

Abstract
The Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiologic agent of Kaposi's sarcoma (KS)—the most common tumor associated with HIV infection and an important cause of morbidity and mortality in this patient population. The majority of patients with KS exhibit little or no clinical response to existing therapies. The nuclear factor-kappaB (NF-κB) family of transcription factors plays a critical role in facilitating cancer pathogenesis associated with oncogenic viruses, and a better understanding of how cellular factors regulate NF-κB activation in the context of KSHV infection may facilitate development of new therapies for KS. Existing data implicate heat shock protein-90 associated with the cell surface (csHsp90) as a co-factor in cancer cell migration and invasion, and we recently reported that csHsp90 serves as a co-factor for mitogen-activated protein kinase (MAPK) activation during de novo KSHV infection. However, whether csHsp90 regulates NF-κB activation, or cellular pathogenesis associated with KS, has not been established. We have found that csHsp90 serves as an important co-factor for canonical NF-κB activation by KSHV during de novo infection of primary human cells relevant to KS. Furthermore, our correlative functional studies reveal that csHsp90 inhibition suppresses KSHV-induced, NF-κB-dependent secretion of the pro-migratory factors interleukin-8 and vascular endothelial growth factor as well as invasiveness for primary cells following de novo infection. These data implicate csHsp90 in KSHV-mediated activation of NF-κB and associated pathogenesis, and support the potential utility of targeting csHsp90 as a therapeutic approach for KS.
Keywords: Cancer, Kaposi's sarcoma, NF-κB, KSHV, heat shock protein, invasion, transcription factors, signal, trans-duction
Introduction
The Kaposi's sarcoma-associated herpesvirus (KSHV) is one of the most common etiologic agents for cancers arising in the setting of HIV infection, including primary effusion lymphoma (PEL) [1], multicentric Castleman's disease (MCD)[2], and Kaposi's sarcoma (KS)[3]. Although highly active antiretroviral therapy (HAART) has successfully reduced the incidence of HIV-associated KS[4], KS still represents the most common HIV/AIDS-associated malignancy and an important cause of morbidity and mortality in the modern era [5,6]. Moreover, KS has been increasingly recognized in patients exhibiting successful suppression of HIV replication with HAART [7-10]. Notably, KS also represents an important cause of morbidity and mortality in patients receiving solid organ transplants [11].
Standard therapy for KS includes cytotoxic che-motherapeutic agents administered alone or in combination. Unfortunately, the majority of patients exhibit little or no clinical response with this approach, and progression-free intervals are often less than one year [12,13]. Moreover, these agents incur many side effects compounding those caused by HAART [4,14]. During de novo infection, KSHV proteins initiate signal transduction resulting in activation of pro-migratory cytokine responses and induction of cell motility and angiogenesis [15-22]. These data suggest that suppression of KSHV-induced cancer pathogenesis through targeting of signal transduction initiated by KSHV may offer an appealing therapeutic strategy. However, existing approaches targeting signal transduction pathways activated by KSHV are not widely used in clinical practice due to their inability to achieve remission for many existing tumors, or to their well-characterized toxicities (due in part to non-specificity of drug targets) [23-26]. Identification of cellular co-factors regulating signal transduction initiated during KSHV infection may lead to identification of new and safer-therapeutic targets for KS.
NF-κB represents a highly conserved family of transcription factors, including RelA (p65), RelB, c-Rel, p50 (NF-κB1) and p52 (NF-κB2), that share an N-terminal Rel homology domain responsible for DNA binding, dimerization and nuclear translocation [27,28]. Canonical (a.k.a. classical) activation of NF-κB occurs in response to inflammatory mediators and involves IκB kinase (IKKβ) phosphorylation of IκBα, leading to IκBα ubiquitination and degradation and NF-κB nuclear translocation [29]. KSHV-encoded proteins, including the viral G-protein coupled receptor (vGPCR) and viral FLICE inhibitory protein (vFLIP), induce gene transcription, secretion of soluble mediators of cell migration and invasion, and endothelial cell transformation through canonical activation of NF-κB [30-26]. It follows that inhibition of NF-κB may interfere with migration and invasion for KSHV-infected tumor cells. Although under evaluation in clinical trials, small molecule inhibitors of NF-κB also incur toxicities that may limit their clinical utility [37]. Therefore, a better understanding of alternative mechanisms for KSHV activation of NF-κB might facilitate development of novel strategies for targeting this pathway.
Heat shock proteins (Hsp) modulate a wide variety of intracellular processes through the stabilization or regulation of protein folding [38], and Hsp90 plays an important role in the activation of signaling proteins relevant to cell migration and invasion [39]. Furthermore, Hsp90 inhibitors have proven beneficial for reducing solid tumor burden, and their utility is under evaluation in clinical trials for a variety of cancers [40]. Recent identification of Hsp90 on the cell surface (csHsp90) [41] has led to the observation that csHsp90 serves as a co-factor in the activation of specific intracellular signal transduction pathways in a manner distinct from the intracellular form of the protein [42]. Moreover, we have recently demonstrated a role for csHsp90 in KSHV induction of MAPK activation during de novo infection [43]. In the present study, we sought to determine whether csHsp90 regulates KSHV-initiated NF-κB activation and whether targeting csHsp90 interferes with NF-κB-associated cell migration and invasion for KSHV -infected cells.
Materials and methods
Cell culture and infection assays
KSHV-infected body cavity-based lymphoma (BCBL-1) cells were maintained in RPMI 1640 media (Mediatech) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES (pH 7.5), 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 0.05 mM β-mercaptoethanol, and 0.02% (wt/vol) sodium bicarbonate. To obtain purified KSHV for infection experiments, BCBL-1 cells were incubated with 0.6 mM valproic acid for 6 days and purified virus concentrated from culture supernatants as described previously [43]. For negative controls using ultraviolet light-inactivated KSHV (UV-KSHV), viral aliquots were exposed to 1200 J/cm2 UV light for 10 min using a CL-1000 Ultraviolet Crosslinker. Human primary dermal microvascular endothelial cells (pDMVEC) were maintained according to the manufacturer's instructions (Lonza). HeLa and human foreskin fibroblasts (HFF) were maintained in Dulbecco's modified Eagle's medium (DMEM, Mediatech) supplemented with 10% FBS, 10 mM HEPES (pH 7.5), 100 U/mL of penicillin, and 100 μg/mL streptomycin. Human umbilical vein endothelial cells (HUVEC) were maintained in Dulbecco's modified Eagle's medium/Ham's F12 50/50 mix (DMEM F12, Mediatech) supplemented with 1 μg/mL puromycin, 10 mM HEPES (pH 7.5), and 5% FBS. Infectious titers were determined using both pDMVEC and HeLa cells and previously described methods [43].
Hsp90/ NF-κB inhibition
The previously characterized csHsp90 inhibitor DMAG-N-oxide (DNo) [42] and the NF-κB inhibitor Bay11-7082 (Sigma-Aldrich) were dissolved in DMSO and aliquots frozen at -80°C. Cells were incubated with DNo for 16 h at 37°C, Bay11-7082 for 1 h at 37°C or with equivalent volumes of DMSO (vehicle) for negative controls prior to subsequent analyses. Cells were also incubated with 20 ng/mL TNF-α (Cell Signaling) for 20 minutes to induce canonical NF-κB activation for some experiments. In parallel experiments, cells were incubated for 12-16 h with 15 μg/mL of an anti-Hsp90 monoclonal antibody (Stressgen) or a control rat IgG2A isotype antibody (Invitrogen).
Cell viability assays
Cell viability was assessed using a standard MTT assay as previously described [42]. A total of 5×103 cells were incubated in individual wells in a 96-well plate for 24 h. Serial dilutions of DNo or Bay11-7082 were added and cells subsequently incubated in 1 mg/mL MTT solution (Sigma-Aldrich) at 37°C for 3 h followed by 50% DMSO overnight. Optical density was determined thereafter at 570 nm using a spectrophotometer (Thermo Labsystems).
Transfection assays
pFLAG-CMV2-p65 constructs were used in transfection assays to overexpress p65, and pFLAG-CMV2 empty vectors were used as negative controls. For other experiments, cells were transfected with luciferase-based NF-κB reporter constructs for quantification of endogenous NF-κB activation. Cells were transfected in 12-well plates for 24-48 h using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For luciferase expression assays, cells were lysed with 100 μL of lysis buffer (Promega), and 20 μL aliquots from each lysate were analyzed for luciferase activity using a Berthold FB12 luminometer. Light units were normalized to total protein levels for each sample using the BCA protein assay kit (Pierce) according to the manufacturer's instructions to determine relative luciferase units (RLU). Transfection efficiency was assessed through co-transfection of a lacZ reporter construct, and β-galactosidase activity was determined using a commercially available β-galactosidase enzyme assay system according to the manufacturer's instructions (Promega). Three independent transfections were performed for each experiment, and all samples were analyzed in triplicate for each transfection.
Immunoblotting
Cells were lysed in buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% NP40, 1 mM EDTA, 5 mM NaF and 5 mM Na3VO4. Lysates were resolved by 10% SDS-PAGE and transferred to nitrocellulose membranes. Proteins were identified using antibodies recognizing total and phosphorylated proteins as follows: phospho-NF-κB p65 (Ser536), phospho-IKKα/β (Ser176/180), NF-κB p65 (C22B4), IKKα, IKKβ (L570), IκBα (L35A5), Ref1 (Cell Signaling), α-Tubulin (Sigma), and β-Actin (Sigma) for loading controls. Immunoreactive bands were developed using an enhanced chemiluminescence reaction (Perkin-Elmer). Nuclear protein fractions were isolated using a Nuclear Extract Kit (Active Motif) as previously described [31], and the nuclear origin of these extracts was verified using anti-Ref1 antibodies. Anti-α-tubulin antibodies were also used to exclude contamination of nuclear extracts with extranuclear proteins. Protein concentrations were determined using a bicinchoninic acid reagent (Pierce Biotechnology) and stored at -80° C.
ELISA
Concentrations of IL-8 and VEGF in culture supernatants were determined using human IL-8 (Becton Dickinson) and VEGF (Pierce Biotechnology) ELISA kits according to the manufacturers' instructions.
Transwell invasion assays
After incubation, Matrigel Invasion Chambers (Becton Dickinson) were hydrated for 2 h at 37° C with appropriate media. After hydration, fresh media was added to the bottom of the well, then 5 × 104 HFF or 1-1.5 × 104 HUVEC were plated in the top of the chamber. After 24 h, the cells were fixed with 3% paraformaldehyde in PBS for 15 min at room temperature and the chambers were rinsed in PBS and stained with 0.2% crystal violet for 10 minutes. After washing the chambers 5 times with dH2O, the cells at the top of the Matrigel membrane were removed with cotton swabs. “Invading” cells at the bottom of the membrane were counted using a phase contrast microscope. Relative invasion for cells in experimental groups was calculated as follows: relative invasion = # invading cells in experimental group/ # invading cells in control groups.
Statistical analysis
Significance for differences between experimental and control groups was determined using the Student's two-tailed t-test (Excel 8.0). P values less than 0.05 or 0.01 were considered significant or highly significant, respectively.
Results
Canonical activation of NF-κB by KSHV is csHsp90-dependent
KSHV induces canonical activation of NF-κB during de novo infection [30,31,32]. We recently demonstrated that targeting csHsp90 suppressed KSHV activation of MAPK during de novo infection [43]. However, ectopic MAPK overexpression during csHsp90 inhibition only partially rescued KSHV gene expression [43], suggesting that multiple signaling pathways initiated by KSHV may be csHsp90-dependent. Therefore, we sought to determine whether KSHV induction of NF-κB during de novo infection was csHsp90-dependent using two complimentary methods for specifically inhibiting csHsp90 function that we and others have employed in previous studies [38,39,43]: pharmacologic inhibition of csHsp90 using a membrane non-permeable compound (DMAG-N-oxide, or DNo) targeting the extracellular ATP-binding pocket of the Hsp90-alpha subunit located in the N-terminal region of the protein; and targeting of an N-terminal epitope within the Hsp90-alpha subunit using a monoclonal antibody. First, we confirmed that DNo induced minimal cytotoxicity over a range of concentrations and exposure times for two primary cell types relevant to KS pathogenesis, primary dermal microvascular endothelial cells (pDMVEC) and human foreskin fibroblasts (HFF) (Figure 1A, B). KSHV initiates canonical NF-κB activation through phosphorylation of p65 and an increase p65/p50 dimerization [31, 32, 44]. Immunoblotting experiments using whole cell lysates revealed that csHsp90 inhibition suppressed KSHV induction of p65 phosphorylation (Figure 1C, D). To confirm these results and to assess the role of csHsp90 in KSHV-induced activation of NF-κB binding to gene promoters, we employed NF-κB reporter assays in which binding of endogenous NF-κB to an ectopically expressed promoter induces luciferase expression. These assays showed that csHsp90 inhibition significantly suppressed KSHV-induced activation NF-κB (Figure 1E-H). The degree of NF-κB inhibition observed with csHsp90 targeting in our immunoblotting and reporter assays approximated that observed with direct NF-κB inhibition using a well-characterized NF-κB inhibitor (Figure 1C, E-H).
Figure 1.
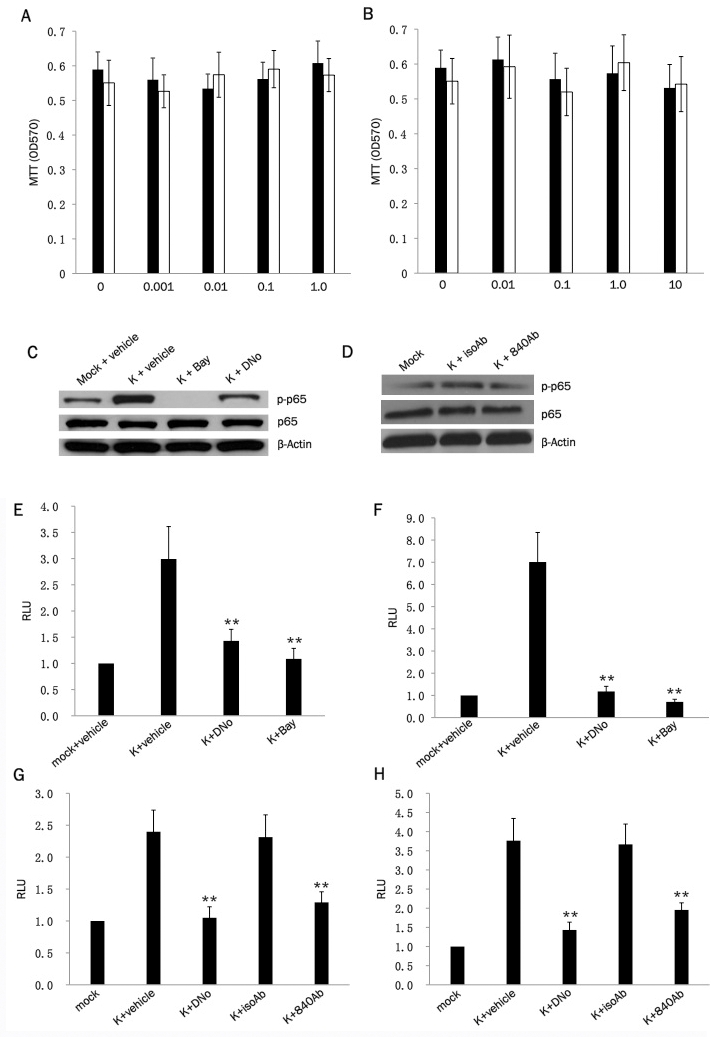
KSHV activation of NF-κB is cs-Hsp90-dependent. (A, B) pDMVEC (black bars) and HFF (white bars) were incubated with the indicated concentrations of DNo (μM) for 16 h (A) or Bay11-7082 (Bay) for 1 h (B), and cell viability was determined thereafter using a standard MTT assay according to the manufacturer's instructions. (C, D) Following their incubation with KSHV for 2 h, pDMVEC (C) were incubated with DMSO (vehicle control), DNo or Bay as above. HFF (D) were incubated with 15mg/mL monoclonal anti-Hsp90 antibody (840Ab) or 15mg/mL antibody control (isoAb) for 16 h. Immunoblots were performed to quantify total or phosphorylated signaling intermediates and β-actin as an internal loading control. (E, F) pDMVEC (E) and HFF (F) were transiently transfected with a luciferase-based NF-κB reporter construct and, 48 h later, incubated with KSHV for 2 h followed by either DMSO, 1 μM DNO for 16 h, or 10μM Bay for 1 h prior to assessment of NF-κB activity (RLU) as described in Methods. (G, H) pDMVEC (G) and HFF (H) were transiently transfected as in (E) and (F) and incubated with KSHV for 2 h prior to their incubation with either DMSO, 1 μM DNO, 15mg/mL monoclonal anti-Hsp90 antibody (840Ab) or 15mg/mL antibody control (isoAb) for 16 h prior to assessment of NF-κB activity (RLU) as described in Methods. Error bars represent the S.E.M. for three independent experiments. * = p<0.05, ** = p<0.01.
TNF-α initiation of canonical NF-κB activation is well-characterized [45,46] and may play an important role in facilitating KS pathogenesis, possibly through paracrine upregulation of NF-κB following interactions between TNF-α and cell surface receptors [47-51]. HeLa cells have been used previously to investigate the role of canonical NF-κB activation in KSHV pathogenesis [52,53]. Therefore, we used HeLa cells to determine whether csHsp90 regulates TNF-α and/or KSHV induction of canonical NF-κB activation. TNF-α induced phosphorylation of p65 and IKKα/β and the degradation of IκBα (consistent with canonical NF-κB activation) in HeLa cells as previously demonstrated, and csHsp90 inhibition suppressed these effects to a degree similar to that achieved with direct NF-κB inhibition (Figure 2A). Nuclear translocation of p65 is a key step in canonical NF-κB-mediated gene transactivation [28], and we confirmed that csHsp90 inhibition significantly reduced TNF-α-mediated nuclear translocation of p65 (Figure 2B). In subsequent de novo infection experiments, we observed that KSHV increased phosphorylation of p65 and IKKα/β as well as nuclear translocation of p65 as anticipated, and that csHsp90 inhibition suppressed these effects to an extent similar to that observed with direct NF-κB inhibition (Figure 2C, D).
Figure 2.
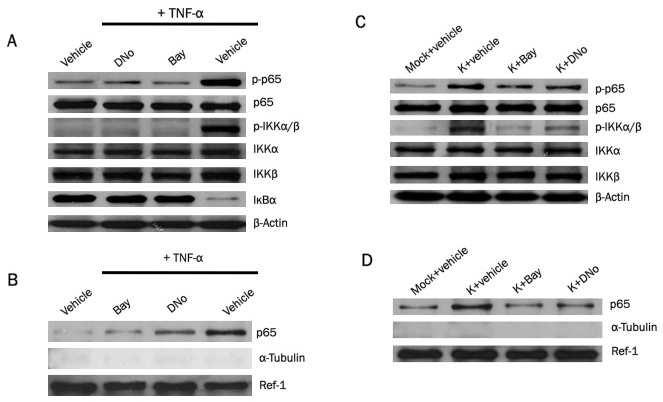
csHsp90 mediates KSHV induction of canonical NF-κB activation. (A) HeLa cells were incubated with DMSO (vehicle control), 1.0 μM DNO for 16 h, or 10 μM Bay for 1 h followed by 20 ng/mL TNF-α for 20 minutes. Immunoblots were performed 12 h later to quantify total or phosphorylated signaling intermediates or β-Actin as an internal control. (B) HeLa cells were incubated with DNo, Bay and TNF-α as in (A), then nuclear fractions isolated as described in Methods. Immunoblots were performed to detect signaling molecules, Ref-1 as a positive internal control for nuclear protein expression/loading, and α-tubulin to exclude the possibility of contamination of nuclear fractions with extranuclear proteins. (C) Following their incubation with KSHV for 2 h, HeLa cells were incubated with DNo or Bay as in (A), then immunoblots performed 12 h after viral incubation to detect expression of signaling molecules as above. (D) Following their incubation with KSHV, HeLa cells were incubated with DNo or Bay as in (A), then nuclear fractions isolated after 12 h and immunoblots performed as above.
csHsp90 regulates KSHV-initiated/NF-κB-dependent secretion of pro-migratory factors and cell invasiveness
Previous studies have demonstrated that NF-κB initiates expression and secretion of soluble mediators of tumor cell invasion and angiogenesis, including VEGF and IL-8, within a variety of cancer cells [54-56]. Neovascular proliferation and proliferation of KSHV-infected cells are cardinal pathologic features of KS, and VEGF and IL-8 are upregulated by KSHV-induced NF-κB activation during de novo infection of endothelial cells [57-60]. In addition, KSHV induces cell migration and invasion following de novo infection [61,62]. We found that targeting csHsp90 significantly reduced KSHV-initiated secretion of VEGF and IL-8 during infection of pDMVEC and HFF, approximating the effect seen with direct inhibition of NF-κB (Figure 3A-F). Next, we used transwell invasion assays to determine whether csHsp90 facilitates KSHV-mediated invasiveness for human umbilical vein endothelial cells (HUVEC) and HFF. We used HUVEC rather than pDMVEC since we find that pDMVEC do not efficiently pass through the matrigels used in our transwell assays. Of note, we observed no apparent toxicity for HUVEC incubated with DNo over the range of concentrations and exposure times in our assays (Figure 4A). We found that csHsp90 inhibition significantly suppressed KSHV-induced invasiveness for both HUVEC and HFF (Fig. 4B-I). In subsequent experiments, we transfected HUVEC with a construct encoding p65 (Figure 5A) and found that NF-κB overexpression restored secretion of VEGF and IL-8, as well as invasiveness, for KSHV-infected cells in the presence of csHsp90 inhibition (Figure 5B-H), implicating csHsp90 regulation of NF-κB as a mechanism for KSHV induction of cell migration and invasion following de novo infection.
Figure 3.
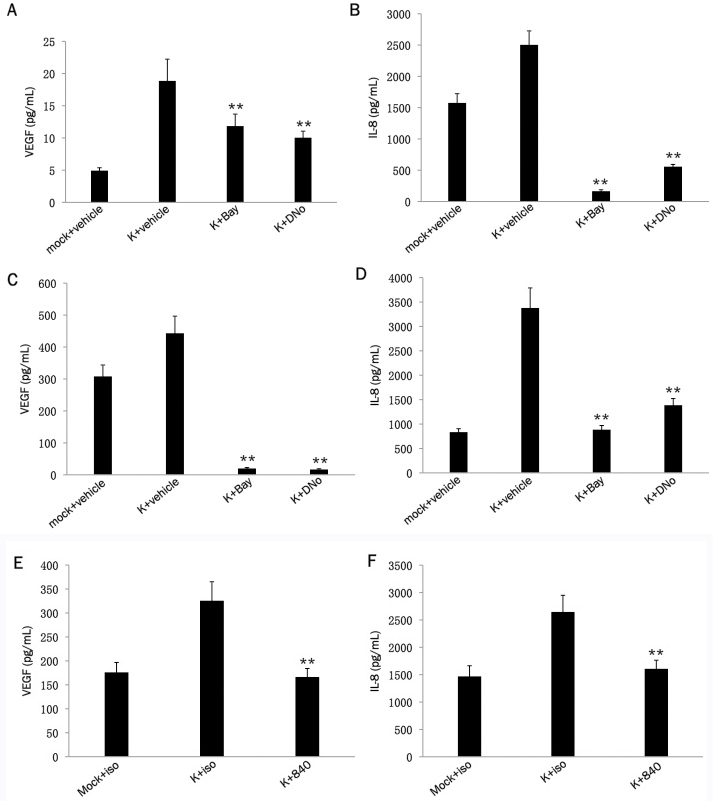
KSHV-induced secretion of pro-migratory factors is csHsp90-dependent. Following their incubation with either media (mock) or KSHV (K) for 2 h, pDMVEC (A, B) or HFF (C, D) were incubated with DMSO (vehicle control), 10 μM Bay for 1 h, or 1.0 μM DNO for 16 h. In separate experiments, KSHV-infected pDMVEC (E) and HFF (F) were incubated with 15mg/mL of a monoclonal anti-Hsp90 antibody (840) or 15mg/mL of an isotype control antibody (iso). VEGF and IL-8 were quantified within culture supernatants 20 h later using a commercial ELISA kit as described in Methods. Error bars represent the S.E.M. for three independent experiments. ** = p<0.01 relative to K + vehicle (A-D) or K + iso (E-F).
Figure 4.
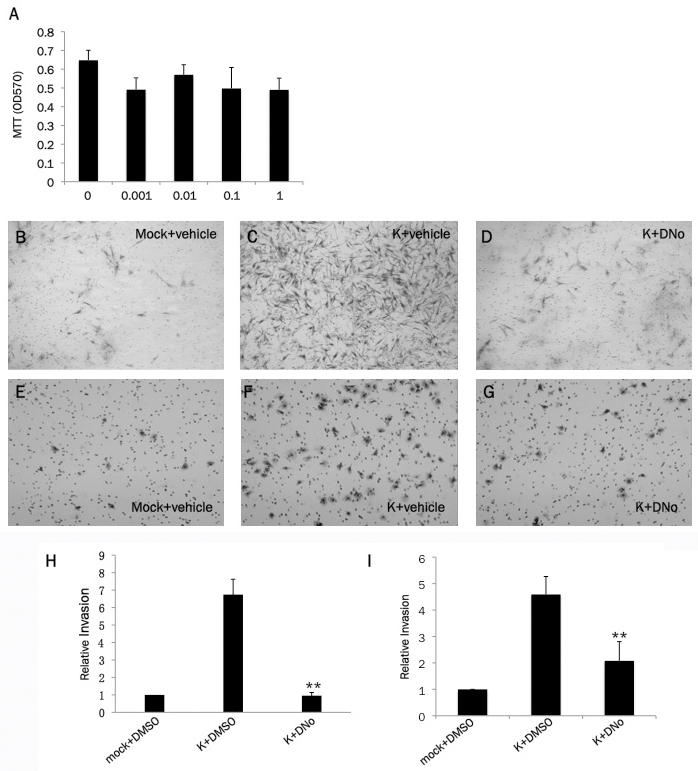
KSHV-induced invasiveness is csHsp90-dependent. (A) HUVEC were incubated with the indicated concentrations of DNo (in μM) for 16 h and cell viability determined using a standard MTT assay according to the manufacturer's instructions. (B-G) Following a 2 h incubation with either media (mock) or KSHV (K), HFF (B-D) or HUVEC (E-G) were incubated with DMSO (vehicle control) or 1.0 μM DNO prior to assessment of cell invasiveness using matrigel invasion assays as described in Methods. Representative images from one of three independent experiments are shown. (H, I) Relative invasiveness for groups B-G was determined as described in Methods. Error bars represent the S.E.M. for three independent experiments. ** = p<0.01.
Figure 5.
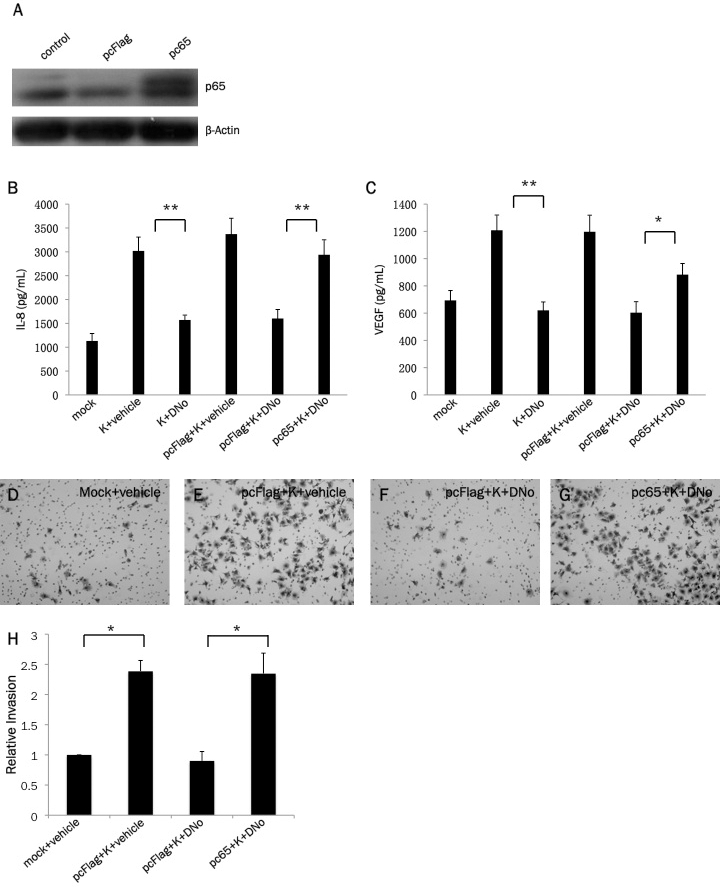
csHsp90 regulation of KSHV-initiated pathogenesis is mediated through csHsp90 regulation of NF-κB. (A) HUVEC were transfected with a construct encoding p65 (pc65) or empty control vector (pcFLAG), and 24 h later im-munoblots were used to identify expression of p65 and β-Actin for loading controls as described in Methods. (B, C) HUVEC were incubated with media (mock) or KSHV (K) for 2 h in the presence or absence of p65 (or control vector) transfection as in (A), then incubated with either DMSO (vehicle control) or 1.0 μM DNO for 16 h. IL-8 and VEGF were then quantified within culture supernatants 20 h later using a commercial ELISA kit as described in Methods. (D-G) HUVEC were treated as above, and matrigel invasion assays used to assess invasiveness as described in Methods. Representative images from one of three independent experiments are shown. (H) Relative invasiveness for groups D-G was quantified as described in Methods. Error bars represent the S.E.M. for three independent experiments. * = p<0.05, ** = p<0.01.
Discussion
We recently demonstrated that csHsp90 serves as a co-factor for KSHV-induced MAPK activation and viral gene expression [43]. In that study, we found that MAPK overexpression only partially restored KSHV gene expression in the presence of csHsp90 inhibition, suggesting that csHsp90 regulates multiple signal transduction pathways activated during KSHV infection. NF-κB transcriptional activation plays a key role in facilitating KSHV gene expression as well as cell proliferation and survival of transformed cells, and several KSHV-encoded proteins initiate canonical activation of NF-κB [30-32, 36, 44, 53, 60, 63]. Existing data support a role for intracellular Hsp90 in the regulation of NF-κB activation and herpesvirus gene expression [64] as well as NF-κB-mediated survival for KSHV-infected lymphoma cells [65]. Other work has demonstrated that intracellular Hsp90 regulates TNF-associated canonical activation of NF-κB [66], and that a cell permeable Hsp90 inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG), suppresses IκB kinase-dependent IκBα phosphorylation/degradation, NF-κB activation, and cancer cell invasiveness [67]. Although exhibiting promise for reducing solid tumor burden in phase II clinical trials, cell-permeable Hsp90 inhibitors incur toxicities due to off-target effects that may ultimately limit their efficacy [40]. csHsp90 regulates a more limited array of signal transduction pathways relative to the intra-cellular protein [42], justifying additional studies to explore the role of csHsp90 in viral cancer pathogenesis. To our knowledge, there are no prior studies addressing whether csHsp90 regulates virus-associated NF-κB activation and related cancer pathogenesis. Therefore, we sought to extend our prior observations regarding the role of csHsp90 in KSHV-initiated signal transduction, and to identify related functional effects relevant to KS cellular pathogenesis, through determination of whether csHsp90 serves as a co-factor for KSHV activation of NF-κB and KSHV-initiated cell invasion.
We found that targeting csHsp90 reduces both TNF-α and KSHV-induced canonical activation of NF-κB. TNF-α is a well-characterized inducer of canonical NF-κB activation [45,46, 66] and may play an important role in facilitating KS pathogenesis, in part through TNFR-mediated canonical activation of NF-κB [47-51]. Therefore, it is interesting to speculate whether targeting csHsp90 inhibits KS pathogenesis indirectly through a reduction in soluble TNF-α-receptor interactions, or other undetermined mechanisms. Furthermore, since NF-κB regulates KSHV gene expression during de novo infection [31], and since csHsp90 inhibition suppresses MAPK-dependent and MAPK-independent KSHV gene expression [43], we hypothesize that targeting csHsp90 reduces NF-κB-mediated viral gene expression following de novo infection, although this is not addressed in the present studies. Furthermore, our studies do not address whether direct interactions between KSHV and csHsp90 initiate NF-κB activation. This possibility is supported by published data revealing that direct interactions between infectious pathogens and csHsp90 activate NF-κB [68].
NF-κB transactivation of soluble mediators of angiogenesis facilitates pathogenesis related to a wide variety of cancers [69, 70]. Existing data also suggest that NF-κB-dependent induction of angiogenic cytokines and chemokines by KSHV facilitates KS progression [54,57,58,60]. Inhibition of csHsp90 using DNo reduces cell motility in vitro and cancer metastasis in vivo [41,71,72]. Our experiments including primary human endothelial cells and fibroblasts (two cell types present within KS lesions) revealed that targeting csHsp90 effectively suppresses KSHV-induced secretion of VEGF and IL-8, two well-characterized mediators of cell migration and angiogenesis induced following KSHV activation of NF-κB [55, 56]. These results complimented our functional assays wherein targeting csHsp90 effectively suppressed KSHV-induced migration and invasiveness following de novo infection. Furthermore, invasiveness was restored with ectopic p65 expression, suggesting that suppression of invasion during csHsp90 inhibition is mediated by NF-κB and its downstream effectors. These results are in agreement with published data supporting a role for extracellular Hsp90 in the induction of NF-κB-mediated transactivation of IL-8 [73]. While VEGF and IL-8 likely participate in csHsp90-mediated migration for KSHV-infected cells, additional pro-angiogenic factors not examined in our studies are also secreted following KSHV infection, including matrix metalloproteinases [62], and we cannot exclude the possibility that targeting csHsp90 suppresses secretion of these factors as well. In addition, our studies do not address whether interactions between cell surface proteins and csHsp90 in KSHV-infected cells induce cell migration and invasion through NF-κB-independent mechanisms. For example, we recently demonstrated that KSHV induces cell invasiveness through upregulation of the membrane-associated protein emmprin (Extracellular Matrix MetalloPRoteinase IN-ducer) [61], and emmprin associates with Hsp90 and other proteins within lipid rafts at the cell surface [74]. Future studies are needed to explore the potential relationship between csHsp90, surface-localized proteins, and NF-κB activation.
The data presented in this study provide novel observations implicating csHsp90 as a co-factor in KSHV-mediated cell migration and invasion through its regulation of NF-κB activation. These studies provide rationale for determining more precisely the mechanistic role of csHsp90 in KSHV-host cell interactions and whether drugs targeting extracellular Hsp90 inhibit KSHV-mediated angiogenesis and KS progression in vivo.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01-CA142362 to C.H.P. and F30-DE020203 to M.D.), and the MUSC Hollings Cancer Center (P30-CA138313). We would also like to thank Dr. Scott Eblen (Medical University of South Carolina) for providing MEK and ERK expression vectors, Dr. Yusuf Hannun (Medical University of South Carolina) for providing NF-κB luciferase reporter constructs, Dr. Ren Sun (University of California, Los Angeles) for providing NF-κB overexpression vectors, Dr. Darwin Bell (Medical University of South Carolina) for providing HUVEC, and Dr. Charles Smith (Medical University of South Carolina) for providing DNo.
References
- 1.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–91. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 2.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276–80. [PubMed] [Google Scholar]
- 3.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 4.Vanni T, Sprinz E, Machado MW, Santana Rde C, Fonseca BA, Schwartsmann G. Systemic treatment of AIDS-related Kaposi sarcoma: current status and perspectives. Cancer Treat Rev. 2006;32:445–55. doi: 10.1016/j.ctrv.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet F, Lewden C, May T, Heripret L, Jougla E, Bevilacqua S, Costagliola D, Salmon D, Chene G, Morlat P. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101:317–24. doi: 10.1002/cncr.20354. [DOI] [PubMed] [Google Scholar]
- 6.Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, Grigg R, Hylton T, Pawlish KS, McNeel TS, Goedert JJ. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123:187–94. doi: 10.1002/ijc.23487. [DOI] [PubMed] [Google Scholar]
- 7.Mani D, Neil N, Israel R, Aboulafia DM. A restrospective analysis of AIDS-associated Kaposi's sarcoma in patients with undetectable HIV viral loads and CD4 counts greater than 300 cells/mm(3) J Int Assoc Physicians AIDS Care (Chic) 2009;8:279–85. doi: 10.1177/1545109709341852. [DOI] [PubMed] [Google Scholar]
- 8.Acharya S, Ross JD. Kaposi's sarcoma of the recto sigmoid colon in a patient with HIV infection and high CD4 count. Int J STD AIDS. 2007;18:499–500. doi: 10.1258/095646207781147184. [DOI] [PubMed] [Google Scholar]
- 9.Krown SE, Lee JY, Dittmer DP. More on HIV-associated Kaposi's sarcoma. N Engl J Med. 2008;358:535–36. doi: 10.1056/NEJMc072994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurer T, Ponte M, Leslie K. HIV-associated Kaposi's sarcoma with a high CD4 count and low viral load. N Engl J Med. 2007;357:1352–53. doi: 10.1056/NEJMc070508. [DOI] [PubMed] [Google Scholar]
- 11.Lebbe C, Legendre C, Frances C. Kaposi sarcoma in transplantation. Transplant Rev (Orlando) 2008;22:252–61. doi: 10.1016/j.trre.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Sullivan RJ, Pantanowitz L, Dezube BJ. Targeted therapy for Kaposi sarcoma. BioDrugs. 2009;23:69–75. doi: 10.2165/00063030-200923020-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen HQ, Magaret AS, Kitahata MM, Von Rompaey SE, Wald A, Casper C. Persistent Kaposi's sarcoma in the era of highly active antiretroviral therapy: characterizing the predictors of clinical response. AIDS. 2008;22:937–45. doi: 10.1097/QAD.0b013e3282ff6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Von Roenn JH. Clinical presentations and standard therapy of AIDS-associated Kaposi's sarcoma. Hematol Oncol Clin North Am. 2003;17:747–62. doi: 10.1016/s0889-8588(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 15.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. ERK ½ and MEK ½ induced by Kaposi's Sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J Virol. 2005;79:10308–29. doi: 10.1128/JVI.79.16.10308-10329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinkmann MM, Glen M, Rainbow L, Kieser A, Henke-Gendo C, Schulz TF. Activation of mito-gen-activated protein kinase and NF-kappaB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membran protein. J Virol. 77:9346–58. doi: 10.1128/JVI.77.17.9346-9358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez CM, Wong EL, Bowser BS, Hong GK, Kenney S, Damania B. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J Virol. 2006;80:3062–70. doi: 10.1128/JVI.80.6.3062-3070.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, Gutkind JS. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen activated protein kinase and p38 pathways acting on hypoxia inducible factor 1alpha. Cancer Res. 2000;60:4873–80. [PubMed] [Google Scholar]
- 19.Vart RJ, Nikitenko LL, Lagos D, Trotter MW, Cannon M, Bourboulia D, Gratrix F, Takeuchi Y, Boshoff C. Kaposi's sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin -2 expression in lymphatic endothelial cells. Cancer Res. 2007;67:4042–51. doi: 10.1158/0008-5472.CAN-06-3321. [DOI] [PubMed] [Google Scholar]
- 20.Pan H, Xie J, Ye F, Gao SJ. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J Virol. 2006;80:5371–82. doi: 10.1128/JVI.02299-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie J, Ajibade AO, Ye K, Kuhne K, Gao SJ. Reactivation of Kaposi's sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology. 2008;371:139–54. doi: 10.1016/j.virol.2007.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie J, Pan H, Yoo S, Gao SJ. Kaposi's sarcomaassociated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J Virol. 2005;79:15027–37. doi: 10.1128/JVI.79.24.15027-15037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, Ranieri E, Gesualdo L, Schena FP, Grandaliano G. Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med. 2005;352:1317–23. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]
- 24.Koon HB, Bubley GJ, Pantanowitz L, Masiello D, Smith B, Crosby K, Proper J, Weeden W, Miller TE, Chatis P, Egorin MJ, Tahan SR, Dezube BJ. Imatinib-induced regression of AIDSrelated Kaposi's sarcoma. J Clin Oncol. 2005;23:982–9. doi: 10.1200/JCO.2005.06.079. [DOI] [PubMed] [Google Scholar]
- 25.Dezube BJ, Sullivan R, Koon HB. Emerging targets and novel strategies in the treatment of AIDS-related Kaposi's sarcoma: bidirectional translational science. J Cell Physiol. 2006;209:659–62. doi: 10.1002/jcp.20795. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 27.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 28.Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-kappaB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17:281–93. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 30.Emuss V, Lagos D, Pizzey A, Gratrix F, Henderson SR, Boshoff C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009;5:e1000616. doi: 10.1371/journal.ppat.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sadagopan S, Sharma-Walia N, Veettil MV, Raghu H, Sivakumar R, Bottero V, Chandran B. Kaposi's sarcoma-associated herpesvirus induces sustained NF-kappaB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J Virol. 2007;81:3949–68. doi: 10.1128/JVI.02333-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin D, Galisteo R, Ji Y, Montaner S, Gutkind JS. An NF-kappaB gene expression signature contributes to Kaposi's sarcoma virus vGPCR-induced direct and paracrine neoplasia. Oncogene. 2008;27:1844–52. doi: 10.1038/sj.onc.1210817. [DOI] [PubMed] [Google Scholar]
- 33.Montaner S, Sodhi A, Pece S, Mesri EA, Gutkind JS. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001;61:2641–8. [PubMed] [Google Scholar]
- 34.Schwarz M, Murphy PM. Kaposi's sarcomaassociated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J Immunol. 2001;167:505–13. doi: 10.4049/jimmunol.167.1.505. [DOI] [PubMed] [Google Scholar]
- 35.Matta H, Mazzacurati L, Schamus S, Yang T, Sun Q, Chaudhary PM. Kaposi's sarcomaassociated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IkappaB kinase complex to selectively activate NF-kappaB without JNK activation. J Biol Chem. 2007;282:24858–65. doi: 10.1074/jbc.M700118200. [DOI] [PubMed] [Google Scholar]
- 36.Pati S, Foulke JS, Jr, Barabitskaya O, Kim J, Nair BC, Hone D, Smart J, Feldman RA, Reitz M. Human herpesvirus 8-encoded vGPCR activates nuclear factor of activated T cells and collaborates with human immunodeficiency virus type 1 Tat. J Virol. 2003;77:5759–73. doi: 10.1128/JVI.77.10.5759-5773.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin Y, Bai L, Chen W, Xu S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets. 2010;14:45–55. doi: 10.1517/14728220903431069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsutsumi S, Neckers L. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 2007;98:1536–9. doi: 10.1111/j.1349-7006.2007.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bohonowych JE, Gopal U, Isaacs JS. Hsp90 as a gatekeeper of tumor angiogenesis: clinical promise and potential pitfalls. J Oncol. 2010;2010:412985. doi: 10.1155/2010/412985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramalingam SS, Egorin MJ, Ramanathan RK, Remick SC, Sikorski RP, Lagattuta TF, Chatta GS, Friedland DM, Stoller RG, Potter DM, Ivy SP, Belani CP. A phase I study of 17-allylamino-17-demethoxygeldanamycin combined with paclitaxel in patients with advanced solid malignancies. Clin Cancer Res. 2008;14:3456–61. doi: 10.1158/1078-0432.CCR-07-5088. [DOI] [PubMed] [Google Scholar]
- 41.Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, Lain B, Torella C, Henning SW, Beste G, Scroggins BT, Neckers L, Ilag LL, Jay DG. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. 2004;6:507–14. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- 42.Tsutsumi S, Scroggins B, Koga F, Lee MJ, Trepel J, Felts S, Carreras C, Neckers L. A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene. 2008;27:2478–87. doi: 10.1038/sj.onc.1210897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin Z, DeFee M, Isaacs JS, Parsons C. Extracellular Hsp90 serves as a co-factor for MAPK activation and latent viral gene expression during de novo infection by KSHV. Virology. 2010;403:92–102. doi: 10.1016/j.virol.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-kappaB induces apoptosis of KSHVinfected primary effusion lymphoma cells. Blood. 2000;96:2537–42. [PubMed] [Google Scholar]
- 45.Bista P, Zeng W, Ryan S, Bailly V, Browning JL, Lukashev ME. TRAF3 controls activation of the canonical and alternative NF-kappaB by the lymphotoxin beta receptor. J Biol Chem. 2010;285:12971–8. doi: 10.1074/jbc.M109.076091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnston DA, Dong B, Hughes CC. TNF induction of jagged-1 in endothelial cells is NFkappaB- dependent. Gene. 2009;435:36–44. doi: 10.1016/j.gene.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barillari G, Franzese O, Comandini A, Bonmassar E, Ensoli B. Spindle cells from AIDSassociated Kaposi's sarcoma lesions express telomerase activity that is enhanced by Kaposi's sarcoma progression factors. Oncol Rep. 2010;24:219–23. doi: 10.3892/or_00000849. [DOI] [PubMed] [Google Scholar]
- 48.Monini P, Colombini S, Sturzl M, Goletti D, Cafaro A, Sgadari C, Butto S, Franco M, Leone P, Fais S, Melucci-Vigo G, Chiozzini C, Carlini F, Ascherl G, Cornali E, Zietz C, Ramazzotti E, Ensoli F, Andreoni M, Pezzotti P, Rezza G, Yarchoan R, Gallo RC, Ensoli B. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi's sarcoma. Blood. 1999;93:4044–58. [PubMed] [Google Scholar]
- 49.Murakami-Mori K, Mori S, Bonavida B, Nakamura S. Implication of TNF receptor-Imediated extracellular signal-regulated kinases 1 and 2 (ERK1/2) activation in growth of AIDS-associated Kaposi's sarcoma cells: a possible role of a novel death domain protein MADD in TNF-alpha-induced ERK1/2 activation in Kaposi's sarcoma cells. J Immunol. 1999;162:3672–9. [PubMed] [Google Scholar]
- 50.Samaniego F, Markham PD, Gendelman R, Gallo RC, Ensoli B. Inflammatory cytokines induce endothelial cells to produce and release basic fibroblast growth factor and to promote Kaposi's sarcoma-like lesions in nude mice. J Immunol. 1997;158:1887–94. [PubMed] [Google Scholar]
- 51.Samaniego F, Markham PD, Gendelman R, Watanabe Y, Kao V, Kowalski K, Sonnabend JA, Pintus A, Gallo RC, Ensoli B. Vascular endothelial growth factor and basic fibroblast growth factor present in Kaposi's sarcoma (KS) are induced by inflammatory cytokines and synergize to promote vascular permeability and KS lesion development. Am J Pathol. 1998;152:1433–43. [PMC free article] [PubMed] [Google Scholar]
- 52.Shepard LW, Yang M, Xie P, Browning DD, Voyno-Yasenetskaya T, Kozasa T, Ye RD. Constitutive activation of NF-kappa B and secretion of interleukin-8 induced by the G proteincoupled receptor of Kaposi's sarcomaassociated herpesvirus involve G alpha(13) and RhoA. J Biol Chem. 2001;276:45979–87. doi: 10.1074/jbc.M104783200. [DOI] [PubMed] [Google Scholar]
- 53.Seo T, Park J, Lim C, Choe J. Inhibition of nuclear factor kappaB activity by viral interferon regulatory factor 3 of Kaposi's sarcomaassociated herpesvirus. Oncogene. 2004;23:6146–55. doi: 10.1038/sj.onc.1207807. [DOI] [PubMed] [Google Scholar]
- 54.Martin D, Gutkind JS. Kaposi's sarcoma virally encoded, G-protein-coupled receptor: a paradigm for paracrine transformation. Methods Enzymol. 2009;460:125–50. doi: 10.1016/S0076-6879(09)05206-9. [DOI] [PubMed] [Google Scholar]
- 55.Golovine K, Uzzo RG, Makhov P, Crispen PL, Kunkle D, Kolenko VM. Depletion of intracellular zinc increases expression of tumorigenic cytokines VEGF, IL-6 and IL-8 in prostate cancer cells via NF-kappaB-dependent pathway. Prostate. 2008;68:1443–9. doi: 10.1002/pros.20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allen CT, Ricker JL, Chen Z, Van Waes C. Role of activated nuclear factor-kappaB in the pathogenesis and therapy of squamous cell carcinoma of the head and neck. Head Neck. 2007;29:959–71. doi: 10.1002/hed.20615. [DOI] [PubMed] [Google Scholar]
- 57.Bais C, Van Geelen A, Eroles P, Mutlu A, Chiozzini C, Dias S, Silverstein RL, Rafii S, Mesri EA. Kaposi's sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR. Cancer Cell. 2003;3:131–43. doi: 10.1016/s1535-6108(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 58.Shin YC, Joo CH, Gack MU, Lee HR, Jung JU. Kaposi's sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxiainducible factor-1 alpha to induce vascular endothelial growth factor expression. Cancer Res. 2008;68:1751–9. doi: 10.1158/0008-5472.CAN-07-2766. [DOI] [PubMed] [Google Scholar]
- 59.Lane BR, Liu J, Bock PJ, Schols D, Coffey MJ, Strieter RM, Polverini PJ, Markovitz DM. Interleukin-8 and growth-regulated oncogene alpha mediate angiogenesis in Kaposi's sarcoma. J Virol. 2002;76:11570–83. doi: 10.1128/JVI.76.22.11570-11583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun Q, Matta H, Lu G, Chaudhary PM. Induction of IL-8 expression by human herpesvirus 8 encoded vFLIP K13 via NF-kappaB activation. Oncogene. 2006;25:2717–26. doi: 10.1038/sj.onc.1209298. [DOI] [PubMed] [Google Scholar]
- 61.Qin Z, Dai L, Slomiany MG, Toole BP, Parsons C. Direct activation of emmprin and associated pathogenesis by an oncogenic herpesvirus. Cancer Res. 2010;70:3884–9. doi: 10.1158/0008-5472.CAN-09-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qian LW, Xie J, Ye F, Gao SJ. Kaposi's sarcoma -associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J Virol. 2007;81:7001–10. doi: 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Konrad A, Wies E, Thurau M, Marquardt G, Naschberger E, Hentschel S, Jochmann R, Schulz TF, Erfle H, Brors B, Lausen B, Neipel F, Sturzl M. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-kappaB activation. J Virol. 2009;83:2563–74. doi: 10.1128/JVI.01512-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Basha W, Kitagawa R, Uhara M, Imazu H, Uechi K, Tanaka J. Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir Chem Chemother. 2005;16:135–46. doi: 10.1177/095632020501600206. [DOI] [PubMed] [Google Scholar]
- 65.Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. KSHV vFLIP binds to IKKgamma to activate IKK. J Cell Sci. 2003;116:3721–8. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- 66.Lewis J, Devin A, Miller A, Lin Y, Rodriguez Y, Neckers L, Liu ZG. Disruption of hsp90 function results in degradation of the death domain kinase, receptor-interacting protein (RIP), and blockage of tumor necrosis factorinduced nuclear factor-kappaB activation. J Biol Chem. 2000;275:10519–26. doi: 10.1074/jbc.275.14.10519. [DOI] [PubMed] [Google Scholar]
- 67.Kim MS, Kwak HJ, Lee JW, Kim HJ, Park MJ, Park JB, Choi KH, Yoo H, Shin SH, Shin WS, Song ES, Lee SH. 17-Allylamino-17- demethoxygeldanamycin down-regulates hyaluronic acid-induced glioma invasion by blocking matrix metalloproteinase-9 secretion. Mol Cancer Res. 2008;6:1657–65. doi: 10.1158/1541-7786.MCR-08-0034. [DOI] [PubMed] [Google Scholar]
- 68.Jin S, Song YC, Emili A, Sherman PM, Chan VL. JlpA of Campylobacter jejuni interacts with surface-exposed heat shock protein 90alpha and triggers signalling pathways leading to the activation of NF-kappaB and p38 MAP kinase in epithelial cells. Cell Microbiol. 2003;5:165–74. doi: 10.1046/j.1462-5822.2003.00265.x. [DOI] [PubMed] [Google Scholar]
- 69.Brown M, Cohen J, Arun P, Chen Z, Van Waes C. NF-kappaB in carcinoma therapy and prevention. Expert Opin Ther Targets. 2008;12:1109–22. doi: 10.1517/14728222.12.9.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li ZW, Chen H, Campbell RA, Bonavida B, Berenson JR. NF-kappaB in the pathogenesis and treatment of multiple myeloma. Curr Opin Hematol. 2008;15:391–9. doi: 10.1097/MOH.0b013e328302c7f4. [DOI] [PubMed] [Google Scholar]
- 71.Becker B, Multhoff G, Farkas B, Wild PJ, Landthaler M, Stolz W, Vogt T. Induction of Hsp90 protein expression in malignant melanomas and melanoma metastases. Exp Dermatol. 2004;13:27–32. doi: 10.1111/j.0906-6705.2004.00114.x. [DOI] [PubMed] [Google Scholar]
- 72.Li W, Li Y, Guan S, Fan J, Cheng CF, Bright AM, Chinn C, Chen M, Woodley DT. Extracellular heat shock protein-90alpha: linking hypoxia to skin cell motility and wound healing. EMBO J. 2007;26:1221–33. doi: 10.1038/sj.emboj.7601579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chung SW, Lee JH, Choi KH, Park YC, Eo SK, Rhim BY, Kim K. Extracellular heat shock protein 90 induces interleukin-8 in vascular smooth muscle cells. Biochem Biophys Res Commun. 2009;378:444–9. doi: 10.1016/j.bbrc.2008.11.063. [DOI] [PubMed] [Google Scholar]
- 74.Ghatak S, Misra S, Toole BP. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J Biol Chem. 2005;280:8875–83. doi: 10.1074/jbc.M410882200. [DOI] [PubMed] [Google Scholar]