

Abstract
11β-hydroxysteroid dehydrogenase type 1 (11β-HSD-1) intracellularly regenerates active corticosterone from circulating inert 11-dehydrocorticosterone (11-DHC) in specific tissues. The hippocampus is a brain structure particularly vulnerable to glucocorticoid neurotoxicity with aging. In intact hippocampal cells in culture, 11β-HSD-1 acts as a functional 11β-reductase reactivating inert 11-DHC to corticosterone, thereby potentiating kainate neurotoxicity. We examined the functional significance of 11β-HSD-1 in the central nervous system by using knockout mice. Aged wild-type mice developed elevated plasma corticosterone levels that correlated with learning deficits in the watermaze. In contrast, despite elevated plasma corticosterone levels throughout life, this glucocorticoid-associated learning deficit was ameliorated in aged 11β-HSD-1 knockout mice, implicating lower intraneuronal corticosterone levels through lack of 11-DHC reactivation. Indeed, aged knockout mice showed significantly lower hippocampal tissue corticosterone levels than wild-type controls. These findings demonstrate that tissue corticosterone levels do not merely reflect plasma levels and appear to play a more important role in hippocampal functions than circulating blood levels. The data emphasize the crucial importance of local enzymes in determining intracellular glucocorticoid activity. Selective 11β-HSD-1 inhibitors may protect against hippocampal function decline with age.
Much evidence in rodents, nonhuman primates, and humans suggests that prolonged elevation of blood glucocorticoid levels impairs cognitive function, an effect that becomes more marked with aging (1–3). Chronic glucocorticoid excess has deleterious actions on brain neuronal biochemistry and electrophysiological function. This occurs particularly in the hippocampus, where glucocorticoid excess inhibits the formation of long-term potentiation (4), reduces dendritic structural complexity (5), and may eventually produce neuronal loss (6, 7). Manipulations that keep glucocorticoid levels low throughout life in rats [including adrenalectomy with low-dose glucocorticoid replacement, and neonatal handling (8, 9)] prevent the emergence of watermaze learning deficits with age. However, such approaches are not appropriate for human therapy.
Recently, it has been recognized that tissue glucocorticoid concentrations are determined not only by plasma hormone levels, but also by intracellular 11β-hydroxysteroid dehydrogenases (11β-HSDs), which locally interconvert active glucocorticoids (corticosterone in rats and mice, cortisol in humans) and inert 11-keto forms [11-dehydrocorticosterone (11-DHC), cortisone (10)]. The importance of such enzymatic interconversion is clearly demonstrated by the action of 11β-HSD type 2 (an exclusive dehydrogenase), which potently inactivates glucocorticoids thus allowing selective access for aldosterone to intrinsically nonselective mineralocorticoid receptors (MR) in the kidney (11, 12). The observation of 11β-HSD in neurons in the brain prompted suggestions that the enzyme might attenuate the deleterious effects of chronic glucocorticoid excess on neuronal function and survival (13). However, the predominant isoform in the brain, 11β-HSD type 1 (11β-HSD-1), which is located in lumen of the endoplasmic reticulum (14), appears to function as a predominant 11β-reductase (regenerating active glucocorticoids from inert 11-DHC) in intact cells in primary culture (15), including neurons (16), and in the liver in vivo (17). This reductase activity, far from protecting neurons against glucocorticoid excess, would be anticipated to increase intraneuronal glucocorticoid levels, potentiating neurotoxicity. Indeed, in vitro, otherwise inert 11-DHC potentiates kainate-induced neurotoxicity in hippocampal neurons in culture, an effect lost in the presence of an 11β-HSD inhibitor (16). If such regeneration of glucocorticoids within neurons is important in vivo, inhibition of 11β-reductase might attenuate neuronal dysfunction and the consequent watermaze task performance decline with aging. To test this hypothesis, we examined the effects of aging on hippocampal function (by using the Morris watermaze) in mice homozygous for targeted disruption of the 11β-HSD-1 gene (17).
Materials and Methods
Experimental Animals.
Male mice with a targeted disruption of the 11β-HSD-1 gene were inbred on the original 129/Ola strain background, as were wild-type controls (17). Young (4–7 months old) and aged (18–20 months old) mice were housed in individual cages on a 12-h light–dark cycle (light on 7 a.m. to 7 p.m.). Both genotypes had similar survival rates up to 23 months. All animals (n = 28) were tested between 9 a.m. and 5 p.m. Animal care was in strict accordance with Home Office and institutional guidelines.
Watermaze.
The watermaze was a circular tank (1.8 m diameter, 0.60 m height) filled to a depth of 35 cm with water (25 ± 1°C) made opaque with latex liquid. The paths taken by the mice were monitored with an overhead video camera connected to an image analyzer (HVS Image, Hampton, U.K.) and an Acorn Archimedes (Edinburgh) running software that sampled the coordinates on-line at 10 Hz for subsequent automated data analysis. For all training trials, the computer calculated the escape latency, percentage of time spent within 15 cm of the side walls of the pool, and the average swim speed of the mice. The day before training, all mice were given a 60-s swim and allowed to mount a 30-cm platform submerged just below the water surface.
Mice were tested in the proximally cued version of the maze where the platform position was indicated by a visible flag. This behavioral task requires the animal to associate the flag with escape from the water. The target platform was submerged just below the water surface and located under a flag (5 cm high × 8 cm wide) attached to the platform 10 cm above the water. Curtains were drawn around pool to exclude extramaze cues. Training consisted of four trials per day for 5 days, each trial lasting until the mouse reached the platform (escape latency) or swam for 60 s. After each trial the mice were allowed to remain on the platform for 30 s. Mice were released in the pool from one of four equally spaced start positions along the perimeter of the pool in a predetermined, pseudorandom order. On the first day of training, a larger platform (30 cm diameter) was used to increase the chance for the mice to find the platform. From the second day onward the diameter of the platform was 20 cm. The location of the submerged platform was varied randomly from trial to trial. Mice were returned to drying cages and kept warm under a heat lamp between trials (intertrial interval 10 min). The performance of all mice were videotaped in a single blind analysis. Mice that tended to float rather than swim were excluded.
Ibotenate Lesions of Hippocampus.
Adult male 129/Ola mice (25 g) were anaesthetized with Hypnorm/Hypnovel (0.8 ml/kg, i.p.) and placed in a stereotaxic frame. Hippocampal lesions were made by 20 discrete injections of 0.03 μl ibotenic acid (10 mg/ml; Sigma) at 0.06 μl/min; the needle was left in place for 2 min following injection to avoid spread of the toxin up the needle tract. Stereotaxic coordinates were those used by Jarrard (18), modified for mice. Sham operated controls underwent similar surgical procedures except that no drug was infused. Six weeks after surgery, mice were tested in the cued version of the watermaze task, as described above. Following behavioral testing, mice were anaesthetized and perfused intracardially with saline and 4% paraformaldehyde. The brains were stored in the perfusate for 1 week, then frozen, sectioned coronally (30 μm), and stained with cresyl violet to determine cell loss.
Corticosterone, 11-DHC, Corticosteroid Binding Globulin (CBG), and Glucose Measurements.
Blood samples were obtained by cardiac puncture immediately after killing mice by cervical dislocation in the morning. Plasma corticosterone levels were measured by RIA (19) modified for microtiter plate scintillation proximity assay. The intra- and interassay coefficients of variation were 9.4 and 9.2%, respectively. Plasma 11-DHC was measured by the above RIA, but using [3H]-11-DHC [synthesized by using kidney homogenates, NAD, and [3H]corticosterone (20)] and the antibody for 11-DHC. Plasma glucose levels were measured by using the infinity glucose reagent from Sigma.
For the measurement of CBG, endogenous steroids were removed by diluting plasma samples 1:100 in dextran-coated charcoal in PBS-gelatin (DCC-G) and incubation for 1 h at room temperature followed by centrifugation at 3,000 g for 15 min. Aliquots (100 μl) of the supernatant were then incubated in buffer (PBS-gelatin) containing a saturating 10-nM concentration of [3H]corticosterone for 1 h at room temperature. Nonspecific binding was defined in parallel incubations by the addition of 2,000-fold excess cold corticosterone. The samples were then incubated with DCC-G at 4°C for 10 min and centrifuged at 3,000 g for 15 min at 4°C to separate bound from free. Scintillation fluid was added to 900 μl of supernatant in mini vials and counted. Protein content was determined by the method of Bradford (21).
Distribution of Infused Corticosterone in Brain Tissues of Wild-Type and 11β-HSD-1 Knockout Mice.
Male mice aged 18 months were infused s.c. with [3H]corticosterone (6.56Ci/d, [1,2,6,7-3H]corticosterone, specific activity 80Ci/mmol, plc, Amersham Pharmacia; 1 Ci = 37 GBq) in saline:ethanol (9:1) via Alzet miniosmotic infusion pumps (model 2001, Alza, Palo Alto, CA). Groups of six wild-type and knockout mice were infused for 7 d and then killed in the morning. Brains were removed, dissected, and snap frozen. The content of [3H]corticosterone in each region was measured by solvent extraction with chromatographic separation of steroids. Briefly, tissues were homogenized in >9 volumes of ice-cold Tris⋅HCl buffer (pH 6.5). Homogenates were incubated for 16 h at 37°C with acetone:ethanol (9:1), extracts filtered and evaporated to dryness, reconstituted in buffer, washed with hexane, and reextracted with ethyl acetate. Samples were separated by TLC by using UV absorption of an internal standard to localize corticosterone and radioactivity measured.
Histopathologic Comparisons of Retinas from Aged and Young Mice.
At autopsy, the eyes were removed and stored in paraformaldehyde at 4°C. Following removal of the lens, the eyes were embedded in paraffin. Tissue blocks were sectioned (7 μm) and stained with Harris' hematoxylin and eosin. Slides were coded before histopathologic evaluation at 40× magnification by an ophthalmologist unaware of age or genotype.
Data Analysis.
Data were assessed by ANOVA followed by Scheffé post hoc tests. Regression analysis was by the Pearson correlation matrix method. Significance was set at P < 0.05. Values are means ± SEM.
Results
Performance in Cued Platform Watermaze Task.
During a 5-day training period, young (4–7 months old) wild-type mice showed a progressive reduction in the latency to escape onto a flagged hidden platform in the watermaze [mean of the four trials per day; F(5,24) = 5.3, P < 0.005; Fig. 1A]. Although on initial entry into the pool (trial 1, day 1) the escape latency did not differ between the young and aged (18–20 months old) mice [35.8 1 ± 5.2 s (all young); 34.0 ± 4.9 s (all old)], the reduction in latency with training did not occur in the aged wild-type controls as a group [F(7,32) = 1.6, P = 0.2]; their average escape latencies (trials 1–4) for each day of training were significantly longer than young wild-type mice (P < 0.05). Young 11β-HSD-1−/− mice performed as well as young wild-type controls in the proximally cued platform watermaze task (Fig. 1A). Strikingly, aged 11β-HSD-1−/− mice also learned the task well, showing decreased escape latencies to find the platform with increasing days of training [F(7,32) = 17, P < 0.001]. ANOVA of mean escape latencies for each day of training showed that the performance of the aged 11β-HSD-1−/− mice was not significantly different from the aged wild-type mice initially, but then improved rapidly to the level of young wild-type mice, showing a significant age X genotype effect [ day 1, F(1,24) = 0.67, P = 0.4; day 2, F(1,24) = 0.7, P = 0.4; day 3, F(1,24) = 0.3, P = 0.6; day 4, F(1,24) = 3.9, P = 0.06; day 5, F(1,24) = 8.8, P < 0.01].
Figure 1.

(A) Impaired learning of aged wild-type, but not 11β-HSD-1−/− mice in the watermaze. Mice were trained for 5 consecutive days (four trials per day) to find and escape onto a randomly located flagged hidden platform. Young, 4-month-old wild type (open circles), young 7-month-old transgenics (open triangle), aged 18–20-month-old wild type (closed circle), and aged 18–20-month-old transgenics (open square). Escape latency (mean ± SEM) are shown for six young mice per genotype and eight aged mice per genotype, all on an isogenic 129/Ola strain background. Each point represents the average four trials per day. *, P < 0.01 compared with both young wild-type and young 11β-HSD-1−/− mice and P < 0.05 compared with old 11β-HSD-1−/− mice. (B) Failure to learn the proximally cued watermaze task following hippocampal lesions in young 129/Ola mice. Hippocampal lesions were made stereotaxically by multiple injections of ibotenic acid. Mice were trained for 5 consecutive days (four trials per day) to find and escape onto a randomly located flagged hidden platform. Escape latency (mean ± SEM) are shown for five mice per group. (C) High-power photomicrographs (×40 objective) showing Cresyl violet-stained hippocampal neurons of a sham-operated mouse and a hippocampal lesioned mouse. Note the loss of cells following the ibotenate lesion.
The differences between aged wild-type and 11β-HSD-1 null mice in the cued platform watermaze task did not appear to be due to swim speeds or visual processing or thigmotaxis (i.e., staying near the side walls). Although the significantly longer escape latencies shown by the aged mice (both genotypes) at the end of the first day of training (P < 0.05) (Fig. 1A) may partly reflect a slower swim speed (P < 0.01) [wild type: 0.24 m/s (young), 0.17 m/s (old); 11β-HSD-1−/−: 0.22 m/s (young), 0.14 m/s (old)], the average swim speed did not vary significantly across the last four days of training for individual mice (P = 0.98). Therefore, changes of escape latency associated with consecutive training trials reflect learning of the task. Visual processing abilities can also affect learning, because a direct correlation between the severity of photoreceptor degeneration and performance of aged rats in the watermaze has been reported (22). However, histopathological examination of the eyes of young and aged mice from both genotypes showed normal retinal integrity (all had retinas with an outer nuclear layer composed of ≈10 rows of photoreceptor nuclei, a five-cell-layer-thick inner nuclear layer, and a single cell layer of ganglion cells; not shown). All of the mice showed a similar degree of thigmotaxis (percentage of time near the side walls of the pool). ANOVA of mean thigmotaxis from all of the trials follows [wild type: 25.7 ± 1.2% (young), 26.3 ± 2.8% (old); 11β-HSD-1−/−: 20.4 ± 1.7% (young), 23.1 ± 2.3% (old)] revealed no significant effect of age [F(1,24) = 0.48, P = 0.5], genotype [F(1,24) = 3.39, P = 0.08], or age X genotype interaction [F(1,24) = 0.2, P = 0.6].
129/Ola Wild-Type Mice Performance in the Watermaze Following Hippocampal Lesions.
To determine whether the proximally cued platform watermaze task is hippocampus-dependent in 129/Ola mice, we selectively lesioned the whole hippocampus by using ibotenate acid. The escape latency to find the platform on initial entry into the pool (trial 1, day 1) did not differ between the sham and hippocampal lesioned mice (50.1 ± 6.1 s and 49.0 ± 6.7 s, respectively, P = 0.9), but by the end of first day of testing (trial 4) and across the remaining days, the hippocampal lesioned mice showed significantly longer escape latencies than the sham controls (P < 0.001, Fig. 1B). In contrast with the sham controls, hippocampal lesioned mice were unable to learn the task, showing no decrease in escape latency across the four days of training (F4,20 = 0.38, P = 0.8; Fig. 1B). Cresyl violet staining of brain sections confirmed selective removal of hippocampal cells with no evidence of damage to extrahippocampal areas (Fig. 1C).
Plasma Corticosterone, 11-DHC, CBG, and Glucose Levels.
Basal (morning) plasma corticosterone levels were low in young wild-type controls, but became markedly elevated with age (P < 0.001; Table 1). Young 11β-HSD-1−/− mice had significantly higher basal plasma corticosterone levels than young wild-type controls (P < 0.001; Table 1), confirming previous data (17). There was no further rise of plasma corticosterone in 11β-HSD-1−/− mice with age, so both genotypes had similarly elevated corticosterone levels when aged (Table 1). In wild-type controls, plasma corticosterone levels correlated directly with the escape latency to find the flagged hidden platform (Table 2 and Fig. 2). There was no correlation between plasma glucocorticoid levels and watermaze performance in 11β-HSD-1−/− mice (Table 2).
Table 1.
Basal (09.00–11.00 h) plasma corticosterone (cort), calculated ‘free’ corticosterone, and 11-DHC and CBG levels
| Mice (n) | cort, μg/dl | ‘free’ cort, μg/dl | 11-DHC, μg/dl | CBG, pmol/mg |
|---|---|---|---|---|
| Young wild type (5) | 2.5 ± 0.5 | 0.13 ± 0.03 | 0.9 ± 0.3 | 2.41 ± 0.28 |
| Young 11β-HSD-1−/− (5) | 13.4 ± 2.0* | 0.67 ± 0.10* | 3.3 ± 0.4** | 2.74 ± 0.41 |
| Old wild type (8) | 13.7 ± 1.5* | 0.68 ± 0.08* | 1.05 ± 0.15 | 2.62 ± 0.47 |
| Old 11β-HSD-1−/− (8) | 11.2 ± 3.5* | 0.56 ± 0.18* | 2.6 ± 0.5** | 3.30 ± 0.13 |
Values are means ± SEM. *, P < 0.001 compared to young wild-type mice. **, P < 0.05 compared to age-matched wild-type mice.
Table 2.
Correlation coefficients between plasma corticosterone levels and watermaze performance in the proximally cued platform learning (mean escape latency times of the four trials) for each day of training in combined young and old wild-type (n = 14) and 11β-HSD-1−/− mice (n = 13)
| Day | Wild type | 11β-HSD-1−/− |
|---|---|---|
| 1 | 0.24 (ns) | −0.47 (ns) |
| 2 | 0.71** | −0.44 (ns) |
| 3 | 0.55* | −0.16 (ns) |
| 4 | 0.69** | −0.58 (ns) |
| 5 | 0.77** | −0.26 (ns) |
, P < 0.05; **, P < 0.01; ns, nonsignificant.
Figure 2.

Scatter plot showing correlation between morning plasma corticosterone levels and escape latency to find a flagged hidden platform on the fifth day of training for individual wild-type mice (young and old combined). The open squares represent individual young mice and the filled squares represent individual aged mice.
Plasma 11-DHC levels in the wild-type mice correlated with plasma corticosterone levels (r = 0.65, P < 0.02). 11β-HSD-1−/− mice had significantly elevated 11-DHC levels [which they cannot activate to corticosterone (17); P < 0.05; Table 1 and Fig. 3]. 11-DHC in 11β-HSD-1−/− mice did not correlate with plasma corticosterone. Plasma CBG levels were not significantly altered by genotype or age (Table 1). Plasma glucose levels [wild-type: 6.55 ± 1.38 mM (young), 7.00 ± 0.83 mM (old); 11β-HSD-1−/−, 6.80 ± 1.09 mM (young), 5.29 ± 1.30 mM (old)] were not significantly altered by genotype or age.
Figure 3.
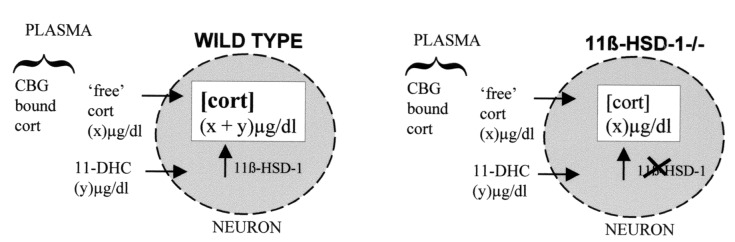
Schematic diagram showing contribution of 11-DHC to intraneuronal corticosterone levels in wild-type and 11β-HSD-1-null mice. ‘free’ cort reflects 5% of total corticosterone (i.e., excludes 95% bound to CBG). Intraneuronal cort is the sum of free corticosterone and 11-DHC in wild-type mice, but only free corticosterone in 11β-HSD-1-null mice, which cannot regenerate corticosterone from 11-DHC.
Distribution of Infused [3H]-Labeled Corticosterone in Brain Tissues of Wild-Type and 11β-HSD-1 Knockout Mice.
To determine whether effective intracerebral glucocorticoid levels in the hippocampus were altered in aged 11β-HSD-1−/− mice, [3H]corticosterone was infused under equilibrium conditions. The amount of [3H]corticosterone retained in the hippocampus was significantly lower in 18-month-old 11β-HSD-1−/− mice (64% decrease, P < 0.05; Fig. 4). [3H]corticosterone levels were also lower in the brainstem (61% decrease, P < 0.05) and showed a similar nonsignificant trend for lower levels in cerebellum, both tissues highly expresses 11β-HSD-1. There were no significant differences between genotypes in cortex that expresses 11β-HSD-1 predominantly in a single layer (ref. 23; Fig. 4).
Figure 4.

Comparison of distribution of infused [3H]corticosterone in brain tissues of wild-type and 11β-HSD-1−/− mice. Aged mice (18 months old) were infused s.c. with [3H]corticosterone via Alzet miniosmotic infusion pumps for 7 days. The content of [3H]corticosterone in each brain region was measured by solvent extraction with chromatographic separation of steroids. n = 6 per genotype. *, P < 0.05 compared with wild-type controls. Values are means ± SEM.
Discussion
Strikingly, in the face of increased plasma levels of active corticosterone throughout life, the 11β-HSD-1−/− mice resist glucocorticoid-associated learning impairments with aging, as shown in the flagged hidden platform watermaze task. All of the mice (young and aged, either genotype) failed to learn the classical hippocampus-dependent spatial task where the hidden platform is located by using extramaze distal cues around the experimental room (data not shown). This is not unusual because mice of the 129 strain generally perform poorly in the distally cued version of the watermaze task (24, 25). The assumption that learning of the proximally cued platform watermaze task is partly hippocampus-independent is based on findings in rats. However, this interpretation may be species-dependent, because hippocampal lesions in some strains of mice impair learning in both distally and proximally cued platform versions of the watermaze task (26). Recent studies have shown that both spatial and nonspatial information can be encoded within the hippocampus in a manner consistent with the mnemonic demands of the task (27). The severe impairment of the hippocampal lesioned 129/Ola mice to learn the proximally cued platform watermaze task suggests hippocampal dependence, at least in this strain.
Differences in sensorimotor or visual factors could have a secondary effect on the rate of learning in the aged mice. However, examination of the swim speeds and retinas suggests that the learning deficit in the aged wild-type mice are not simply due to motor or visual impairments. In addition, the degree of thigmotaxis (i.e., the percentage of time spent near the side walls of the pool), thought to be an indication of “emotionality,” did not differ significantly with age or genotype. The performance of the aged mutant mice in the watermaze was similar to the aged wild-type mice initially, but the old 11β-HSD-1-null mice then rapidly improved to match the learning performance of young wild types. This suggests that whatever other impairments exist in old mice, they are overcome by the mutation, and argues against this reflecting a generalized performance effect, and for more specific learning/memory effects of aging. Further studies are required to evaluate any other behavioral differences in the mutant mice.
Glucose is the major energy substrate for the brain and alterations in glucose availability can alter neuronal function and cognitive performance. Moreover, young 11β-HSD-1 null mice resist hyperglycemia on stress or obesity, although basal glucose levels are similar to wild type (17). In this study plasma glucose levels were not significantly affected by age or genotype, suggesting that at least gross differences in glucose homeostasis do not underlie the cognitive differences by genotype in aged mice.
Plasma Glucocorticoid Levels.
Recent findings that show that corticosterone levels in 18-month-old rats may predict spatial learning in the watermaze 6 months later (J. L. W. Yau and J. R. Seckl, unpublished data), emphasizes the causal link between corticosterone and hippocampal function in individual animals and echoing analogous prospective data in humans (2). In the present study, basal (morning) plasma corticosterone levels were low in young wild-type mice, but became markedly elevated with age, consistent with findings in rats and humans (2, 28, 29). Plasma corticosterone levels in wild-type controls correlated directly with the escape latency to find the flagged hidden platform. This is consistent with studies in rats where elevated corticosterone levels correlate with specific age-associated learning impairments in the watermaze (1, 3). Interestingly, the correlation between plasma corticosterone levels and watermaze performance is lost in 11β-HSD-1−/− mice, presumably because local tissue corticosterone levels in the brain regions involved in the learning task are lower than in wild-type controls because of the lack of 11-DHC reactivation and fail to reach the threshold for performance decline. This contention is supported by the requirement for clearly elevated glucocorticoid levels before inhibition of long-term potentiation or atrophy of hippocampal dendritic structure (5, 30, 31). Indeed, correlations between corticosterone and maze performance trended negative (although not significant) in the 11β-HSD-1-null mice. We speculate that the absence of 11-DHC reactivation produces lower intraneuronal corticosterone levels in sites such as the hypothalamic paraventricular nuclei attenuating feedback on the hypothalamic–pituitary–adrenal (HPA) axis, thus increasing plasma corticosterone levels.
It has been assumed that the learning deficits in the aged wild-type mice is an effect of chronically elevated glucocorticoid levels, because measures that prevent the long-term, age-associated rise in plasma corticosterone preserve learning and memory in aged rats (8, 9). However, elevated corticosterone levels can also have acute effects on hippocampal function (32–35). Whether the improved learning in aged 11β-HSD-1-null mice, which have reduced intrahippocampal corticosterone levels, is partly due to acute effects of lower corticosterone remains to be determined, but seems unlikely because acute blockade of corticosteroid receptors has either detrimental effects or no effect on learning (35). Indeed, the subtlety of the model is that glucocorticoid levels in tissues expressing 11β-HSD-1 are reduced but not absent, obviating the hazards of complete adrenal steroid depletion that produce neuronal loss and learning impairments.
Tissue Glucocorticoid Levels.
In young wild-type mice, 11-DHC levels are approximately half those of total corticosterone. However, around 95% of corticosterone is bound to plasma proteins [largely CBG (36)] and therefore unable to enter cells, so “free” corticosterone levels are less than 11-DHC levels, which is little bound by CBG (see Fig. 3). Thus, in young wild-type mice a substantial proportion of intracellular active glucocorticoid is likely to be derived from circulating 11-DHC, activated within cells to corticosterone by 11β-HSD-1, the only murine enzyme catalyzing 11β-reduction (17). Aged wild-type mice have elevated plasma corticosterone levels, but 11-DHC is still likely to contribute substantially to intracellular corticosterone (see Fig. 3). In contrast, although 11β-HSD-1−/− mice have elevated 11-DHC levels (presumably a consequence of reduced clearance due to loss of metabolism by 11β-HSD-1), these cannot be converted to corticosterone within cells. Increased plasma corticosterone levels in the null mice are likely to reflect both increased adrenal production to compensate for increased clearance (due to a lack of regeneration by 11β-HSD-1) and a loss of 11β-reductase in central nervous system areas responsible for glucocorticoid negative feedback on the HPA axis. The hippocampus, hypothalamus, and pituitary all express 11β-HSD-1 (23, 37). Loss of glucocorticoid regeneration in such sites would be anticipated to attenuate feedback and thus increase HPA activity. Indeed, 11β-HSD-1-null mice are less sensitive to exogenous cortisol suppression of HPA activation (38). Thus, in the null mice the only source of intracellular glucocorticoid is circulating corticosterone.
The aged 11β-HSD-1-null mice show improved learning in the flagged hidden platform watermaze task despite increased plasma levels of active corticosterone throughout life. This strongly implicates lower intraneuronal corticosterone levels and suggests that 11β-reductase regeneration of corticosterone from 11-DHC is an important determinant of effective intraneuronal glucocorticoid action. Plasma CBG levels were similar in 11β-HSD-1−/− and wild-type mice and were not significantly altered with aging. As 5% of corticosterone is “free” and assuming near complete conversion of 11-DHC to corticosterone by 11β-HSD-1 in wild-type neurons, then “intraneuronal” corticosterone levels are likely to be lower in aged 11β-HSD-1-null mice than in aged wild-type controls, which have the additional burden of corticosterone regenerated from 11-DHC (see Fig. 3). In support of this contention, retention of [3H]corticosterone in the hippocampus of aged 11β-HSD-1−/− mice was less than half that of aged wild-type controls, in the face of similar plasma corticosterone levels at this age. Taken together, the data suggest that 11β-HSD-1 contributes substantially to active corticosterone levels within the hippocampus in vivo and hence to the deleterious effects of glucocorticoids with aging.
The key question addressed here is whether the effects of active glucocorticoids are in any way influenced by the local conversion of inert circulating 11-DHC by the activity of 11β-reductase within the central nervous system. The data suggest that regeneration of active corticosterone by 11β-HSD-1 within the brain is indeed important, because glucocorticoid-associated learning deficits with aging are significantly improved when the enzyme is absent. Although prolonged, cumulative glucocorticoid actions seem most likely, our results do not exclude the possibility that the learning performance changes in the aged mice are the result of the acute actions of glucocorticoids. It remains to be determined whether hippocampal neuronal structural changes and other features of long-term glucocorticoid excess with aging are also prevented in the 11β-HSD-1−/− mice. Nonselective 11β-HSD inhibition increases hepatic insulin sensitivity in humans (39), in line with the hepatic phenotype of the 11β-HSD-1−/− mouse (17). Clearly, selective inhibitors of 11β-reductase could act as useful agents to prevent/ameliorate glucocorticoid associated learning deficits with age. 11β-HSD-1 represents a plausible therapeutic target to attenuate effective glucocorticoid action in the brain in vivo.
Acknowledgments
We thank Richard G. M. Morris for advice on the watermaze protocol for mice and critical reading of the manuscript, Mark Ramsay for advice on the watermaze protocol for mice, Alasdair MacLullich for helpful discussions, and Dr. John Stokes for ophthalmological assessments. This work was funded by a Program grant from the Wellcome Trust (to J.R.S. and J.J.M.), a Wellcome Senior Clinical Fellowship (to J.R.S.), and a research development grant from SHEFC (to J.R.S.). J.J.M. acknowledges support as a Principal Research Fellow from the Wellcome Trust.
Abbreviations
- 11β-HSD-1
11β-hydroxysteroid dehydrogenase type 1
- 11-DHC
11-dehydrocorticosterone
- CBG
corticosteroid binding globulin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Issa A M, Rowe W, Gauthier S, Meaney M J. J Neurosci. 1990;10:3247–3254. doi: 10.1523/JNEUROSCI.10-10-03247.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lupien S J, de Leon M, de Santi S, Convit A, Tarshish C, Nair N P V, Thakur M, McEwen B S, Hauger R L, Meaney M J. Nat Neurosci. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- 3.Yau J L W, Olsson T, Morris R G M, Meaney M J, Seckl J R. Neuroscience. 1995;66:571–581. doi: 10.1016/0306-4522(94)00612-9. [DOI] [PubMed] [Google Scholar]
- 4.Kerr D S, Huggett A M, Abraham W C. Psychobiology. 1994;22:123–133. [Google Scholar]
- 5.Woolley C, Gould E, McEwen B S. Brain Res. 1990;531:225–231. doi: 10.1016/0006-8993(90)90778-a. [DOI] [PubMed] [Google Scholar]
- 6.Landfield P W. Prog Brain Res. 1987;72:279–300. doi: 10.1016/s0079-6123(08)60215-0. [DOI] [PubMed] [Google Scholar]
- 7.Landfield P W, McEwen B S, Sapolsky R M, Meaney M J. Science. 1996;272:1249–1251. [PubMed] [Google Scholar]
- 8.Landfield P W, Baskin R K, Pitler T A. Science. 1981;214:581–584. doi: 10.1126/science.6270791. [DOI] [PubMed] [Google Scholar]
- 9.Meaney M J, Aitken D H, van Berkel C, Bhatnagar S, Sapolsky R M. Science. 1988;239:766–768. doi: 10.1126/science.3340858. [DOI] [PubMed] [Google Scholar]
- 10.White P C, Mune T, Agarwal A K. Endocr Rev. 1997;18:135–156. doi: 10.1210/edrv.18.1.0288. [DOI] [PubMed] [Google Scholar]
- 11.Funder J W, Pearce P T, Smith R, Smith A I. Science. 1988;242:583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- 12.Edwards C R W, Stewart P M, Burt D, Brett L, McIntyre M A, Sutanto W S, de Kloet E R, Monder C. Lancet. 1988;ii:986–989. doi: 10.1016/s0140-6736(88)90742-8. [DOI] [PubMed] [Google Scholar]
- 13.Low S C, Moisan M-P, Edwards C R W, Seckl J R. J Neuroendocrinol. 1994;6:285–290. doi: 10.1111/j.1365-2826.1994.tb00584.x. [DOI] [PubMed] [Google Scholar]
- 14.Ozols J. J Biol Chem. 1995;270:2305–2312. doi: 10.1074/jbc.270.5.2305. [DOI] [PubMed] [Google Scholar]
- 15.Jamieson P M, Chapman K E, Edwards C R W, Seckl J R. Endocrinology. 1995;136:4754–4761. doi: 10.1210/endo.136.11.7588203. [DOI] [PubMed] [Google Scholar]
- 16.Rajan V, Edwards C R W, Seckl J R. J Neurosci. 1996;16:65–70. doi: 10.1523/JNEUROSCI.16-01-00065.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kotelevtsev Y, Holmes M C, Burchell A, Houston P M, Schmoll D, Jamieson P M, Best R, Brown R, Edwards C R W, Seckl J R, et al. Proc Natl Acad Sci USA. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jarrard L E. J Neurosci Methods. 1989;29:251–259. doi: 10.1016/0165-0270(89)90149-0. [DOI] [PubMed] [Google Scholar]
- 19.Al Dujaili E A S, Williams B C, Edwards C R W. Steroids. 1981;37:157–176. doi: 10.1016/s0039-128x(81)80015-3. [DOI] [PubMed] [Google Scholar]
- 20.Brown R W, Chapman K E, Edwards C R W, Seckl J R. Endocrinology. 1993;132:2614–2621. doi: 10.1210/endo.132.6.8504762. [DOI] [PubMed] [Google Scholar]
- 21.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 22.Osteen W K, Spencer R L, Bare D J, McEwen B S. Behav Brain Res. 1995;68:151–158. doi: 10.1016/0166-4328(94)00168-f. [DOI] [PubMed] [Google Scholar]
- 23.Moisan M-P, Seckl J R, Edwards C R W. Endocrinology. 1990;127:1450–1455. doi: 10.1210/endo-127-3-1450. [DOI] [PubMed] [Google Scholar]
- 24.Montkowski A, Poettig M, Mederer A, Holsboer F. Brain Res. 1997;762:12–18. doi: 10.1016/s0006-8993(97)00370-3. [DOI] [PubMed] [Google Scholar]
- 25.Gerlai R. Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- 26.Logue S F, Paylor R, Wehner J M. Behav Neurosci. 1997;111:104–113. doi: 10.1037//0735-7044.111.1.104. [DOI] [PubMed] [Google Scholar]
- 27.Hampson R E, Simeral J D, Deadwyler S A. Nature (London) 1999;402:610–614. doi: 10.1038/45154. [DOI] [PubMed] [Google Scholar]
- 28.Seckl J R, Olsson T. J Endocrinol. 1995;145:201–211. doi: 10.1677/joe.0.1450201. [DOI] [PubMed] [Google Scholar]
- 29.Lupien S, Lecours A R, Lussier I, Schwartz G, Nair N, Meaney M J. J Neurosci. 1994;14:2893–2903. doi: 10.1523/JNEUROSCI.14-05-02893.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diamond D M, Bennett M C, Fleshner M, Rose G M. Hippocampus. 1992;2:421–430. doi: 10.1002/hipo.450020409. [DOI] [PubMed] [Google Scholar]
- 31.Joels M, de Kloet E R. Trends Neurosci. 1992;15:25–30. doi: 10.1016/0166-2236(92)90345-9. [DOI] [PubMed] [Google Scholar]
- 32.de Quervain D J, Roozendaal B, McGaugh J L. Nature (London) 1998;394:787–790. doi: 10.1038/29542. [DOI] [PubMed] [Google Scholar]
- 33.Roozendaal B, Bohus B, McGaugh J L. Psychoneuroendocrinology. 1996;21:681–693. doi: 10.1016/s0306-4530(96)00028-5. [DOI] [PubMed] [Google Scholar]
- 34.Lupien S J, McEwen B S. Brain Res Rev. 1997;24:1–27. doi: 10.1016/s0165-0173(97)00004-0. [DOI] [PubMed] [Google Scholar]
- 35.Conrad C D, Lupien S J, McEwen B S. Neurobiol Learn Mem. 1999;72:39–46. doi: 10.1006/nlme.1998.3898. [DOI] [PubMed] [Google Scholar]
- 36.Dunn J F, Nisula B C, Rodbard D. J Chem. 1981;53:58–68. doi: 10.1210/jcem-53-1-58. [DOI] [PubMed] [Google Scholar]
- 37.Sakai R R, Lakshmi V, Monder C, McEwen B S. J Neuroendocrinol. 1992;4:101–106. doi: 10.1111/j.1365-2826.1992.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 38.Harris H J, Kotelevtsev Y, Mullins J J, Seckl J R, Holmes M C. Endocrinology. 2001;142:114–120. doi: 10.1210/endo.142.1.7887. [DOI] [PubMed] [Google Scholar]
- 39.Walker B R, Connacher A A, Lindsay R M, Webb D J, Edwards C R W. J Clin Endocrinol Metab. 1995;80:3155–3159. doi: 10.1210/jcem.80.11.7593419. [DOI] [PubMed] [Google Scholar]