

Abstract
Huntington disease (HD) is a neurodegenerative disorder involving preferential loss of striatal GABAergic medium spiny neurons. Adenosine A2A receptors (A2ARs) are present in the striatum at both presynaptic and post-synaptic levels. Blocking pre-synaptic A2ARs, localized in glutamatergic terminals that contact striatal GABAergic dynorphinergic neurons, reduces glutamate release, which could be beneficial in HD. On the other hand, blockade of post-synaptic A2ARs, localized in striatal GABAergic enkephalinergic neurons, could exacerbate the motor dysfunction. To evaluate the function of pre- or post-synaptic A2ARs in HD we used selective antagonists for these receptors in a transgenic rat model of HD. Locomotor activity after systemic administration of the postsynaptic A2AR antagonist KW-6002 was used to investigate the function of post-synaptic A2ARs. The role of pre-synaptic A2ARs was instead evaluated by measuring the reduction of the electromyographic response of mastication muscles during electrical stimulation of the orofacial motor cortex after the systemic administration of the presynaptic A2AR antagonist SCH-442416. The ability of KW-6002 to produce locomotor activation was lost at 6 and 12 month-old of age in heterozygous and homozygous transgenic rats, but not in wild-type littermates. Nevertheless, no significant changes were observed up to 12 months of age in the potency of SCH-442416 to decrease the electromyographic response after cortical electrical stimulation. These results agree with a selective impairment of the striatal GABAergic enkephalinergic neuronal function during pre-symptomatic stages in HD. Since presynaptic A2AR function is not impaired, this receptor could probably be used as a target for the symptomatic treatment of the disease.
Keywords: Huntington disease, transgenic rat, adenosine A2A receptor, locomotor activity, cortical electrical stimulation
Introduction
Huntington disease (HD) is a neurodegenerative disorder characterized by progressive chorea, cognitive impairments and emotional disturbances. HD is caused by expansion of a CAG repeat within exon 1 of the huntingtin gene on chromosome 4. The mechanism by which the mutant huntingtin may cause neurodegeneration is unknown and even controversial (Wellington et al., 2000; Yang et al., 2002, Ravikumar et al., 2004). There have been many attempts to generate animal models of HD, using excitotoxins that may replicate some of the histological changes as well as some of the motor signs, but given that neurodegeneration is progressive, these models are at least questionable. The use of transgenic animals is more useful to study neuropathological mechanisms, especially a transgenic rat model generated recently that bears a human HD mutation with 51 CAG repeats and shows progressive neurological, neurochemical and neuropathological features similar to the slowly progressing disease (von Hörsten et al., 2003; Nguyen et al., 2006).
The most vulnerable neurons in HD are the GABAergic enkephalin-containing neurons of the basal ganglia (Mitchell et al., 1999). It has been described that the GABAergic enkephalinergic medium spiny neurons (MSN) of the striatum express both adenosine A2A receptors (A2ARs) and D2 dopamine receptors (D2Rs) (Ferré et al., 1993, Rosin et al., 2003, Quiroz et al., 2009; Schiffmann et al., 2007). It is known that these two receptors may postsynaptically interact at least in two different ways, modulating the effects of glutamate and dopamine on the MSNs. On one hand, A2AR and D2R may form heteromers, (Ferré et al., 2008) and the A2AR in these heteromers counteracts the Ca2+ influx through NMDA receptors inhibited by D2R activation (Azdad et al., 2009; Higley and Sabatini, 2010). On the other hand, and in addition to the above described mechanism, a second biochemical signaling mechanism exists in the GABAergic MSN, controlled by D2Rs; these receptors exert an inhibitory effect on A2ARs (not forming heteromers) and their positive coupling to the adenylyl cyclase second messenger pathway (Hillion, 2002; Hakansson et al., 2006), resulting in an increased activity of the neuron under basal conditions. These two antagonistic mechanisms (A2ARs over D2Rs and vice versa) coexist in the same neuron and their balance contributes to determine the neuronal response (Ferré et al., 2008).
In addition to these two populations of receptors, A2ARs are also localized presynaptically in the glutamatergic neurons that contact the other type of striatal MSN, the GABAergic dynorphin-containing neuron (Rosin et al., 2003; Quiroz et al., 2009) where they form heteromers with the adenosine A1 receptor (A1R) and participate in the regulation of glutamate release (Ciruela et al., 2006, Quiroz et al., 2009). In this case, activation of A2ARs in the A1R-A2AR heteromer produces an increase in glutamate release. Previous studies in an animal model of HD disease (3-nitropropionic acid-induced striatal lesions in rodents) showed glutamate-dependent neuroprotective effects of A2ARs antagonists (Blum et al., 2003a). On the other hand, in the same animal model, blockade of postsynaptic A2ARs seemed to increase neuronal death (Blum et al., 2003a), suggesting that presynaptic A2ARs could be considered as targets for the pathogenetic treatment of HD (Blum et al., 2003b; Popoli et al., 2007). However, according to the predominant localization of presynaptic A2ARs, GABAergic dynorphinergic MSNs should be more vulnerable than GABAergic enkepalinergic MSNs. Nevertheless, according to the most accepted model of basal ganglia circuitry (DeLong and Wichmann, 2007), selective blockade of presynaptic striatal A2ARs could be useful in the symptomatic treatment of HD.
We have recently shown that the affinity of A2AR antagonists is different depending on the heteromerization of A2ARs with different receptors (Orrú et al., 2011a). In this sense, SCH-442416 binds with more affinity to A2ARs forming heteromers with A1Rs or not forming heteromers, while KW-6002 shows better affinity for A2ARs when co-expressed with D2Rs. We have also shown that in vivo (and using A2AR antagonists with different pharmacological profile), the potency at blocking the motor output (electromyographic response by mastication muscles) induced by cortical electrical stimulation (in the premotor orofacial area) and the potency at inducing locomotor activation can be used to distinguish between pre- and postsynaptic activities, respectively.
Using this experimental approach and transgenic HD rats, we have studied the response to a preferentially postsynaptic A2AR antagonist and the induced activation of locomotor activity, and to a preferentially presynaptic A2AR antagonist and its potency in blocking the motor output induced by cortical electrical stimulation (Orrú et al., 2011a). Our study compared the responses in the wild type, the heterozygous and homozygous rats at different ages, when they still are asymptomatic for the disease or when some symptoms have already been described (von Hörsten et al., 2003; Nguyen et al., 2006). We report here that the locomotor response induced by an A2AR antagonist is present in all groups of rats at an early age (3 months), but is lost at a later age (6 months), when symptoms are still not apparent, only in the transgenic HD rats. Also, the motor output during cortical electrical stimulation is maintained in wild type and transgenic rats at least until 12 months of age.
Materials and methods
Animals and drugs
Transgenic HD rats expressing 727 aminoacids of the HD gene with 51 CAG repeats were used (von Hörsten et al., 2003). The present study used male homozygous, heterozygous and wild-type littermate control rats from litters all born within a few days of each other. Rats of 3, 6, and 12 months of age from each group (homozygous (HOM) (+/+), heterozygous (HET) (+/-) and wild-type controls (-/-) were used for the studies. Animals were housed in pairs per cage and kept on a 12/12 dark/light cycle with food and water available ad libitum. All animals used in the study were maintained in accordance with the guidelines of National Institutes of Health animal care, and the animal research conducted to perform the study was approved by the NIDA IRP Animal Care and Use Committee (protocol #: 09-BNRB-73). The A2AR antagonists SCH-442416 and KW-6002 were provided by CHDI Foundation Inc. (Los Angeles, CA). The doses used in the study were selected based on a previous study (Orrú et al., 2011a). SCH-442416 was suspended in 5% DMSO and 5% Tween-80, and KW-6002 in 8% Tween-80. Compounds were administered intraperitoneally with an injection volume of 2 ml/kg body weight.
Locomotor activity
Locomotor acivity was measured in open field soundproof chambers (50×50 cm, Med Associates Inc., VT). The animals were not habituated to the experimental cages and recording of the locomotor activity started immediately after administering the A2AR antagonists or the vehicle, by placing the animals in the boxes. A lamp inside each chamber remained lit during the 90 min test. Locomotion was measured by counting the number of breaks in the infrared beams of the chamber during consecutive periods of 10 min. Results were analyzed as means ± S.E.M. of the average of 10-min period transformed counts (square root) during the 90-min period of observation. All the animals were tested only once. The effect of different doses of the A2AR antagonist on locomotor activity was analyzed suing a one-way analysis of variance (ANOVA), followed by Newman-Keuls’ post-hoc test.
Surgical procedures
Rats were anesthetized with 3 ml/kg of Equithesin (NIDA Pharmacy, Baltimore, MD) and implanted unilaterally with bipolar stainless steel electrodes, 0.15 mm in diameter (Plastics One, Roanoke, VA), into the orofacial area of the lateral agranular motor cortex (3 mm anterior, 3 and 4 mm lateral, and 4.2 mm below bregma), as described elsewhere (Quiroz et al., 2009). Briefly, the electrodes and a head holder were fixed on the skull with screws and acrylic resin. Electrodes were also implanted in mastication muscles. Two 5 mm-long incisions were made in the skin on the upper and lower jaw areas to expose the masseter and the lateral pterygoid muscles. Two silicon rubber-coated coiled stainless steel recording electrodes (Plastics One, Roanoke, VA) were slipped below the skin from the incision in the skull until the tips showed up from the incisions in the jaw. The bare tips of the electrodes were then held in contact with the masseter and the lateral pterygoid muscles and the skin was closed with surgical staples. The other end of the recording electrodes was encased in a molded plastic pedestal with a round threaded post which is attached to an electrical swivel for stimulation and recording. The pedestal was secured to the skull with dental cement together with the stimulation electrodes.
Electromyographic recording and power correlation analysis
Five days after surgery rats were placed in individual bowl chambers as described elsewhere (Quiroz et al, 2009). Both stimulation and recording electrodes were attached to an electrical swivel. Stimulation electrodes were connected to constant current isolation units (PSIU6X, Grass Instruments, West Warwick, RI) driven by an electrical stimulator (Grass S88X, Grass Instruments, West Warwick, RI). The recording electrodes were connected to a differential amplifier (Grass LP511, Grass Instruments, West Warwick, RI). This configuration allows the rat to move freely. After 60 min of habituation, biphasic current pulse trains (pulse of 0.1 ms at 120–200 μA; 100 Hz, 160 ms trains repeating once per 2 seconds) were delivered. Animals that failed to show a positive electromyographic (EMG) response (less than 10%) with stimulation intensities of 200 μA were discarded from the experiments (7 animals out of 128 tested). Both stimulator monitoring and the amplified EMG signal were directed to analog-to-digital converter for recording (NI 9215, National Instruments, Austin, TX). Off-line, both signals were processed and a Pearson’s correlation -based analysis between the stimulation and jaw EMG response signals was calculated for each subject. Decrease in the power correlation coefficient (PCC) between these two signals indicates a decrease in the efficacy of the transmission in the neural circuit. The effects of the different doses of compounds on PCC were analyzed by one-way ANOVA, followed by Dunnett’s post-hoc test.
Results
Effect of preferential postsynaptic A2AR antagonism on locomotor activity
Similar to what it has been previously shown (Orrú et al., 2011a), the preferential postsynaptic A2AR antagonist KW-6002 produced a strong locomotor activation at doses of 1 and 3 mg/kg i.p. in 3 months old rats. The effect was very similar in 6 and 12 months old wild-type rats (Figure1). The two transgenic rat groups (heterozygous and homozygous) showed a similar profile of locomotor activation as the wild-type littermates at 3 months of age (Figure 1). However, the locomotor activation by KW-6002 was not maintained in 6 and 12 months old transgenic rats (both heterozygous and homozygous). A lack of locomotor activation was seen in these groups even using higher doses of KW-6002 (up to 10 mg/kg, i.p.) (Figure 1). These results indicate a dysfunction of striatal postsynaptic A2ARs in HD transgenic rats starting in the asymptomatic stages.
Figure 1.
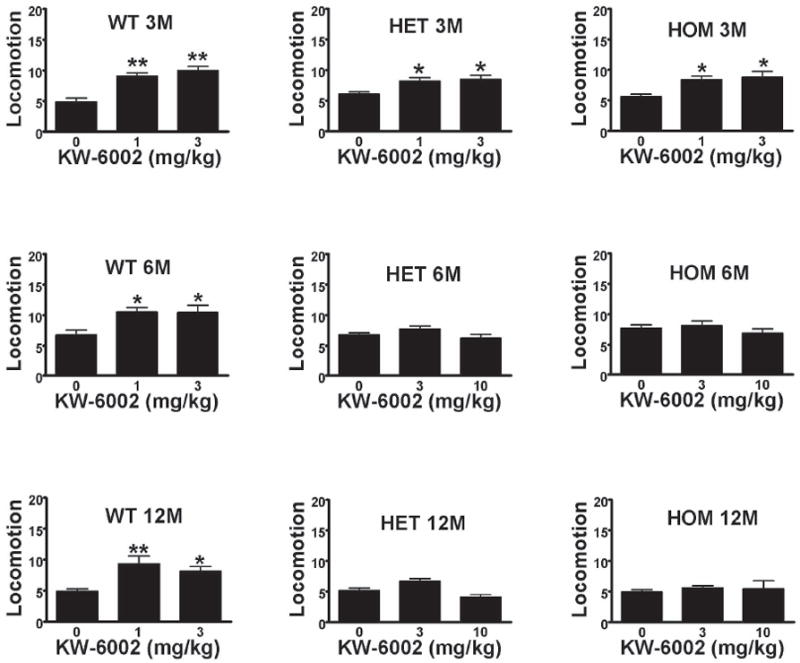
Locomotor activity in wild type (WT) heterozygous (HET) and homozygous (HOM) transgenic rats at 3 months (3M), 6 months (6M) and 12 months (12M) of age induced by the systemic administration of KW-6002. Results represent means ± S.E.M. (n=6-8 per group) of the average of 10-min period transformed counts (square root) during the 90-min period of observation. *p<0.05, **p<0.01, in comparison to vehicle (VEH)-treated animals (ANOVA with post hoc Newman–Keuls comparisons, p<0.5 and p<0.01, respectively). All animals were used only once.
Effect of preferential presynaptic A2AR antagonism on power correlation analysis
It has been previously described that SCH-442416, a presynaptic A2AR preferentially-acting antagonist is able to reduce PCC at doses with no effect on locomotor activity (Orrú et al., 2011a). Administration of SCH-442416 to 3 months old wild-type and HD transgenic rats induced a significant reduction in PCC of similar magnitude (Figure 2). This response was observed across time, with similar reductions of PCC in the 6 and 12 months old rats in the three genetically distinct groups (Figure 2). In a previous study, and using in vivo microdialysis (Orrú et al., 2011a) we showed that reducing PCC by SCH-442416 parallels with a reduction in striatal glutamate release induced by cortical stimulation. These results indicate a preserved function of striatal presynaptic A2ARs in HD transgenic rats.
Figure 2.
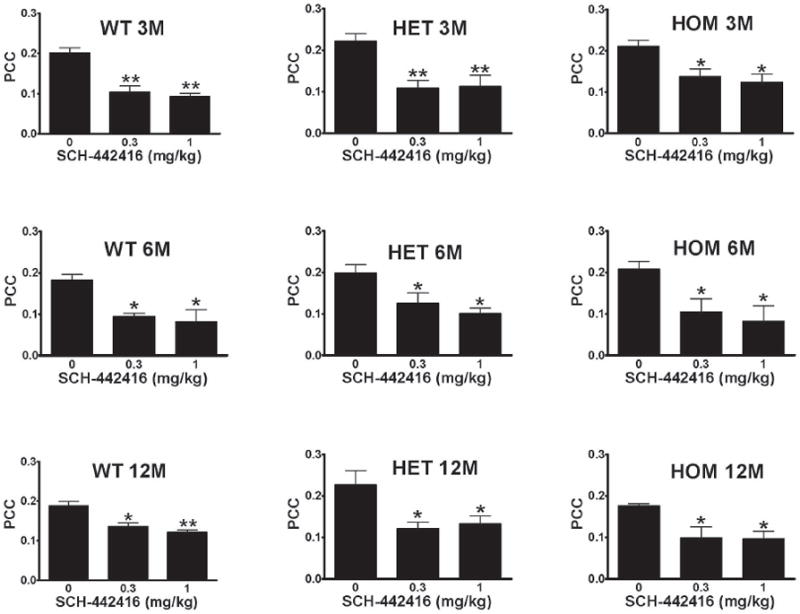
Dose-dependent decrease in the power correlation coefficient (PCC) in wild type (WT) heterozygous (HET) and homozygous (HOM) transgenic rats at 3 months (3M), 6 months (6M) and 12 months (12M) of age induced by the systemic administration of SCH-442416. Results represent means ± SEM (n=5-6 per group). *p<0.05, **p<0.01, in comparison to vehicle (VEH)-treated animals (ANOVA with post hoc Newman–Keuls comparisons, p<0.5 and p<0.01, respectively). All animals were used only once.
Discussion
The findings of the present study indicates that the use of different A2AR antagonists previously shown to have different pre- and postsynaptic profiles (Orrú et al., 2011a) may reveal functional changes in HD transgenic rats when they are still asymptomatic. We have studied the locomotor response and the PCC response to the A2AR antagonists KW-6002 and SCH-442416, respectively, in HD rats at 3, 6 and 12 months of age. The preferentially postsynaptic A2AR antagonist KW-6002 was used at doses that have been described to increase locomotor activity without affecting PCC or modifying striatal glutamate release induced by cortical stimulation. Similarly, the preferentially presynaptic A2AR antagonist SCH-442416 was used at doses that reduce PCC without affecting locomotor activity (Orrú et al., 2011a).
According to previous studies (van Hörsten et al., 2003, Nguyen et al., 2006), at 10 months of age HD rats may start showing some cognitive decline signs and it is not until the age of 10 to 15 months that progressive motor impairment and striatal atrophy appear. Consequently and based on these studies, 3 and 6 months of age can be considered as presymptomatic stages, although some anxiety-related symptoms could be observed already at 2 months of age (Nguyen et al., 2006). Our study shows that both heterozygous and homozygous HD rats developed insensitivity to the locomotor activating effects of the A2AR antagonist KW-6002 some time between 3 and 6 months of age. This insensitivity was still observed in 12-month old rats, and the effect was not observed in wild-type animals at any age. Surprisingly, the ability of the A2AR antagonist SCH-442416 to reduce PCC was preserved across at least 12 months of age and regardless the different genotype, with similar reductions in homozygous, heterozygous and wild-type HD rats. These results indicate that the HD rats develop a significant and selective impairment of striatal postsynaptic A2AR function, which agrees with the possibility of a selective impairment of the GABAergic striatopallidal neuronal function long before overt motor symptoms of HD appear (Popoli et al., 2007). In fact, A2AR density has been found to be significantly decreased in early stages of HD (Glass et al., 2000). Interestingly, a genetic variation of the A2AR has been shown to decrease the age of onset in HD (Dhaenens et al., 2009; Taherzadeh-Fard et al., 2010). Furthermore, an aberrant A2ARs signaling has been found in an in vitro model of the disease and in peripheral circulating cells of HD patients (Varani et al., 2001, 2003).
Previous results on a mouse model of HD (R6/2 mice) have been controversial, and both a downregulation and an upregulation of A2ARs have been reported during early presymptomatic stages (Cha et al., 1999; Tarditi et al., 2006). Cha et al. (1999) reported a significant decrease of the striatal density of A2ARs, while Tarditi et al. (2006) found a transient A2AR upregulation associated with an increased biochemical response (cAMP accumulation) to A2AR agonists. The present study shows that a striatal A2AR dysfunction is also present in the transgenic HD rat model, which is believed to be a more appropriate animal model of HD than R6/2 mice (see von Hörsten et al., 2003). Our results fit better with those by Cha et al. (1999) and indicate that there is a permanent functional downregulation of postsynaptic striatal A2ARs. In a previous study, a significant reduction in the density of A2ARs was found in the striatum of 2-year old HD transgenic rats, but correlating with degeneration of the GABAergic striatopallidal neurons (Bauer et al., 2005). Additional biochemical studies in this model should provide a better understanding of a putative A2AR dysfunction in the pre-symptomatic stages of the disease. Importantly, the fact that in our study presynaptic A2AR function seems not to be impaired (at least up to 12 months of age), still makes possible to think of this receptor as a target for the symptomatic treatment of the disease.
Although in the present study we only focused on two subpopulations of striatal A2ARs, there is evidence for the existence of other subpopulations in the striatum, such as the postsynaptic A2AR also localized in GABAergic enkephalinergic MSNs that do not form heteromers with D2Rs (see Introduction). We have recently shown that this subpopulation is, in fact, also sensitive to the blocking properties of SCH-442416 (Orru et al., 2011b). Therefore, it would be of importance to evaluate which subpopulation of striatal postsynaptic A2ARs are functionally altered in the transgenic rat model of HD. And there are still other subpopulations of striatal A2AR, such as those forming heteromers with cannabinoid CB1 receptors and those localized in cholinergic interneurons, which should also be studied (for a recent review, see Ferre et al., 2011). Finally, it would also be of importance to analyze the function of extrastriatal A2ARs, such as those found in the cortex and hippocampus. Although their expression is much lower that those in the striatum, if dysfunctional they could be involved in the cognitive impairment in HD.
Highlights.
>Striatal adenosine A2A receptors are both presynaptic and post-synaptic. >We studied their functional activity in a transgenic rat model of Huntington disease. >Postsynaptic A2A receptor function was impaired between 3 and 6 months of age. >Presynaptic A2A receptor function was not impaired up to 12 months of age. >Results suggest an impairment of A2A receptor function during pre-symptomatic stages.
Acknowledgments
Work funded by the intramural funds of NIDA and by CHDI Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Azdad K, Gall D, Woods AS, Ledent C, Ferré S, Schiffmann SN. Dopamine D2 and adenosine A2A receptors regulate NMDA-excitation in accumbens neurons through A2A-D2 receptor heteromerization. Neuropsychopharmacology. 2009;34:972–986. doi: 10.1038/npp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer A, Zilles K, Matusch A, Holzmann C, Riess O, von Hörsten S. Regional and subtype selective changes of neurotransmitter receptor density in a rat transgenic for the Huntington’s disease mutation. J Neurochem. 2005;94:639–650. doi: 10.1111/j.1471-4159.2005.03169.x. [DOI] [PubMed] [Google Scholar]
- Blum D, Galas MC, Pintor A, Brouillet E, Ledent C, Muller CE, Bantubungi K, Galluzzo M, Gall D, Cuvelier L, Rolland AS, Popoli P, Schiffmann SN. A dual role of adenosine A2A receptors in 3-nitropropionic acid-induced striatal lesions: implications for the neuroprotective potential of A2A antagonists. J Neurosci. 2003a;23:5361–5269. doi: 10.1523/JNEUROSCI.23-12-05361.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum D, Hourez R, Galas MC, Popoli P, Schiffmann SN. Adenosine receptors and Huntington’s disease: implications for pathogenesis and therapeutics. Lancet Neurol. 2003b;2:366–374. doi: 10.1016/s1474-4422(03)00411-3. [DOI] [PubMed] [Google Scholar]
- Cha JH, Frey AS, Alsdorf SA, Kerner JA, Kosinski CM, Mangiarini L, Penney JB, Jr, Davies SW, Bates GP, Young AB. Altered neurotransmitter receptor expression in transgenic mouse models of Huntington’s disease. Philos Trans R Soc Lond B Biol Sci. 1999;354:981–989. doi: 10.1098/rstb.1999.0449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Casado V, Rodrigues RJ, Luján R, Burgueno J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortés A, Canela EI, López-Giménez JF, Milligan G, Lluis C, Cunha RA, Ferré S, Franco R. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol. 2007;64:20–24. doi: 10.1001/archneur.64.1.20. [DOI] [PubMed] [Google Scholar]
- Dhaenens CM, Burnouf S, Simonin C, Van Brussel E, Duhamel A, Defebvre L, Duru C, Vuillaume I, Cazeneuve C, Charles P, Maison P, Debruxelles S, Verny C, Gervais H, Azulay JP, Tranchant C, Bachoud-Levi AC, Dürr A, Buée L, Krystkowiak P, Sablonnière B, Blum D. A genetic variation in the ADORA2A gene modifies age at onset in Huntington’s disease. Neurobiol Dis. 2009;35:474–476. doi: 10.1016/j.nbd.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Ferré S, O’Connor WT, Fuxe K, Ungerstedt U. The striatopallidal neuron: a main locus for adenosine-dopamine interactions in the brain. J Neurosci. 1993;13:5402–5406. doi: 10.1523/JNEUROSCI.13-12-05402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Quiroz C, Woods AS, Cunha R, Popoli P, Ciruela F, Lluis C, Franco R, Azdad K, Schiffmann SN. An update on adenosine A2A-dopamine D2 receptor interactions. Implications for the function of G protein-coupled receptors. Curr Pharm Des. 2008;14:1468–1474. doi: 10.2174/138161208784480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Quiroz C, Orru M, Guitart X, Navarro G, Cortés A, Casadó V, Canela EI, Lluis C, Franco R. Adenosine A(2A) Receptors and A(2A) Receptor Heteromers as Key Players in Striatal Function. Front Neuroanat. 2011;5:36. doi: 10.3389/fnana.2011.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RL. The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience. 2000;97:505–519. doi: 10.1016/s0306-4522(00)00008-7. [DOI] [PubMed] [Google Scholar]
- Håkansson K, Galdi S, Hendrick J, Snyder G, Greengard P, Fisone G. Regulation of phosphorylation of the GluR1 AMPA receptor by dopamine D2 receptors. J Neurochem. 2006;96:482–488. doi: 10.1111/j.1471-4159.2005.03558.x. [DOI] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, Canela EI, Zoli M, Agnati LF, Ibanez CF, Lluis C, Franco R, Ferré S, Fuxe K. Coaggregation, cointernalization and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem. 2002;277:18091–18097. doi: 10.1074/jbc.M107731200. [DOI] [PubMed] [Google Scholar]
- Mitchell IJ, Cooper AJ, Griffiths MR. The selective vulnerability of striatopallidal neurons. Prog Neurobiol. 1999;59:691–719. doi: 10.1016/s0301-0082(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Nguyen HP, Kobbe P, Rahne H, Wörpel T, Jäger B, Stephan M, Pabst R, Holzmann C, Riess O, Korr H, Kántor O, Petrasch-Parwez E, Wetzel R, Osmand A, von Hörsten S. Behavioral abnormalities precede neuropathological markers in rats transgenic for Huntington’s disease. Hum Mol Genet. 2006;15:3177–3194. doi: 10.1093/hmg/ddl394. [DOI] [PubMed] [Google Scholar]
- Orrú M, Bakešová J, Brugarolas M, Quiroz C, Beaumont V, Goldberg SR, Lluís C, Cortés A, Franco R, Casadó V, Canela EI, Ferré S. Striatal pre- and postsynaptic profile of adenosine A2A receptor antagonists. PLoS One. 2011a;6:e16088. doi: 10.1371/journal.pone.0016088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrú M, Quiroz C, Guitart X, Ferré S. Pharmacological evidence for different populations of postsynaptic adenosine A(2A) receptors in the rat striatum. Neuropharmacology. 2011b doi: 10.1016/j.neuropharm.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli P, Blum D, Martire A, Ledent C, Ceruti S, Abbracchio MP. Functions, dysfunctions and possible therapeutic relevance of adenosine A2A receptors in Huntington’s disease. Prog Neurobiol. 2007;81:331–348. doi: 10.1016/j.pneurobio.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Quiroz C, Lujan R, Uchigashima M, Simoes AP, Lerner TN, Borycz J, Kachroo A, Canas PM, Orrú M, Schwarzschild MA, Rosin DL, Kreitzer AC, Cunha RA, Watanabe M, Ferré S. Key modulatory role of presynaptic adenosine A2A receptors in cortical neurotransmission to the striatal direct pathway. TheScientificWorldJournal. 2009;9:1321–1344. doi: 10.1100/tsw.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Hettinger BD, Lee A, Linden J. Anatomy of adenosine A2A receptors in brain: morphological substrates for integration of striatal function. Neurology. 2003;61:S12–S18. doi: 10.1212/01.wnl.0000095205.33940.99. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Fisone G, Moresco R, Cunha R, Ferré S. Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol. 2007;83:277–292. doi: 10.1016/j.pneurobio.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh-Fard E, Saft C, Wieczorek S, Epplen JT, Arning L. Age at onset in Huntington’s disease: replication study on the associations of ADORA2A, HAP1 and OGG1. Neurogenetics. 2010;11:435–439. doi: 10.1007/s10048-010-0248-3. [DOI] [PubMed] [Google Scholar]
- Tarditi A, Camurri A, Varani K, Borea PA, Woodman B, Bates G, Cattaneo E, Abbracchio MP. Early and transient alteration of adenosine A2A receptor signaling in a mouse model of Huntington disease. Neurobiol Dis. 2006;23:44–53. doi: 10.1016/j.nbd.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Varani K, Rigamonti D, Sipione S, Camurri A, Borea PA, Cattabeni F, Abbracchio MP, Cattaneo E. Aberrant amplification of A2A receptor signaling in striatal cells expressing mutant huntingtin. FASEB J. 2001;15:1245–1247. [PubMed] [Google Scholar]
- Varani K, Abbrachio MP, Cannella M, Cislaghi G, Giallonardo P, Mariotti C, Cattabriga E, Borea PA, Squitieri F, Cattaneo E. Aberrant A2A receptor function in peripheral blood cells in Huntington’s disease. FASEB J. 2003;17:2148–2150. doi: 10.1096/fj.03-0079fje. [DOI] [PubMed] [Google Scholar]
- von Hörsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Bader M, Pabst R, Kobbe P, Krotova J, Stiller D, Kask A, Vaarmann A, Rathke-Hartlieb S, Schulz JB, Grasshoff U, Bauer I, Vieira-Saecker AM, Paul M, Jones L, Lindenberg KS, Landwehrmeyer B, Bauer A, Li XJ, Riess O. Transgenic rat model of Huntington’s disease. Hum Mol Genet. 2003;12:617–624. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Leavitt BR, Hayden MR. Huntington disease: new insights on the role of huntingtin cleavage. J Neural Transm Suppl. 2000;58:1–17. doi: 10.1007/978-3-7091-6284-2_1. [DOI] [PubMed] [Google Scholar]
- Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]