

Abstract
Herein we report the discovery and SAR of a novel series of M1 agonists based on the MLPCN probe, ML071. From this, VU0364572 emerged as a potent, orally bioavailable and CNS penetrant M1 agonist with high selectivity, clean ancillary pharmacology and enantiospecific activity.
Keywords: Muscarinic acetylcholine receptor 1, mAChR1 (M1), ML071, Allosteric agonist
The muscarinic acetylcholine receptors, mAChRs or M1–M5, are members of the class A G protein-coupled receptors (GPCRs) that mediate a broad range of actions of the neurotransmitter acetylcholine (ACh) in the central nervous system and other tissues.1–3 Previous attempts to develop compounds that are highly selective for M1 have failed because of the high conservation of the orthosteric ACh binding site and difficulty in developing truly specific compounds, not only for M1 (versus M2–M5) but also other biogenic amine receptors (BARs).4–6 Despite these major shortcomings, multiple orthosteric M1 ‘preferring’ agonists have provided proof of concept in Phase II and III clinical trials for both Alzheimer’s disease (AD) and schizophrenia, generating great enthusiasm for selective M1 activation.7–14
Current efforts are focused on the selective activation of M1 by targeting less conserved allosteric sites, and this approach is proving highly successful for multiple GPCRs.4,5,15,16 Allosteric and/or bi-topic M1 partial agonists first appeared, such as 1–4, with improved mAChR selectivity in many instances, but a general lack of selectivity versus BARs (D2, 5HT2c, 5HT2B, opiate, etc.) and poor DMPK properties precluded their utility as tools to probe selective M1 activation in vivo (Fig. 1).6,17–20 Recently, numerous additional M1 full and partial agonists have been reported with improved properties and efficacy in cognition models.21–23 Positive allosteric modulators (PAMs) from three structural classes, 5–7, have also been reported, with exquisite selectivity for M1 versus M2–M5 and BARs, allowing limited in vivo proof of concept studies to be conducted.24–26,28,29,31,32 For all of these PAMs, both DMPK properties and CNS penetration are barely adequate for in vivo studies; 24–26,28,29,31,32 however, two scaffolds variants of the BQCA PAM scaffold have overcome the DMPK issues and afforded PAMs with good properties and CNS exposure.27,30
Figure 1.

Prototypical ‘M1-preferring’ allosteric (bi-topic) agonists 1–4, and M1 PAM series 5–7.
Based on the need for additional M1 tools, we recently performed a functional M1 HTS of the 65,000 compound MLPCN library using a high-expressing M1 rat CHO cell line.33,34 From this effort, we identified M1 agonists, antagonists and PAMs, which were quickly optimized to afford the M1 selective antagonist ML012 (VU0255035),33 M1 PAMs ML137 (VU0366369)31 and ML169 (VU0405562)32 as well as the M1 agonist ML071 (VU0357017).34 ML071 (8) proved to be a highly selective M1 partial agonist (EC50 = 200 nM, 81% ACh Max, >30 µM versus M2–M5) with clean ancillary pharmacology (no activity greater than 50% in a 10 µM radioligand binding panel of 68 GPCRs, ion channels and transporters, including the BARs) and favorable DMPK properties and CNS penetration (Fig. 2).31 ML071 demonstrated that selective M1 activation potentiated NMDA receptor currents, provided a significant increase in soluble APP (sAPPα) in cell culture, and dose-dependently reversed scopolamine-induced disruption of contextual fear conditioning responses.34 However, ML071 also displayed functional D2 antagonism (IC50 = 4.5 µM), which we hoped to eliminate through chemical optimization. As previously detailed, SAR for ML071 was shallow, with few modifications tolerated.34 In this Letter, we describe the introduction of cyclic constraints to impart improved potency, efficacy and DMPK properties (Fig. 2).
Figure 2.

Prior SAR for ML071 and proposal to introduce cyclic constraints.
A number of cyclic constraints ((R)- and (S)-3-aminopyrroldines (9–12), piperazine (13), [2.2.1] (14) and [3.3.0] (15) congeners) were synthesized (Fig. 3A) and evaluated, affording less than 30% M1 activation at 10 µM (Fig. 3B). However, the (R)-aminopiperidine constraint 17 (VU0364572) afforded full activation of M1, while the (S)-enantiomer 16 was inactive, providing the first reported example of enantioselective M1 activation. VU0364572 proved to be highly selective for M1 (Fig. 3C) with an EC50 of 110 nM and ~95% ACh Max.
Figure 3.

A) Structures of cyclic constrained analogs of 8. B) 10 µM M1 single point screen of cyclic constraint variants of ML071. C) Full CRCs at M1–M5 for VU0364572 (17).
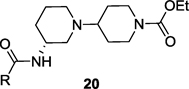
This finding led us to synthesize libraries of analogs around both 16 and 17 wherein we varied the amide moiety; interestingly, all (S)-enantiomers 16 were inactive (M1 EC50s >10 µM). Analogs 17 were synthesized according to Scheme 1. As shown in Table 1, SAR was shallow, as with ML071,34 with only 12 of 36 analogs possessing M1 potencies below 5 µM; moreover, 17 proved to be the best compound within this series in terms of M1 potency, ACh Max and mAChR selectivity. Efforts now focused on full characterization of VU0364572, 17.
Scheme 1.

Reagents: (a) (R)-3-amino-3-N-Boc-piperidine, NaBH(OAc)3, DCE, 95%; (b) HCl, dioxane, rt, 96%; (c) RCOCl, DIEA, DCM, rt, 65–95%.
Table 1.
Structures and activities of M1 agonist analogs 20a–k.
 | ||||
|---|---|---|---|---|
| Cmpd | R | M1 EC50 (µM)a |
ACh Max (%)a |
M2–M5 EC50s (µM)a |
| 8 | 0.2 | 81 | >30 | |
| 17 | 0.11 | 96 | >30 | |
| 20a | 0.06 | 77 | >10 | |
| 20b | 0.13 | 91 | >10 | |
| 20c | 0.14 | 92 | >10 | |
| 20d | 0.25 | 91 | >10 | |
| 20e | 0.31 | 84 | >10 | |
| 20f | ||||
| 20g | 0.89 | 41 | >10 | |
| 20h | 0.69 | 44 | >10 | |
| 20i | 1.3 | 61 | >10 | |
| 20j | 0.96 | 63 | >10 | |
| 20k | 2.6 | 57 | >10 | |
| 5.0 | 35 | >10 | ||
Average of at least three determinations in our rat HTS high-expressing CHO cell line
We then evaluated VU0364572 (17) in a radioligand binding panel of 68 GPCRs, ion channels and transporters (including all the BARs),35 and no significant activities were reported (no inhibition >30% @10 µM). Importantly, 17 showed minimal activity at hERG (22%@10 µM), despite possessing a classical hERG pharmacophore. Furthermore, based on the previously observed weak functional D2 antagonism of ML071 (D2 IC50 = 4.5 µM), we evaluated 17, and were delighted to find no functional activity at the D2 receptor (Fig. 4). Based on this, VU0364572 is a highly selective M1 agonist with exceptionally clean ancillary pharmacology, and a valuable small molecule tool to dissect the role of direct and selective M1 activation.1–20
Figure 4.

A) CRC for ML071 for D2 antagonism, affording an IC50 of 4.5 µM. B) CRC for VU0364572 for D2 antagonism, affording an IC50 > 30 µM.
Further in vitro characterization followed, evaluating the ability of 17 to shift APP processing towards the non-amyloidogenic pathway. Employing our standard model20,22,28,29 in TREx293-hM1 cells, carbachol (CCh) (10 µM) affords a significant increase in soluble APP (sAPPα), whereas an identical concentration of 17 provides a more robust increase (~3-fold) in sAPPα. Based on this data, selective activation of M1, via 17, may have a disease modifying role in AD.
Activation of NMDA receptor currents by M1 is postulated to play a critical role in the cholinergic regulation of cognitive function and circuitry that underlie the efficacy of mAChR agonists in schizophrenia (the NMDA receptor hypofunction hypothesis of schizophrenia)36 and AD. As shown in Figure 5, VU0364572 (17), was found to potentiate NMDA receptor currents in hippocampal CA1 pyramidal cells, further validating selective M1 activation as a means to promote synaptic plasticity.
Figure 5.

VU0364572 potentiates NMDA receptor currents in hippocampal CA1 pyramidal cells. A) Representative whole cell traces of NMDA-evoked currents and B) time course of normalized amplitude of NMDAR currents before, during and after application of 30 µM VU0364572 (17). Notably, even after washout, the potentiation persists for 10 min. Error bars represent mean ± SEM for five independent determinations.
The in vitro data was tremendously exciting, prompting the evaluation of 17 in our tier 1 DMPK battery. 17 displayed low plasma protein binding for both human (fu = 5.8%) and rat fu = 14.9%) and had a clean CYP profile (3A4, 2C9, 1A2 and 2D6; IC50 >25 µM). Intrinsic clearance experiments (rat CLINT = 23.4 mL/min/kg and human CLINT = 11.2 mL/min/kg) suggested 17 would be a low to moderate clearance compound, and rat PK confirmed a good in vitro/in vivo correlation. A standard rat IV(1 mg/kg)/PO (10 mg/kg) study found 17 to possess a CL of 14.7 mL/min/kg, with a Vss of 0.98 L/kg and a t1/2 of 46 min. Importantly, 17 was orally bioavailable with a %F of 37 (AUCIV = 1.1 µg*hr/mL, AUCPO = 4.2 µg*hr/mL). In parallel, we performed an oral plasma:brain level (PBL) study with 17 in male Sprague-Dawley rats (Table 2). At a dose of 10 mg/kg with a 90 minute endpoint, 17 achieved an average BrainAUC/PlasmaAUC of 1.35, providing excellent CNS exposure.
Table 2.
10 mg/kg Oral Plasma:Brain Level Study with 17.
| Dose (mg/Kg) |
Animal | Plasma (µM) |
Brain (µM) |
Brain:Plasma |
|---|---|---|---|---|
| 10 | 1 | 1.45 | 2.23 | 1.54 |
| 10 | 2 | 1.62 | 1.89 | 1.17 |
As it became time to profile 17 on human M1–M5, our research team had moved away from the high-expressing HTS lines and developed both rat and human M1–M5 with expression more closely resembling native expression levels. In these new cell lines, both 17 and ML071 experienced an ~10-fold right shift in potency (EC50s of 1.3 µM and 2.3 µM, respectively) with slightly lower ACh Max, yet still remained highly selective (M2–M5 EC50s >30 µM). As M1 expression levels vary amongst neuronal tissues and brain regions, compounds such as 17 may behave as full agonists in one population and as weak, partial agonists in another. The ramifications of this, and further in depth in vitro and in vivo pharmacological studies are in progress and will be reported shortly.37
In summary, the chemical lead optimization of the MLPCN M1 agonist probe ML071 (VU0357017), led to the discovery of VU0364572 (17), an M1 agonist with high selectivity (versus M2–M5 as well as the BARs), exceptional PK and brain exposure, robust effects on APP processing and potentiation of NMDA receptor currents and enantiospecific M1 activation. Further in depth in vitro and in vivo pharmacological studies with VU0364572 (17) are in progress and will be reported shortly.
Acknowledgments
The authors thank the NIH and NIMH for support of our M1 muscarinic program (1RO1MH082867-01). Vanderbilt is a Specialized Chemistry Center within the MLPCN (U54MH084659), and ML071 is an MLPCN probe, freely avaialble upon request.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Levey AI. Life Sci. 1993;52:441–448. doi: 10.1016/0024-3205(93)90300-r. [DOI] [PubMed] [Google Scholar]
- 2.Abrams P, Andersson KE, Buccafusco JJ, Chapple C, Chet de Groat W, Fryer AD, Kay G, Laties A, Nathanson NM, Pasricha PJ, Wein A. Br. J. Pharmacol. 2006;148:565–578. doi: 10.1038/sj.bjp.0706780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Volpicelli LA, Levey AI. Prog. Brain Res. 2004;145:59–66. doi: 10.1016/S0079-6123(03)45003-6. [DOI] [PubMed] [Google Scholar]
- 4.Conn PJ, Jones CK, Lindsley CW. Trends Pharmacol. Sci. 2009;30:148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conn PJ, Christopoulos A, Lindsley CW. Nat. Rev. Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heinrich JN, Butera JA, Carrick T, Kramer A, Kowal D, Lock T, Marquis KL, Pausch MH, Popiolek M, Sun SC, Tseng E, Uveges AJ, Mayer SC. Eur. J. Pharmacol. 2009;605:53–56. doi: 10.1016/j.ejphar.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 7.Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Am. J. Psych. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- 8.Mirza NR, Peters D, Sparks RG. CNS Drug Rev. 2003;9(2):159–186. doi: 10.1111/j.1527-3458.2003.tb00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sur C, Mallorga PJ, Wittmann M, Jacobson MA, Pascarella D, Williams JB, Brandish PE, Pettibone DJ, Scolnick EM, Conn PJ. PNAS. 2003;100:13674–13679. doi: 10.1073/pnas.1835612100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pakrasi S, Colloby SJ, Firbank MJ, Perry EK, Wyper DJ, Owens J, McKeith IG, Williams ED, O’Brien JT. J. Neurol. 2003;254:907–913. doi: 10.1007/s00415-006-0473-8. [DOI] [PubMed] [Google Scholar]
- 11.Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, Hurley DJ, Potter WZ, Paul SM. Arch. Neurol. 1997;54:465–473. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- 12.Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. (2006) M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Mol. Psych. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 14.Fisher A. Neurodegen. Diseases. 2008;5:237–240. [Google Scholar]
- 15.Lewis J, Lebois EP, Lindsley CW. Curr. Opin. Chem. Biol. 2008;12:269–280. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Bridges TM, Lindsley CW. ACS Chem. Biol. 2008;3:530–541. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 17.Langmead CJ, Fry VAH, Forbes IT, Branch CL, Christopoulos AC, Wood MD, Herdon HJ. Mol. Pharm. 2006;69:236–246. doi: 10.1124/mol.105.017814. [DOI] [PubMed] [Google Scholar]
- 18.Langmead CJ, Austin NE, Branch CL, Brown JT, Buchanan KA, Davies CH, Forbes IT, Fry VAH, Hagan JJ, Herdon JJ, Jones GA, Jeggo R, Kew JNC, Mazzali A, Melarange R, Patel N, Pardoe J, Randall AD, Roberts C, Roopun A, Starr KR, Teriakidis A, Wood MD, Whittington M, Wu Z, Watson J. Br. J. Pharmacol. 2008;154:1104–1115. doi: 10.1038/bjp.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradley SR, Lameh J, Ohrmund L, Son T, Bajpai A, Nguyen D, Friberg M, Burstein ES, Spalding TA, Schiffer HH, Tabatabaei A, McFarland K, Davis RE, Bonhaus DW. Neuropharmacology. 2010;58:365–373. doi: 10.1016/j.neuropharm.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, Peterson TE, Ansari MW, Baldwin RM, Kessler RM, Deutch AY, Lah JJ, Levey AI, Lindsley CW, Conn PJ. J. Neurosci. 2008;28:10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watt ML, Schober DA, Hitchcock S, Liu B, Chesterfield AK, McKinzie D, Felder CC. J. Pharm. Exp. Ther. 2011;338:622–632. doi: 10.1124/jpet.111.182063. [DOI] [PubMed] [Google Scholar]
- 22.Budzik B, Garzya V, Shi D, Walker G, Wooley-Roberts M, Pardoe J, Lucas A, Tehan B, Rivero RA, Langmead CJ, Watson J, Wu Z, Forbes IT, Jin J. ACS Med. Chem. Lett. 2010;1:244–248. doi: 10.1021/ml100105x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson DJ, Forbes IT, Watson SP, Garzya V, Stevenson GI, Walker GR, Mudhar HS, Flynn ST, Wyman PA, Smith PA, Murkitt GS, Lucas AJ, Mookherjee CR, Watson JM, Garlton JE, Bradford AM, Brown F. Bioorg. Med. Chem. Lett. 2010;20:5434–5438. doi: 10.1016/j.bmcl.2010.07.097. [DOI] [PubMed] [Google Scholar]
- 24.Ma L, Seager M, Wittman M, Bickel N, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danzinger A, Regan C, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Jacobson M, Sur C, Kinney G, Seabrook GR, Ray WJ. Proc. Natl. Acad Sci. USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Jadhav SB, Menon U, Christain EP, Doherty JJ, Quirk MC, Snyder DH, Levey AI, Watson ML, Nicolle MM, Lindsley CW, Conn PJ. J. Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittman M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:531–536. doi: 10.1016/j.bmcl.2009.11.100. [DOI] [PubMed] [Google Scholar]
- 27.Kuduk SD, Chang RK, Di Marco CN, Ray WJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. ACS Med. Chem. Lett. 2010;1:263–267. doi: 10.1021/ml100095k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuduk SD, Di Marco CN, Chang RK, Ray WJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. Bioorg. Med. Chem. Lett. 2010;20:2533–2537. doi: 10.1016/j.bmcl.2010.02.096. [DOI] [PubMed] [Google Scholar]
- 29.Kuduk SD, Chang RK, Di Marco CN, Ray WJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. Bioorg. Med. Chem. Lett. 2011;21:1710–1715. doi: 10.1016/j.bmcl.2011.01.094. [DOI] [PubMed] [Google Scholar]
- 30.Kuduk SD, Chang RK, Di Marco CN, Pitts DR, Greshock TJ, Ma L, Wittman M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT, Ray WJ. J. Med. Chem. 2011;54:4773–4780. doi: 10.1021/jm200400m. and references therein. [DOI] [PubMed] [Google Scholar]
- 31.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reid PR, Bridges TM, Sheffler DA, Cho HP, Lewis LM, Days E, Daniels JS, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Wood MR. Bioorg. Med. Chem. Lett. 2011;21:2697–2701. doi: 10.1016/j.bmcl.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheffler DJ, Williams R, Bridges TM, Lewis LM, Xiang Z, Zheng F, Kane AS, Byum NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Conn PJ. Mol. Pharmacol. 2009;76:356–368. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lebois EP, Bridges TM, Dawson ES, Kennedy Jp, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. ACS Chemical Neurosci. 2010;1:104–121. doi: 10.1021/cn900003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.For information on the Ricerca Lead Profiling Screen, see: www.ricerca.com
- 36.Lindsley CW, Shipe WD, Wolkenberg SE, Theberge CR, Williams DL, Jr, Sur C, Kinney GG. Curr. Topics in Med. Chem. 2006;8:771–784. doi: 10.2174/156802606777057599. [DOI] [PubMed] [Google Scholar]
- 37.Manuscripts in preparation.