor, enter lymphatic and blood vessels and circulate through the bloodstream, they extravasate and colonize the microenvironment of the distant organ in which they grow into a new tumor [11, 12, 14, 15]. Colonization of distant organs is a rate-limiting step to the formation of metastatic tumors [65]. This process depends greatly on the new microenvironment and on the dynamic and reciprocal interactions that take place between the tumor cells and the stromal cells of the metastatic organ [5, 9, 66]. Unfortunately, what happens during the early stages of development of a metastatic tumor is still unknown. The interactions between disseminated cancer cells and their microenvironment during metastasis have been mostly based on correlation and logically inferred since most animal models of cancer provide only end-point outcomes. Transgenic mouse models or methods such as tail vein injection of metastatic cells (a model for lung metastasis) do not permit the detection of micrometastases or the measurement of tumor growth in real time, and the metastatic lesions can be analyzed only after the mice are sacrificed. Indeed, it is impossible to predict where metastasis will arise and metastatic tumors are detected only after they are well-established, precluding the study of the early steps of the process. Consequently it is impossible, for example, to tell whether a small tumor in the bone at the time of necropsy is new and aggressive or old and relatively slow-growing. Data must be inferred indirectly and on a statistical basis. Certain inferences are impractical to achieve because the mouse has to be sacrificed for each observation, and therefore can only be done on an average basis. IVM has the potential to solve this problem almost completely by continuous monitoring of individual tumors, thereby eliminating uncertainties in interpolation.
Ex-vivo metastasis assays use fluorescent metastatic cancer cells orthotopically implanted in the relevant organ, which then lodge into secondary organs. Cells can also be injected into the tail-vein of mice to lodge in the lungs. Animals are usually sacrificed and the organs resected for IVM measurements as in [67]. Other laboratories use tissue perfusion and direct injection of tumor cells into the perfusion system to extend the time of observation [68]. Using a similar model, Gassman et al. demonstrated that CXCL12, expressed in the endothelial cells of the liver blood vessels, interacted with CXCR4 expressed in the tumor cells, and promoted tumor cell adhesion to the liver blood vessels and extravasation into the liver [69]. A study in which the lungs were perfused and sections of tissue placed on agarose-based medium for up to 21 days, allowing the monitoring of metastatic tumor growth, showed that the survival of cells arriving in lung was the limiting step of metastatic efficiency [70]. In these models, the tissue architecture is maintained, with stromal cells and relevant ECM components. However, some aspects of metastatic tumor growth are lacking such as vascularization or infiltration with immune cells.
This problem is solved, for brain metastasis, by the use of cranial windows that were designed to provide visual access and to examine the microcirculation in the brain environment following implantation of glioma cells or breast cancer cells, thus mimicking a relevant environment for brain tumors or breast cancer metastases to the brain [40, 71]. For example, Monsky et al. used the cranial window to compare physiological parameters including vascularization of breast cancer cells implanted in mammary windows (orthotopic environment) or in cranial windows (brain metastasis environment), showing among other things that the expression of pro-angiogenic growth factors in a tumor depends on its microenvironment [72].
The pseudo-orthotopic model partially remedies the limitation of organ accessibility. In theory, tumors can be grown on any grafted tissue, to the extent that the pseudo-orthotopic tissue retains the properties of the native tissue, and analysis of several steps of metastatic tumor growth and adaptation can be followed by IVM. Thus, we have compared the adaptation and the growth of breast cancer cells H2B-GFP/N202 in several pseudo-organs relevant to breast cancer metastasis. To generate a pseudo -orthotopic metastasis model, minced mammary fat pad, liver, lung and skin obtained from lactating female mice were implanted into dorsal skinfold chambers of nude mice. H2B-GFP/N202 cells were then implanted on these tissues. The importance of the microenvironment for the growth of N202 tumors was apparent, since tumors grew poorly when co-implanted with tissues other than mammary fat pad (manuscript in preparation). This model thus allows one to study many biological parameters underlying the real-time adaptation of tumor cells to a new environment, particularly in the context of a new host tissue relevant to metastasis.
Testing therapeutics in the chamber model
Because vascular events can be accurately quantified, IVM in the dorsal chamber model is ideally suited to evaluate the efficacy of potential anti-angiogenic drugs. Treatment of vascularized tumors implanted into dorsal skinfold or cranial chambers with neutralizing antibodies against growth factor VEGF induced vascular regression [44, 45, 73, 74]. It was later shown that inhibition of the VEGF receptor (Flk-1) reduced angiogenesis in the peritumoral areas but not in the intratumoral area [75]. High angiogenic activity is required for glioma invasion of adjacent tissue, suggesting that targeting angiogenesis in glioma may reduce metastasis. Indeed, inhibition of Flk-1 reduced invasion as well as angiogenesis [76]. In an attempt to specifically target the tumor vasculature, a thrombogene was linked to the heparin-binding domain of VEGF. The drug localized to tumor blood vessels in vivo, and had potent anti-angiogenic activity [77]. Another work showed that the Notch signaling pathway is involved in angiogenesis because neutralizing one of the Notch ligands blocked angiogenesis and pancreatic tumor growth [78]. Finally, this model is widely used to demonstrate the vascular effects of new anti-angiogenic inhibitors [62, 79-86]. In addition to monitoring the vasculature in real-time, IVM discriminates between the direct effects of a drug on tumor cells and its effects on the vasculature and thus provides a clear picture of its mechanism of action [87-91].
Since orthotopic models are much better suited to predict the effectiveness of chemotherapy than are sub-cutaneous models [54], we argue that pseudo-orthotopic chamber models are very relevant to drug therapy testing [61, 62]. As an example, the effect of COX-2 inhibitor Celecoxib (Celebrex) was investigated in the pseudo-orthotopic dorsal chamber model for prostate cancer using H2B-GFP/TRAMP prostate cancer cells (derived from the tumors of a TRAMP mouse [92]). Celecoxib was administered orally, alone or in combination with castration-induced androgen-withdrawal. In vitro and in vivo experiments indicated that Celecoxib alone had a direct cytostatic effect on tumor cells in which it induced mitotic arrest followed by cell death. The drug had but little effect on vascular parameters in vivo, whereas castration alone caused vascular shrinking, as previously observed [47]. Profound tumor regression was observed with the combination of Celecoxib and androgen-withdrawal, causing a synergistic effect due to decreased vascularization upon androgen withdrawal on the one hand, and growth arrest of tumor cells due mostly to Celecoxib on the other hand. Interestingly, the efficacy of the combination was much better in vivo than in vitro, because of the separate effects of each treatment on distinct biological compartments that are not represented in cultured cell lines.
To conclude, the pseudo-orthotopic dorsal chamber system is a promising model to evaluate novel therapies for the treatment of cancer without animal necropsy, and to determine vascular parameters as well as cytostatic or cytotoxic effects. The strengths of this system lie in the relative speed and mechanistic detail by which therapies can be evaluated. In addition, this model recapitulates the native three-dimensional architecture and microenvironment (cells and ECM) of the tumor, with the advantages of an observation window. It extends the possibilities of the cranial and mammary windows by allowing other types of cancer to be studied.
Future directions
Stromal cells in cancer tissue display different gene expression patterns than stromal cells in normal tissue, a result of the bi-directional interactions that take place between stroma and tumor [93, 94]. So far, very few transgenic animals have been generated that display tissue-specific alterations of cancer microenvironments. Such models would be extremely useful in exploring novel therapeutic strategies. The pseudo-orthotopic model, in addition to MIW and cranial chambers, will be used to study the stroma at early stages of metastatic tumor development and to examine the effects of therapeutic treatments targeting the tumor microenvironment that could have synergistic effects when combined with current therapies that target the tumor cells. As tumor growth parameters are tracked in real time, tumors can be harvested when they undergo major adaptive transitions. Thus, it becomes possible to study changes in gene expression, signaling pathways, etc.
Finally, the use of donor animals with defined genetic modifications (such as knock-out and transgenic mice) as source of grafted organ will open new avenues of investigation since it will be possible to examine the role of defined signaling pathways in the survival and growth of metastatic tumors.
Conclusion
New probes and whole-body imaging technologies are a major step for real-time measurements of tumor growth and for the detection of metastases, but still do not accurately visualize early events because they do not provide a way to access information (or, in case of early metastasis, a way of even knowing where the information is). IVM goes beyond whole-body imaging to measure single-cell mechanisms and early events with very high resolution. Thus, it has provided us with new, unexpected information regarding the mechanisms of angiogenesis, lymphogenesis, cell motility and invasion processes relevant to early steps of dissemination. Animal models in which IVM can be used are evolving too, allowing a more accurate representation of human cancer. Chamber models, through the design of observations windows at various peripheral locations, have become relevant to the study of stroma-tumor interactions and of later steps of metastasis such as colonization of target organs, which, until recently, were not amenable to direct observation.
Acknowledgments
This work was supported by UC/CBRP grant 151B-0133 and DOD-OCRP grant W81XWH-09-1-0280 (PB) and by NIH/NCI grant CA133638 (JW).
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- 4.Hu M, Polyak K. Microenvironmental regulation of cancer development. Curr Opin Genet Dev. 2008;18:27–34. doi: 10.1016/j.gde.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer. 2009;9:285–293. doi: 10.1038/nrc2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhowmick NA, Moses HL. Tumor-stroma interactions. Curr Opin Genet Dev. 2005;15:97–101. doi: 10.1016/j.gde.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukino K, Shen L, Matsumoto S, Morrison CD, Mutter GL, Eng C. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004;64:7231–7236. doi: 10.1158/0008-5472.CAN-04-2866. [DOI] [PubMed] [Google Scholar]
- 8.Eng C, Leone G, Orloff MS, Ostrowski MC. Genomic alterations in tumor stroma. Cancer Res. 2009;69:6759–6764. doi: 10.1158/0008-5472.CAN-09-0985. [DOI] [PubMed] [Google Scholar]
- 9.Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25:30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 11.Molloy T, van't Veer LJ. Recent advances in metastasis research. Curr Opin Genet Dev. 2008;18:35–41. doi: 10.1016/j.gde.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 13.Loberg RD, Bradley DA, Tomlins SA, Chinnaiyan AM, Pienta KJ. The lethal phenotype of cancer: the molecular basis of death due to malignancy. CA Cancer J Clin. 2007;57:225–241. doi: 10.3322/canjclin.57.4.225. [DOI] [PubMed] [Google Scholar]
- 14.Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70:5649–5669. doi: 10.1158/0008-5472.CAN-10-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 16.Ansieau S, Hinkal G, Thomas C, Bastid J, Puisieux A. Early origin of cancer metastases: dissemination and evolution of premalignant cells. Cell Cycle. 2008;7:3659–3663. doi: 10.4161/cc.7.23.7049. [DOI] [PubMed] [Google Scholar]
- 17.Dong F, Budhu AS, Wang XW. Translating the metastasis paradigm from scientific theory to clinical oncology. Clin Cancer Res. 2009;15:2588–2593. doi: 10.1158/1078-0432.CCR-08-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302–312. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- 19.Kaijzel EL, van der Pluijm G, Lowik CW. Whole-body optical imaging in animal models to assess cancer development and progression. Clin Cancer Res. 2007;13:3490–3497. doi: 10.1158/1078-0432.CCR-07-0402. [DOI] [PubMed] [Google Scholar]
- 20.Dufort S, Sancey L, Wenk C, Josserand V, Coll JL. Optical small animal imaging in the drug discovery process. Biochim Biophys Acta. 2010;1798:2266–2273. doi: 10.1016/j.bbamem.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 21.Luker GD, Luker KE. Optical imaging: current applications and future directions. J Nucl Med. 2008;49:1–4. doi: 10.2967/jnumed.107.045799. [DOI] [PubMed] [Google Scholar]
- 22.Henriquez NV, van Overveld PG, Que I, Buijs JT, Bachelier R, Kaijzel EL, Lowik CW, Clezardin P, van der Pluijm G. Advances in optical imaging and novel model systems for cancer metastasis research. Clin Exp Metastasis. 2007;24:699–705. doi: 10.1007/s10585-007-9115-5. [DOI] [PubMed] [Google Scholar]
- 23.Beerling E, Ritsma L, Vrisekoop N, Derksen PW, van Rheenen J. Intravital microscopy: new insights into metastasis of tumors. J Cell Sci. 2011;124:299–310. doi: 10.1242/jcs.072728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makale M. Intravital imaging and cell invasion. Methods Enzymol. 2007;426:375–401. doi: 10.1016/S0076-6879(07)26016-1. [DOI] [PubMed] [Google Scholar]
- 25.Hoffman RM. The multiple uses of fluorescent proteins to visualize cancer in vivo. Nat Rev Cancer. 2005;5:796–806. doi: 10.1038/nrc1717. [DOI] [PubMed] [Google Scholar]
- 26.Hoffman RM. Imaging cancer dynamics in vivo at the tumor and cellular level with fluorescent proteins. Clin Exp Metastasis. 2009;26:345–355. doi: 10.1007/s10585-008-9205-z. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman RM. Real-time subcellular imaging in live animals: new visible targets for cancer drug discovery. IDrugs. 2006;9:632–635. [PubMed] [Google Scholar]
- 28.Tada H, Higuchi H, Wanatabe TM, Ohuchi N. In vivo real-time tracking of single quantum dots conjugated with monoclonal anti-HER2 antibody in tumors of mice. Cancer Res. 2007;67:1138–1144. doi: 10.1158/0008-5472.CAN-06-1185. [DOI] [PubMed] [Google Scholar]
- 29.Oh P, Borgstrom P, Witkiewicz H, Li Y, Borgstrom BJ, Chrastina A, Iwata K, Zinn KR, Baldwin R, Testa JE, Schnitzer JE. Live dynamic imaging of caveolae pumping targeted antibody rapidly and specifically across endothelium in the lung. Nat Biotechnol. 2007;25:327–337. doi: 10.1038/nbt1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kedrin D, Gligorijevic B, Wyckoff J, Verkhusha VV, Condeelis J, Segall JE, van Rheenen J. Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods. 2008;5:1019–1021. doi: 10.1038/nmeth.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukumura D, Duda DG, Munn LL, Jain RK. Tumor microvasculature and microenvironment: novel insights through intravital imaging in pre-clinical models. Microcirculation. 2010;17:206–225. doi: 10.1111/j.1549-8719.2010.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sckell A, Leunig M. The dorsal skinfold chamber: studying angiogenesis by intravital microscopy. Methods Mol Biol. 2009;467:305–317. doi: 10.1007/978-1-59745-241-0_19. [DOI] [PubMed] [Google Scholar]
- 33.Lunt SJ, Gray C, Reyes-Aldasoro CC, Matcher SJ, Tozer GM. Application of intravital microscopy in studies of tumor microcirculation. J Biomed Opt. 2011;15:011113. doi: 10.1117/1.3281674. [DOI] [PubMed] [Google Scholar]
- 34.MacDonald IC, Chambers AF. Breast cancer metastasis progression as revealed by intravital videomicroscopy. Expert Rev Anticancer Ther. 2006;6:1271–1279. doi: 10.1586/14737140.6.9.1271. [DOI] [PubMed] [Google Scholar]
- 35.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 36.Condeelis J, Singer RH, Segall JE. The great escape: when cancer cells hijack the genes for chemotaxis and motility. Annu Rev Cell Dev Biol. 2005;21:695–718. doi: 10.1146/annurev.cellbio.21.122303.120306. [DOI] [PubMed] [Google Scholar]
- 37.Sahai E. Illuminating the metastatic process. Nat Rev Cancer. 2007;7:737–749. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 38.Madsen CD, Sahai E. Cancer dissemination–lessons from leukocytes. Dev Cell. 2010;19:13–26. doi: 10.1016/j.devcel.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Menger MD, Laschke MW, Vollmar B. Viewing the microcirculation through the window: some twenty years experience with the hamster dorsal skinfold chamber. Eur Surg Res. 2002;34:83–91. doi: 10.1159/000048893. [DOI] [PubMed] [Google Scholar]
- 40.Brown E, Munn LL, Fukumura D, Jain RK. In vivo imaging of tumors. Cold Spring Harb Protoc. 2010;2010 doi: 10.1101/pdb.prot5452. pdb prot5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lehr HA, Leunig M, Menger MD, Nolte D, Messmer K. Dorsal skinfold chamber technique for intravital microscopy in nude mice. Am J Pathol. 1993;143:1055–1062. [PMC free article] [PubMed] [Google Scholar]
- 42.Ushiyama A, Yamada S, Ohkubo C. Microcirculatory parameters measured in subcutaneous tissue of the mouse using a novel dorsal skinfold chamber. Microvasc Res. 2004;68:147–152. doi: 10.1016/j.mvr.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8:377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 44.Borgstrom P, Gold DP, Hillan KJ, Ferrara N. Importance of VEGF for breast cancer angiogenesis in vivo: implications from intravital microscopy of combination treatments with an anti-VEGF neutralizing monoclonal antibody and doxorubicin. Anticancer Res. 1999;19:4203–4214. [PubMed] [Google Scholar]
- 45.Borgstrom P, Hillan KJ, Sriramarao P, Ferrara N. Complete inhibition of angiogenesis and growth of microtumors by anti-vascular endothelial growth factor neutralizing antibody: novel concepts of angiostatic therapy from intravital videomicroscopy. Cancer Res. 1996;56:4032–4039. [PubMed] [Google Scholar]
- 46.Frost GI, Dudouet B, Lustgarten J, Borgstrom P. The roles of epithelialmesenchymal interactions and the innate immune response on the tumorigenicity of human prostate carcinoma cell lines grown in immuno-compromised mice. In Vivo. 2003;17:377–388. [PubMed] [Google Scholar]
- 47.Frost GI, Lustgarten J, Dudouet B, Nyberg L, Hartley-Asp B, Borgstrom P. Novel syngeneic pseudo-orthotopic prostate cancer model: vascular, mitotic and apoptotic responses to castration. Microvasc Res. 2005;69:1–9. doi: 10.1016/j.mvr.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 48.Huang Y, Tyler T, Saadatmandi N, Lee C, Borgstrom P, Gjerset RA. Enhanced tumor suppression by a p14ARF/p53 bicistronic adenovirus through increased p53 protein translation and stability. Cancer Res. 2003;63:3646–3653. [PubMed] [Google Scholar]
- 49.Frost GI, Borgstrom P. Real time in vivo quantitation of tumor angiogenesis. Methods Mol Med. 2003;85:65–78. doi: 10.1385/1-59259-380-1:65. [DOI] [PubMed] [Google Scholar]
- 50.Torres Filho IP, Hartley-Asp B, Borgstrom P. Quantitative angiogenesis in a syngeneic tumor spheroid model. Microvasc Res. 1995;49:212–226. doi: 10.1006/mvre.1995.1017. [DOI] [PubMed] [Google Scholar]
- 51.Hoffman RM. Orthotopic metastatic (MetaMouse) models for discovery and development of novel chemotherapy. Methods Mol Med. 2005;111:297–322. doi: 10.1385/1-59259-889-7:297. [DOI] [PubMed] [Google Scholar]
- 52.Leunig M, Yuan F, Menger MD, Boucher Y, Goetz AE, Messmer K, Jain RK. Angiogenesis, microvascular architecture, microhemodynamics, and interstitial fluid pressure during early growth of human adenocarcinoma LS174T in SCID mice. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 53.Lissbrant IF, Stattin P, Wikstrom P, Damber JE, Egevad L, Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int J Oncol. 2000;17:445–451. doi: 10.3892/ijo.17.3.445. [DOI] [PubMed] [Google Scholar]
- 54.Bibby MC. Orthotopic models of cancer for preclinical drug evaluation: advantages and disadvantages. Eur J Cancer. 2004;40:852–857. doi: 10.1016/j.ejca.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 55.Shan S, Sorg B, Dewhirst MW. A novel rodent mammary window of orthotopic breast cancer for intravital microscopy. Microvasc Res. 2003;65:109–117. doi: 10.1016/s0026-2862(02)00017-1. [DOI] [PubMed] [Google Scholar]
- 56.Funk W, Endrich B, Messmer K. A novel method for follow-up studies of the microcirculation in non-malignant tissue implants. Res Exp Med (Berl) 1986;186:259–270. doi: 10.1007/BF01852303. [DOI] [PubMed] [Google Scholar]
- 57.Langer S, Sinitsina I, Biberthaler P, Krombach F, Messmer K. Revascularization of transplanted adipose tissue: a study in the dorsal skinfold chamber of hamsters. Ann Plast Surg. 2002;48:53–59. doi: 10.1097/00000637-200201000-00008. [DOI] [PubMed] [Google Scholar]
- 58.Laschke MW, Elitzsch A, Vollmar B, Vajkoczy P, Menger MD. Combined inhibition of vascular endothelial growth factor (VEGF), fibroblast growth factor and platelet-derived growth factor, but not inhibition of VEGF alone, effectively suppresses angiogenesis and vessel maturation in endometriotic lesions. Hum Reprod. 2006;21:262–268. doi: 10.1093/humrep/dei308. [DOI] [PubMed] [Google Scholar]
- 59.Laschke MW, Elitzsch A, Scheuer C, Holstein JH, Vollmar B, Menger MD. Rapamycin induces regression of endometriotic lesions by inhibiting neovascularization and cell proliferation. Br J Pharmacol. 2006;149:137–144. doi: 10.1038/sj.bjp.0706857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abedinpour P, Baron VT, Welsh J, Borgstrom P. Regression of prostate tumors upon combination of hormone ablation therapy and celecoxib in vivo. Prostate. 2011 doi: 10.1002/pros.21297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jin H, Aiyer A, Su J, Borgstrom P, Stupack D, Friedlander M, Varner J. A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. J Clin Invest. 2006;116:652–662. doi: 10.1172/JCI24751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang Y, Borgstrom P, Maynard J, Koziol J, Hu Z, Garen A, Deisseroth A. Mapping of angiogenic markers for targeting of vectors to tumor vascular endothelial cells. Cancer Gene Ther. 2007;14:346–353. doi: 10.1038/sj.cgt.7701030. [DOI] [PubMed] [Google Scholar]
- 63.Jiao J, Wang S, Qiao R, Vivanco I, Watson PA, Sawyers CL, Wu H. Murine cell lines derived from Pten null prostate cancer show the critical role of PTEN in hormone refractory prostate cancer development. Cancer Res. 2007;67:6083–6091. doi: 10.1158/0008-5472.CAN-06-4202. [DOI] [PubMed] [Google Scholar]
- 64.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 65.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 66.Chung LW, Baseman A, Assikis V, Zhau HE. Molecular insights into prostate cancer progression: the missing link of tumor microenvironment. J Urol. 2005;173:10–20. doi: 10.1097/01.ju.0000141582.15218.10. [DOI] [PubMed] [Google Scholar]
- 67.Chishima T, Miyagi Y, Wang X, Yamaoka H, Shimada H, Moossa AR, Hoffman RM. Cancer invasion and micrometastasis visualized in live tissue by green fluorescent protein expression. Cancer Res. 1997;57:2042–2047. [PubMed] [Google Scholar]
- 68.Yates C, Shepard CR, Papworth G, Dash A, Beer Stolz D, Tannenbaum S, Griffith L, Wells A. Novel three-dimensional organotypic liver bioreactor to directly visualize early events in metastatic progression. Adv Cancer Res. 2007;97:225–246. doi: 10.1016/S0065-230X(06)97010-9. [DOI] [PubMed] [Google Scholar]
- 69.Gassmann P, Haier J, Schluter K, Domikowsky B, Wendel C, Wiesner U, Kubitza R, Engers R, Schneider SW, Homey B, Muller A. CXCR4 regulates the early extravasation of metastatic tumor cells in vivo. Neoplasia. 2009;11:651–661. doi: 10.1593/neo.09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mendoza A, Hong SH, Osborne T, Khan MA, Campbell K, Briggs J, Eleswarapu A, Buquo L, Ren L, Hewitt SM, Dakir el H, Garfield S, Walker R, Merlino G, Green JE, Hunter KW, Wakefield LM, Khanna C. Modeling metastasis biology and therapy in real time in the mouse lung. J Clin Invest. 2010;120:2979–2988. doi: 10.1172/JCI40252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 72.Monsky WL, Mouta Carreira C, Tsuzuki Y, Gohongi T, Fukumura D, Jain RK. Role of host microenvironment in angiogenesis and microvascular functions in human breast cancer xenografts: mammary fat pad versus cranial tumors. Clin Cancer Res. 2002;8:1008–1013. [PubMed] [Google Scholar]
- 73.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci U S A. 1996;93:14765–14770. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borgstrom P, Bourdon MA, Hillan KJ, Sriramarao P, Ferrara N. Neutralizing antivascular endothelial growth factor antibody completely inhibits angiogenesis and growth of human prostate carcinoma micro tumors in vivo. Prostate. 1998;35:1–10. doi: 10.1002/(sici)1097-0045(19980401)35:1<1::aid-pros1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 75.Vajkoczy P, Thurnher A, Hirth KP, Schilling L, Schmiedek P, Ullrich A, Menger MD. Measuring VEGF-Flk-1 activity and consequences of VEGF-Flk-1 targeting in vivo using intravital microscopy: clinical applications. Oncologist. 2000;5(Suppl 1):16–19. doi: 10.1634/theoncologist.5-suppl_1-16. [DOI] [PubMed] [Google Scholar]
- 76.Vajkoczy P, Menger MD, Goldbrunner R, Ge S, Fong TA, Vollmar B, Schilling L, Ullrich A, Hirth KP, Tonn JC, Schmiedek P, Rempel SA. Targeting angiogenesis inhibits tumor infiltration and expression of the pro-invasive protein SPARC. Int J Cancer. 2000;87:261–268. doi: 10.1002/1097-0215(20000715)87:2<261::aid-ijc18>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 77.El-Sheikh A, Borgstrom P, Bhattacharjee G, Belting M, Edgington TS. A selective tumor microvasculature thrombogen that targets a novel receptor complex in the tumor angiogenic microenvironment. Cancer Res. 2005;65:11109–11117. doi: 10.1158/0008-5472.CAN-05-2733. [DOI] [PubMed] [Google Scholar]
- 78.Oishi H, Sunamura M, Egawa S, Motoi F, Unno M, Furukawa T, Habib NA, Yagita H. Blockade of delta-like ligand 4 signaling inhibits both growth and angiogenesis of pancreatic cancer. Pancreas. 2010;39:897–903. doi: 10.1097/MPA.0b013e3181ce7185. [DOI] [PubMed] [Google Scholar]
- 79.Laird AD, Vajkoczy P, Shawver LK, Thurnher A, Liang C, Mohammadi M, Schlessinger J, Ullrich A, Hubbard SR, Blake RA, Fong TA, Strawn LM, Sun L, Tang C, Hawtin R, Tang F, Shenoy N, Hirth KP, McMahon G, Cherrington SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000;60:4152–4160. [PubMed] [Google Scholar]
- 80.Hoshida T, Sunamura M, Duda DG, Egawa S, Miyazaki S, Shineha R, Hamada H, Ohtani H, Satomi S, Matsuno S. Gene therapy for pancreatic cancer using an adenovirus vector encoding soluble flt-1 vascular endothelial growth factor receptor. Pancreas. 2002;25:111–121. doi: 10.1097/00006676-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 81.Strieth S, Eichhorn ME, Sauer B, Schulze B, Teifel M, Michaelis U, Dellian M. Neovascular targeting chemotherapy: encapsulation of paclitaxel in cationic liposomes impairs functional tumor microvasculature. Int J Cancer. 2004;110:117–124. doi: 10.1002/ijc.20083. [DOI] [PubMed] [Google Scholar]
- 82.Strieth S, Nussbaum CF, Eichhorn ME, Fuhrmann M, Teifel M, Michaelis U, Berghaus A, Dellian M. Tumor-selective vessel occlusions by platelets after vascular targeting chemotherapy using paclitaxel encapsulated in cationic liposomes. Int J Cancer. 2008;122:452–460. doi: 10.1002/ijc.23088. [DOI] [PubMed] [Google Scholar]
- 83.Strieth S, Eichhorn ME, Werner A, Sauer B, Teifel M, Michaelis U, Berghaus A, Dellian M. Paclitaxel encapsulated in cationic liposomes increases tumor microvessel leakiness and improves therapeutic efficacy in combination with Cisplatin. Clin Cancer Res. 2008;14:4603–4611. doi: 10.1158/1078-0432.CCR-07-4738. [DOI] [PubMed] [Google Scholar]
- 84.Jiang J, Goel R, Schmechel S, Vercellotti G, Forster C, Bischof J. Pre-conditioning cryosurgery: cellular and molecular mechanisms and dynamics of TNF-alpha enhanced cryotherapy in an in vivo prostate cancer model system. Cryobiology. 2010;61:280–288. doi: 10.1016/j.cryobiol.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cuadros C, Dominguez AL, Frost GI, Borgstrom P, Lustgarten J. Cooperative effect between immunotherapy and antiangiogenic therapy leads to effective tumor rejection in tolerant Her-2/neu mice. Cancer Res. 2003;63:5895–5901. [PubMed] [Google Scholar]
- 86.Cuadros C, Dominguez AL, Lollini PL, Croft M, Mittler RS, Borgstrom P, Lustgarten J. Vaccination with dendritic cells pulsed with apoptotic tumors in combination with anti-OX40 and anti-4-1BB monoclonal antibodies induces T cell-mediated protective immunity in Her-2/neu transgenic mice. Int J Cancer. 2005;116:934–943. doi: 10.1002/ijc.21098. [DOI] [PubMed] [Google Scholar]
- 87.Natsume T, Watanabe J, Koh Y, Fujio N, Ohe Y, Horiuchi T, Saijo N, Nishio K, Kobayashi M. Antitumor activity of TZT-1027 (Soblidotin) against vascular endothelial growth factorsecreting human lung cancer in vivo. Cancer Sci. 2003;94:826–833. doi: 10.1111/j.1349-7006.2003.tb01526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schacht V, Becker K, Szeimies RM, Abels C. Apoptosis and leucocyte-endothelium interactions contribute to the delayed effects of cryotherapy on tumours in vivo. Arch Dermatol Res. 2002;294:341–348. doi: 10.1007/s00403-002-0346-7. [DOI] [PubMed] [Google Scholar]
- 89.Strelczyk D, Eichhorn ME, Luedemann S, Brix G, Dellian M, Berghaus A, Strieth S. Static magnetic fields impair angiogenesis and growth of solid tumors in vivo. Cancer Biol Ther. 2009;8:1756–1762. doi: 10.4161/cbt.8.18.9294. [DOI] [PubMed] [Google Scholar]
- 90.Al-Jamal KT, Al-Jamal WT, Akerman S, Podesta JE, Yilmazer A, Turton JA, Bianco A, Vargesson N, Kanthou C, Florence AT, Tozer GM, Kostarelos K. Systemic antiangiogenic activity of cationic poly-L-lysine dendrimer delays tumor growth. Proc Natl Acad Sci U S A. 2010;107:3966–3971. doi: 10.1073/pnas.0908401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Debergh I, Van Damme N, Pattyn P, Peeters M, Ceelen WP. The low-molecular-weight heparin, nadroparin, inhibits tumour angiogenesis in a rodent dorsal skinfold chamber model. Br J Cancer. 2010;102:837–843. doi: 10.1038/sj.bjc.6605535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–3330. [PubMed] [Google Scholar]
- 93.Dakhova O, Ozen M, Creighton CJ, Li R, Ayala G, Rowley D, Ittmann M. Global gene expression analysis of reactive stroma in prostate cancer. Clin Cancer Res. 2009;15:3979–3989. doi: 10.1158/1078-0432.CCR-08-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gregg JL, Brown KE, Mintz EM, Piontkivska H, Fraizer GC. Analysis of gene expression in prostate cancer epithelial and interstitial stromal cells using laser capture microdissection. BMC Cancer. 2010;10:165. doi: 10.1186/1471-2407-10-165. [DOI] [PMC free article] [PubMed] [Google Scholar]