

Abstract
Heterochromatin assembly at fission yeast centromeres involves a self-reinforcing loop mechanism wherein chromatin-bound RNAi factors facilitate targeting of Clr4–Rik1 methyltransferase. However, the initial nucleation of heterochromatin has remained elusive. We show that cells lacking Mlo3, a protein involved in mRNP biogenesis and RNA quality control, assemble functional heterochromatin capable of promoting chromosome segregation in RNAi deficient cells. Heterochromatin restoration is linked to RNA surveillance because loss of Mlo3-associated TRAMP also rescues heterochromatin defects of RNAi mutants. Remarkably, mlo3Δ, which causes accumulation of bidirectional repeat-transcripts, restores Rik1 enrichment at repeats, and triggers de novo heterochromatin formation in the absence of RNAi. RNAi-independent heterochromatin nucleation occurs at selected euchromatic loci that show upregulation of antisense RNAs in mlo3Δ cells. We find that the exosome RNA degradation machinery acts parallel to RNAi to promote heterochromatin formation. These results suggest that RNAi-independent mechanisms exploit transcription and non-coding RNAs to nucleate heterochromatin.
Heterochromatin is linked to numerous cellular functions including transcriptional regulation, chromosome segregation, and suppression of recombination1,2. Heterochromatic regions show a distinctive pattern of histone modifications. In addition to deacetylation of histones, heterochromatin in many eukaryotes including the fission yeast Schizosaccharomyces pombe is marked by methylation of histone H3 at lysine 9 (H3K9me)3. These modifications are critical for recruitment of heterochromatin factors including HP1 proteins and for the assembly of repressive chromatin structures1,4.
In S. pombe, heterochromatin is preferentially enriched across large chromosomal domains at centromeres, subtelomeres and the mating type locus3. These heterochromatin domains contain dg and/or dh repeats that are transcribed by RNA polymerase II (RNAPII)1,2. Transcripts generated from dg/dh repeats are processed into siRNAs by the RNAi machinery including Argonaute (ago1), Dicer (dcr1) or RNA-dependent RNA polymerase (rdp1)1,2. Mutations in RNAi factors cause defects in H3K9me at centromeres and loss of siRNAs1. siRNAs are bound by Ago1, a subunit of the RNA-induced transcriptional silencing (RITS) complex that is composed of two additional proteins: Chp1 and Tas31. siRNA-bound Ago1, together with binding of the H3K9me by Chp1, assist in the localization of RITS to heterochromatin5,6. RITS facilitates targeting of Clr4, a homolog of mammalian SUV39h, which methylates H3K9 at heterochromatic loci7,8. Clr4 exists in a multisubunit complex, ClrC, which among other factors contains Rik1 that is critical for RNAi-dependent loading of the complex onto transcribed repeats8. The results showing the involvement of RNAi in targeting H3K9me and the requirement of heterochromatin factors for generation of siRNA have revealed the existence of a positive feedback loop in the assembly of heterochromatin5,9.
H3K9me also recruits HP1 family proteins Chp2 and Swi6, which in turn promote the localization of various effectors including factors involved in chromosome dynamics and gene silencing1. Swi6 is required for the localization of cohesin-loading complex involved in proper segregation of chromosomes10. Chp2 and Swi6 also associate with Snf2–HDAC Repressor Complex (SHREC), Asf1–HIRA histone chaperone and Clr6 histone deacetylase (HDAC) complexes involved in transcriptional silencing10–13. Heterochromatin precludes RNAPII accessibility at target loci, but paradoxically, RNAPII transcription of dg/dh repeats is required to generate siRNA precursors. Centromeric repeats are transcribed preferentially during the S phase of the cell cycle when heterochromatin is more amenable to transcription14,15. Apart from generating siRNA precursors, RNAPII transcription may have more direct roles in heterochromatin formation. Indeed, mutations in RNAPII subunits and RNA splicing factors impair heterochromatic silencing16–18.
RNAPII transcription has been shown to integrate multiple aspects of nuclear metabolism. Elongating RNAPII recruits chromatin-modifying activities to help remodel chromatin19. RNAPII machinery also recruits RNA processing factors, including factors which promote mRNP biogenesis and RNA quality control20,21. RNA quality control in the nucleus is monitored by multiple factors such as the TRAMP complex. TRAMP containing Cid14, a member of the Trf4 family of polyA polymerases, channels RNAs into degradation pathways including the exosome with 3′–5′ exonucleolytic activity22. Both the exosome and TRAMP function to degrade centromeric transcripts in S. pombe23,24.
Despite major advances, the exact cascade of events that leads to the initial nucleation of heterochromatin at centromeric repeats has remained unclear. It has been argued that a new class of small RNAs, termed primal RNAs, which require Ago1 for their biogenesis, nucleate heterochromatin, and that ago1Δ shows H3K9me levels comparable to clr4Δ cells25. In this study, we sought to explore mechanisms that nucleate heterochromatin structures. Our analyses have uncovered an RNAi-independent pathway that exploits RNAPII transcription and non-coding RNAs (ncRNAs) to nucleate heterochromatin at centromeres and other parts of the genome. Factors involved in co-transcriptional processes including that affect RNAPII processivity influence RNAi-independent heterochromatin assembly. We provide evidence that the exosome RNA degradation machinery acts parallel to the RNAi to mediate heterochromatin formation.
RESULTS
Loss of Mlo3 restores centromeric silencing in RNAi mutants
Mutations in RNAPII lead to defective heterochromatic silencing at centromeres16,17. Given that RNAPII is linked to heterochromatin modifications, we investigated if RNAPII coupled processes affect heterochromatin assembly. Loss of Mlo3, a homolog of S. cerevisiae Yra1 and metazoan Ref or Aly that acts at the interface of RNAPII transcription and RNA metabolism21,26, restores centromeric silencing in RNAi deficient cells. Whereas ago1Δ alleviated silencing of a ura4+ reporter inserted at an outer centromeric repeat region (otr1R::ura4+), simultaneous deletions of mlo3 and ago1 restored centromeric silencing (Fig. 1a). The observed suppression required heterochromatin machinery because mlo3Δ failed to suppress the silencing defect in clr4Δ mutant (Supplementary Fig. 1).
Figure 1. mlo3Δ restores functional heterochromatin at centromeres in ago1Δ mutant.
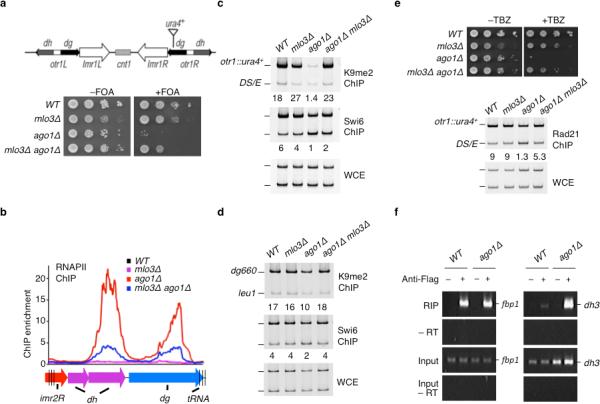
(a) mlo3Δ suppresses the silencing defect at otr1R::ura4+ in ago1Δ cells. Location of ura4+ reporter inserted within pericentromeric region is shown. Serial dilutions of the indicated strains were spotted onto non-selective (-FOA) or counter-selective FOA-containing (+FOA) media to assay ura4+ expression. FOA, 5-fluoroacetic acid. (b) mlo3Δ decreases RNAPII occupancy at centromeric repeats in ago1Δ cells. ChIP-chip using Ser2phospho RNAPII antibody was used to assay RNAPII occupancy. RNAPII levels are plotted in alignment with the right pericentromeric region of cen2. (c) mlo3Δ restores centrometric heterochromatin in ago1Δ cells. ChIP analysis of H3K9me and Swi6 at otr1R::ura4+. DNA isolated from immunoprecipitated chromatin (ChIP) or whole-cell crude extracts (WCE) was analyzed by PCR. Relative fold enrichments depicting the ratios of signals at otr1R::ura4+ locus relative to the euchromatic ura4DS/E (DS/E) locus, between ChIP and WCE, are shown underneath each lane. (d) mlo3Δ restores heterochromatin at dg repeats in ago1Δ cells. H3K9me2 and Swi6 enrichments at dg repeats relative to leu1 were determined by ChIP. (e) mlo3Δ suppresses TBZ sensitivity and restores cohesin localization at centromeres in ago1Δ mutant. Localization of cohesin subunit Rad21 (Rad21-HA) at otr1R::ura4+ was assessed by ChIP (bottom). (f) Mlo3 interacts with dh transcripts. Interaction of Mlo3 with the fbp1 transcripts, used as a control, and dh transcripts was determined by RNA-IP. RNA isolated from immunoprecipitated Mlo3-Flag (Flag RIP) or whole cell extract (input) was analyzed by RT-PCR. –RT, no reverse transcription.
These results suggested that loss of Mlo3 promotes heterochromatic silencing in RNAi deficient cells. We next investigated whether mlo3Δ restores transcriptional repression and localization of heterochromatin factors at centromeres. Chromatin immunoprecipitation (ChIP) analyses across centromere 2 (cen2) showed that the observed changes in silencing correlated with marked reduction in RNAPII occupancy in mlo3Δ ago1Δ cells, as compared to ago1Δ (Fig. 1b). More importantly, mlo3Δ restored H3K9me and Swi6 localization at otr1R::ura4+ and endogenous centromeric repeats in ago1Δ mutant (Fig. 1 c–d). mlo3Δ also restored silencing and heterochromatin formation at centromeres in dcr1Δ mutant (Supplementary Fig. 2a). The rescue of H3K9me in RNAi mutants was not due to changes in histone H3 occupancy (Supplemental Fig. 3a) or restoration of siRNAs (Supplementary Fig. 3b). Together, these results demonstrate that mlo3Δ suppresses heterochromatin defects of RNAi mutants.
mlo3Δ restores functional heterochromatin in RNAi mutants
Heterochromatin facilitates the loading of cohesin essential for sister chromatid cohesion10,27. Defective heterochromatin in RNAi mutants causes impaired centromere cohesion, resulting in chromosome missegregation and sensitivity to the microtubule destabilizing drug thiabendazole (TBZ)28,29. To test whether loss of Mlo3 in RNAi mutant cells restores functional heterochromatin, we measured the TBZ sensitivity of single and double mutants. As expected, cells carrying ago1Δ or dcr1Δ showed severe sensitivity to TBZ, consistent with defective chromosome segregation in these mutants28,29. Combining mlo3Δ with ago1Δ or dcr1Δ suppressed the TBZ sensitivity of RNAi mutants (Fig. 1e, Supplementary Fig. 2a). ChIP analyses showed that loss of TBZ sensitivity correlates with a partial restoration of cohesin localization at centromeres in mlo3Δ ago1Δ mutant (Fig. 1e). Therefore, heterochromatin assembled upon loss of Mlo3 is functional, capable of supporting proper segregation of chromosomes in RNAi deficient cells.
Mlo3 interacts with centromeric transcripts
To determine whether Mlo3 directly functions at centromeres, we tested if it interacts with centromeric transcripts. As expected for a factor involved in mRNP formation26,30, Mlo3 interacted with a euchromatic gene (fbp1) transcript (Fig. 1f). Importantly, Mlo3 also interacted with dh transcript (Fig. 1f), consistent with results of ChIP analyses showing Mlo3 enrichment at transcribing centromeric repeats30. This interaction was greatly enhanced in ago1Δ cells. Therefore, in addition to euchromatic genes, Mlo3 targets heterochromatic repeat transcripts. These data argue that Mlo3 functions at centromeric repeats and that restoration of heterochromatin in RNAi mutant cells may be coupled to its role in co-transcriptional processing of centromeric transcripts30.
TFIIS modulates RNAi-independent heterochromatin assembly
We wondered whether loss of Mlo3, like Yra1, causes defective RNAPII transcription. To test this, we constructed mlo3Δ clr3Δ double mutant. Mutant cells lacking SHREC subunit Clr3 show marked increase in RNAPII occupancy at centromeric repeats10,11,31. Combining mlo3Δ with clr3Δ resulted in small decrease in RNAPII as compared to clr3Δ, albeit levels of H3K9me or Swi6 at repeat elements were comparable in clr3Δ and clr3Δ mlo3Δ (Supplementary Fig. 4a–c). The change in RNAPII levels led us to wonder if defective RNAPII transcription is partially responsible for restoration of heterochromatin in RNAi mutants. We tested this possibility by deleting tfs1 gene encoding the S. pombe homolog of TFIIS, a factor known to affect RNAPII processivity32. Deletion of tfs1, which led to 6-azauracil (6-AU) sensitivity (Fig. 2a)33 and changes in the distribution of RNAPII at body of genes (Supplementary Fig. 3c), resulted in variegated suppression of silencing defects in ago1Δ and dcr1Δ mutants (Fig. 2b and Supplementary Fig. 2b). This suppression was more pronounced in M mating-type (mat1-Msmt0) cells and resulted in decreased RNAPII occupancy at centromeric repeats (Fig. 2c). tfs1Δ-dependent silencing required Clr4 (Supplementary Fig. 1) and was accompanied by an increase in levels of H3K9me and Swi6 at both otr1R::ura4+ and centromeric repeats (Fig. 2d–e and Supplementary Fig. 2b). The increase in H3K9me was not due to changes in histone H3 occupancy or restoration of siRNAs (Supplementary Fig. 3a–b). tfs1Δ suppressesed TBZ sensitivity and partially restored cohesin localization at centromeres in RNAi mutants (Fig. 2f–g and Supplementary Fig. 2b).
Figure 2. tfs1Δ restores centromeric heterochromatin in ago1Δ cells.
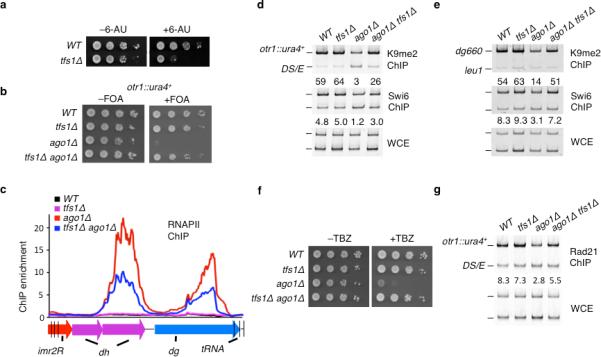
(a) tfs1Δ renders cells sensitive to 6-AU. (b) tfs1Δ suppresses the silencing defect at otr1R::ura4+ in ago1Δ cells. Serial dilutions of WT and mutant strains carrying mat1-Msmt0 mating-type allele were spotted onto the indicated media to assay otr1R::ura4+ expression. (c) tfs1Δ decreases RNAPII occupancy at centromeric repeats in ago1Δ cells. RNAPII occupancy at dh/dg repeats in centromere 2 was determined by ChIP-chip using Ser2 phospho RNAPII antibody. (d) tfs1Δ restores heterochromatin at otr1R::ura4+ in ago1Δ cells. Relative fold enrichments of H3K9me and Swi6 at otr1R::ura4+ were determined by ChIP. e, tfs1Δ restores heterochromatin at dg repeats in ago1Δ cells. H3K9me and Swi6 enrichments at dg repeats were determined by ChIP. (f) tfs1Δ suppresses TBZ sensitivity of ago1Δ cells. (g) tfs1Δ restores cohesin localization to centromeres. Localization of Rad21-HA at otr1R::ura4+ was assessed by ChIP.
These results suggest that cells with compromised RNAPII transcription rescue heterochromatin defects caused by loss of RNAi machinery. Consistently, we found that loss of deubiquitylating enzyme Ubp3, which causes elevated levels of RNAPII ubiquitylation and degradation34, partially suppressed silencing and heterochromatin defects of ago1Δ mutant (Supplementary Fig. 5a–b).
mlo3 and tfs1 differentially affect heterochromatin assembly
Loss of Clr3 and RNAi factors causes a dramatic loss of H3K9me across pericentromeric domains (Fig. 3a–d and Supplementary Fig. 6a–b)31. To gain more insight into effects of mlo3Δ and tfs1Δ on heterochromatin formation in the absence of RNAi, we investigated their effects on heterochromatin modifications in clr3Δ ago1Δ double mutant cells. tfs1Δ failed to restore H3K9me at otr1R::ura4+ in clr3Δ ago1Δ cells (Fig. 3a). In contrast, mlo3Δ resulted in considerable restoration of H3K9me at centromeres in clr3Δ ago1Δ cells (Fig. 3b). These differences in H3K9me were not limited to otr1R::ura4+. ChIP-chip analyses across centromere 2 showed that mlo3Δ, but not tfs1Δ, restored H3K9me across the entire pericentromeric region in clr3Δ ago1Δ cells (Fig. 3c–d, Supplementary Fig. 6a–b). mlo3Δ also decreased in RNAPII occupancy across pericentromeric domains in clr3Δ ago1Δ cells (Supplementary Fig. 6c), while tfs1Δ had only a minor effect (Supplementary Fig. 6d).
Figure 3. tfs1Δ and mlo3Δ differentially suppress heterochromatin defects in clr3Δago1Δ double mutant cells.
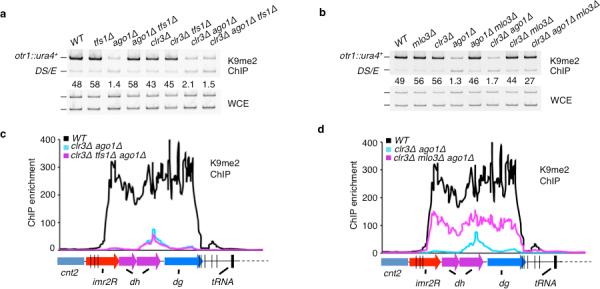
(a–d) mlo3Δ, but not tfs1Δ, restores H3K9me at centromeres in clr3Δ ago1Δ cells. (a–b) Relative fold enrichments of H3K9me at otr1R::ura4+ were determined by ChIP. (c–d) H3K9me levels across pericentromeric domains of cen2 as determined by ChIP-chip.
Differences in mlo3Δ and tfs1Δ mutants were also evident in their effects on hairpin RNA-triggered heterochromatin formation. Despite the prominent role of RNAi in silencing of centromeric repeats, this pathway is constrained and rarely targets detectable levels of heterochromatin in trans35,36. Expression of hairpin complementary to the trp1+ (trp1-HP) failed to elicit H3K9me in tfs1Δ cells but induced H3K9me both in cis and in trans in mlo3Δ cells, dependent on RNAi (Supplementary Fig. 7a–c). Treating cells with 6-AU, which affects RNAPII transcription, caused increased H3K9me both in mlo3Δ and tfs1Δ cells but again the effect was stronger in mlo3Δ (Supplementary Fig.7a–c). Together, these data highlight differential effects of tfs1Δ and mlo3Δ on heterochromatin formation at centromeres and at an ectopic site. Moreover, these analyses suggest that additional RNAi and SHREC independent mechanism(s) nucleate heterochromatin at centromeres.
cid14Δ rescues heterochromatin defects of ago1Δ mutant
Mlo3 forms a complex with TRAMP, which is involved in RNA surveillance and degradation of centromeric transcripts22,24,30. We therefore investigated whether mlo3Δ-mediated suppression of heterochromatin defects in RNAi mutants is linked to defects in RNA surveillance. Remarkably, loss of Cid14 subunit of TRAMP affects RNAi-independent heterochromatin formation in a manner similar to mlo3Δ. Loss of Cid14 restored H3K9me at otr1R::ura4+ and centromeric repeats in ago1Δ mutant (Fig. 4a–b). Moreover, cid14Δ suppressed the silencing defect caused by ago1Δ, as indicated by reduction in the levels of dg/dh transcript in cid14Δ ago1Δ as compared to ago1Δ (Fig. 4c). These results suggest that defects in RNA surveillance mechanisms involving Mlo3-associated TRAMP, promote heterochromatin formation independent of RNAi.
Figure 4. Loss of RNA surveillance factor TRAMP restores centromeric heterochromatin in ago1Δ cells.
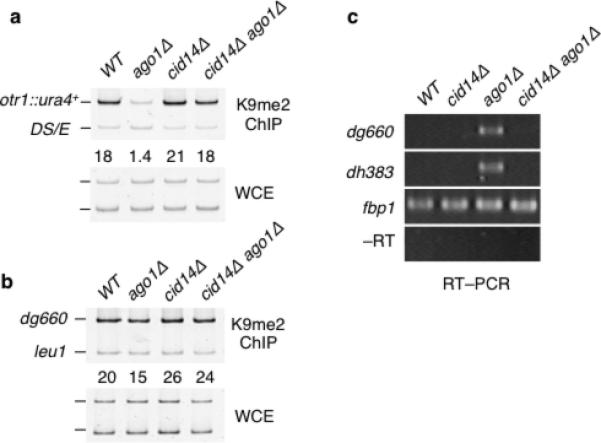
(a–b) cid14Δ restores heterochromatin at otr1R::ura4+ and dg repeats in ago1Δ cells. H3K9me levels at otr1R::ura4+ and dg were determined by ChIP. (c) cid14Δ suppresses centromeric silencing defect in ago1Δ cells. RT-PCR analysis of dg and dh transcripts in the indicated strains is shown. fbp1 transcripts were assayed as an amplification control.
Exosome acts parallel to RNAi to nucleate heterochromatin
As mentioned above, Mlo3–TRAMP channels centromeric transcripts to downstream-acting RNA degradation factors including the exosome24,30. However, these factors share a complex genetic relationship indicative of their diversified functions. Unlike single mutants, double mutants containing null alleles of Mlo3–TRAMP and the exosome subunit rrp6 show synthetic lethality (our unpublished data). In light of these observations and previous results showing that aberrant RNAs sequestered near site of transcription are degraded by the exosome37,38, it was of interest to investigate whether the loss of Rrp6 affects centromeric heterochromatin. Northern and RT-PCR analyses using single and double mutants showed a large increase in centromeric repeat transcripts in rrp6Δ ago1Δ mutant as indicated by both Northern blot and RT-PCR analyses (Fig. 5a–b). This result is in marked contrast to the restoration of silencing observed in cid14Δ ago1Δ mutant (Fig. 4). We next investigated whether rrp6Δ alone or in combination with ago1Δ affects centromeric heterochromatin assembly. Unlike mlo3Δ and cid14Δ, combining rrp6Δ with ago1Δ largely abolished H3K9me levels at otr1R::ura4+ and dg repeats (Fig. 5c). Furthermore, ChIP-chip showed severe cumulative loss of H3K9me across the entire pericentromeric domain in rrp6Δ ago1Δ mutant, as compared to rrp6Δ or ago1Δ (Fig. 5d). Together, these data reveal the nuclear exosome acts parallel to RNAi to mediate heterochromatin assembly at centromeres.
Figure 5. Rrp6 acts parallel to RNAi to mediate heterochromatin formation and silencing at centromeres.
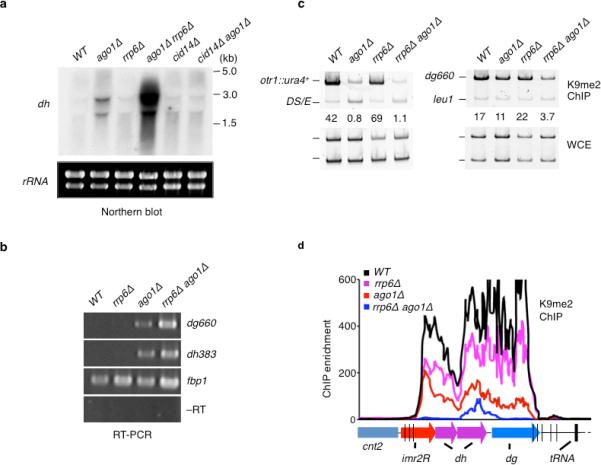
(a) Cid14 and Rrp6 differentially affect dh expression in ago1Δ cells. Northern blot analysis of dh transcripts in WT and mutant cells. rRNA was used as a loading control. (b) rrp6Δ and ago1Δ cause cumulative increase in dg and dh transcript levels. RT-PCR analysis of dg and dh transcript levels in the indicated strains is shown. fbp1 transcripts were assayed as an amplification control. (c) Loss of Rrp6 in ago1Δ cells severely affects H3K9me at centromeres. H3K9me enrichment at otr1R::ura4+ and dg repeats were determined by ChIP. (d) Deletions of rrp6 and ago1 cause cumulative loss in H3K9me across pericentromeric domains, as determined by ChIP-chip.
De novo heterochromatin assembly in mlo3Δ cells
mlo3Δ restored H3K9me both in ago1Δ and clr3Δ ago1Δ cells. We next tested whether mlo3Δ, which causes accumulation of transcripts in the nucleus (Supplementary Fig. 8), can trigger de novo heterochromatin assembly in the absence of RNAi. For this purpose, we employed strains that carry either clr4Δ alone or in combination with ago1Δ or ago1Δ mlo3Δ. Since Clr4 is the sole H3K9 methyltransferase in S. pombe, these mutant strains lack H3K9me (Fig. 6a). We transformed the mutant strains with a plasmid containing clr4+ and monitored H3K9me levels at centromeres by ChIP. Introduction of clr4+ led to restoration of H3K9me at centromeres in clr4Δ single mutant but not in clr4Δ ago1Δ double mutant (Fig. 6a). Remarkably, H3K9me could be readily detected at centromeres in clr4Δ ago1Δ mlo3Δ cells upon introduction of the clr4+ (Fig. 6a). These data demonstrate that loss of Mlo3, involved in processing of centromeric transcripts30, triggers de novo heterochromatin assembly at centromeres, independent of RNAi.
Figure 6. mlo3Δ restores Rik1 enrichment at centromeric repeats and triggers de novo heterochromatin formation in the absence of RNAi.
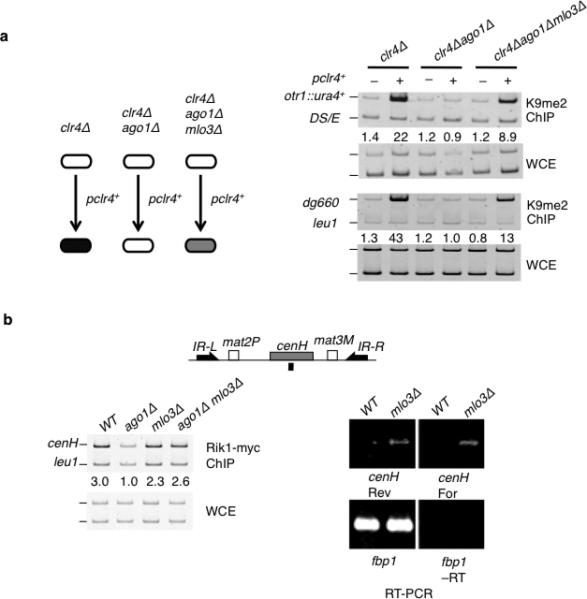
(a) mlo3Δ induces establishment of heterochromatin at centromeres in the absence of Ago1. Indicated mutant strains were transformed with a plasmid containing clr4+ gene. Levels of H3K9me were assayed by ChIP at otr1R::ura4+ and dg repeats in the indicated strains. (b) mlo3Δ causes increased accumulation of bidirectional transcripts and restores Rik1 ChIP enrichment at cenH in ago1Δ mutant. Schematic representation indicating the location of cenH and primers (black bar) used is shown (upper). ChIP analysis of Rik1-myc at cenH (left). RT-PCR analysis of cenH transcripts (right). fbp1 transcripts were assayed as an amplification control.
We next tested if mlo3Δ affects localization of ClrC, which requires RNAi for its targeting to centromeric repeats7,8,14. In particular, ClrC subunit Rik1 is recruited to centromeres during S-phase, when both forward and reverse strands of centromeric repeats are transcribed14. We probed the effect of mlo3Δ on Rik1 enrichment at cenH, which is homologous to dg/dh and serves as an RNAi-dependent heterochromatin nucleation center at mat locus39,40. cenH was selected because defects in RNAi abolish Rik1 ChIP enrichment at this site without affecting heterochromatin structure nucleated by redundant mechanisms8. This regime ensures that Rik1 enrichment is not indirectly altered by changes in heterochromatin modifications in mlo3Δ. As expected, ago1Δ abolished Rik1 ChIP enrichment at cenH (Fig. 6b). However, we found that simultaneous deletion of mlo3 and ago1 restored Rik1 enrichment at cenH (Fig. 6b). Given the connection between transcript levels and Rik1 localization8, we tested if mlo3Δ affects cenH transcripts. Levels of forward and reverse transcripts originating from cenH were elevated considerably in mlo3Δ cells (Fig. 6b). Together, these results demonstrate that mlo3Δ, which causes accumulation of bidirectional cenH transcripts, bypasses the RNAi requirement for targeting Rik1.
mlo3Δ induces H3K9me at euchromatic loci
Since cells lacking Mlo3 accumulate antisense RNAs at euchromatic loci30, we wondered whether heterochromatin could be assembled at euchromatic loci in mlo3Δ cells. Our analyses showed that loss of Mlo3 induces H3K9me at selected euchromatic genes (Fig. 7a), which show increased accumulation of antisense transcripts in mutant cells (Fig. 7b). Notably, H3K9me at these loci was not abolished when mlo3Δ cells was combined with ago1Δ (Fig. 7a). Therefore, the targeting of H3K9me occurs even in the absence of RNAi (Fig. 7a). We also analyzed a locus showing overlapping sense and antisense transcripts that were unaffected by mlo3Δ. However, H3K9me could not be detected at this site (Fig. 7b). One possibility is that the retention of transcripts at transcribed loci such as observed in RNA surveillance mutants37,38, is necessary to generate signals for RNAi-independent heterochromatin formation. Regardless, it is interesting that mlo3Δ, which causes accumulation of antisense transcripts, results in heterochromatin modifications at euchromatic loci.
Figure 7. RNAi-independent heterochromatin formation occurs at euchromatic loci in mlo3Δ cells.
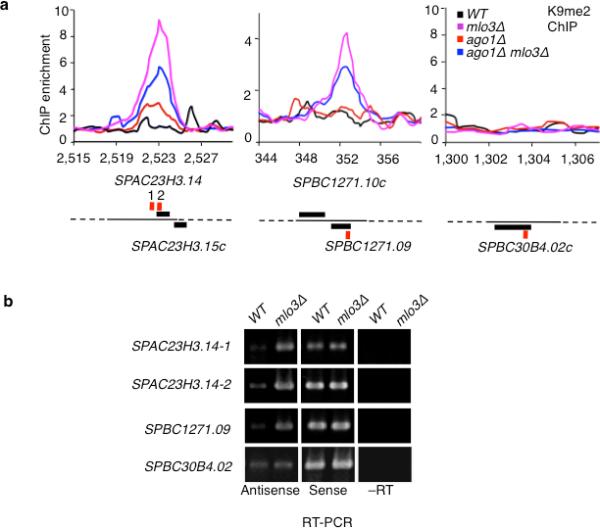
(a) Appearance of H3K9me in mlo3Δ cells correlates with the accumulation of antisense transcripts. Relative fold enrichment of H3K9me was determined by ChIP-chip in WT and mutant strains. Red bars indicate locations of primers used for RT-PCR in ‘b’. (b) RT-PCR analysis of sense and antisense at indicated loci in WT and mlo3Δ cells.
DISCUSSION
Heterochromatin assembly is a complex multistep process that involves a variety of factors1. Studies from diverse systems have suggested a prominent role for transcription and ncRNAs in heterochromatin assembly1,41,42. In S. pombe, RNAPII transcription of centromeric repeats provides scaffolds for heterochromatin formation. This process involves RNAi, which not only processes repeat transcripts but also mediates the loading of heterochromatin factors7,8. Despite the prominent role for RNAi in heterochromatin assembly, loss of RNAi factors does not completely abolish heterochromatin modifications such as H3K9me at centromeric repeats31,43,44. Similarly, evidence from other systems suggest that defects in RNAi has no major effects on heterochromatin modifications45,46, although in some of these cases transcription and ncRNAs could be involved42,47. Our analyses suggest that RNAPII transcription and ncRNAs function to target heterochromatin via an RNAi-independent mechanism.
We demonstrate that defects in cotranscriptional RNA surveillance or factors that affect RNAPII processivity bypass the RNAi requirement to assemble functional heterochromatin. Besides loss of Mlo3 or Cid14, tfs1Δ restores heterochromatin in cells lacking the RNAi machinery. Distinct mechanisms are likely responsible for mlo3Δ- or tfs1Δ-mediated suppression of heterochromatin defect in RNAi mutants, despite similarities in phenotypes. We note that mlo3Δ, but not tfs1Δ, rescues H3K9me in clr3Δ ago1Δ double mutant deficient in heterochromatin modifications at centromeres31. Also, mutations in these factors differentially affect hairpin-induced H3K9me at an ectopic site. Given that the RNAi machinery interacts with RNAPII and modulates transcription in other organisms48,49, defective RNAPII elongation could directly contribute to bypassing of RNAi. Tfs1 may affect heterochromatin by influencing processing of transcripts and/or release of RNAPII. Indeed, cells lacking Ubp3, which affects RNAPII stability34, show partial rescue heterochromatin defects in ago1Δ cells. Another possibility is that impaired transcription affects chromatin dynamics by precluding elongation-coupled turnover of histones and/or their modification state50,51. The binding of factors, such as Clr48, to residual histones decorated with H3K9me could shift the dynamic equilibrium and stabilize and/or propagate heterochromatin in cis40.
Mlo3 interacts with TRAMP30, and these factors are required for processing centromeric transcripts and antisense RNAs23,24,30. It is therefore interesting that loss of either Mlo3 or TRAMP suppresses the heterochromatin defects in ago1Δ or dcr1Δ mutants which are deficient in siRNA production. cid14Δ causes severe reduction in siRNAs without altering H3K9me levels, leading to suggestion that low level of siRNAs are sufficient to nucleate heterochromatin24. Our results suggest that cid14Δ activates RNAi-independent heterochromatin assembly pathway(s), which might also be triggered by the accumulation of RNAs produced by multiple copy sequences in other systems52. We show that the targeting of ClrC subunit Rik1 to dg/dh repeats, which normally requires RNAi8, is restored in mlo3Δ cells showing elevated levels of forward and reverse repeat transcripts. Furthermore, mlo3Δ triggers de novo targeting of heterochromatin to centromeric repeats independent of RNAi. Remarkably, loss of Mlo3 also causes RNAi-independent targeting of H3K9me at selected euchromatic loci, which show accumulation of antisense transcripts in mlo3Δ mutant. These results argue that ncRNAs generated by centromeric repeats and certain euchromatic loci assemble heterochromatin by mechanism(s) independent of RNAi. Consistent with the existence of such pathway(s) that relies of accumulation of RNAs, we have found that the exosome involved in degradation of aberrant RNAs22, acts in a pathway parallel to RNAi to mediate heterochromatin formation at centromeres.
How do accumulations of transcripts caused by loss of Mlo3–TRAMP impact heterochromatin assembly in RNAi mutants? One possibility is that ncRNAs accumulated at the sites of transcription recruit the exosome degradation machinery that in turn facilitates loading of heterochromatin factors in a manner similar to the RNAi, in which targeting of ClrC is linked to the processing of repeat transcripts in cis5. RNAi-dependent nucleation of heterochromatin involves RITS, which interacts with ClrC7,8. Indeed, RITS tethering to transcripts can induce heterochromatin formation53. However, this process still requires Dcr1, suggesting that additional siRNA-dependent steps, perhaps the generation of double stranded RNA (dsRNA) or other RNA structures, is necessary for nucleating heterochromatin. In this regard, loss of Mlo3–TRAMP, acting cotranscriptionally, could bypass RNAi requirement by generating dsRNA or yet undefined RNA signals capable of loading Clr4–Rik1. Such signals would be distinct from the recently described primal RNAs that require Ago1 for their biogenesis25. It is possible that mechanism(s) that trigger H3K9me in mlo3Δ or cid14Δ mutant are activated during S-phase when both strands of dg/dh repeat are transcribed, correlating with targeting of Rik114. Regardless of the mechanism, it is clear that an RNAi-independent pathway(s) exists that relies on ncRNAs to target heterochromatin.
Since transcription and ncRNAs have been linked to epigenetic chromatin modifications in multiple organisms including mammals42,47,54–56, our results may have general significance. In several instances, transcription and ncRNAs can modify chromatin independent of RNAi. For example, RNAPII transcription and ncRNAs trigger chromatin modifications and parental imprinting in mammals54,57 In such cases, RNAs retained in the nucleus act largely in cis. The retention of bidirectional transcripts near their transcription sites might facilitate the localization of chromatin modifying activities. To this end, widespread transcription of eukaryotic genomes41 might allow RNAPII and ncRNAs to function as a molecular sensors that specify certain genomic regions as preferential targets for repressive chromatin assembly. Such domains might include transposons that when uncontrolled can lead to genome instability.
Supplementary Material
ACKNOWLEDGEMENTS
We are thankful to D. Eick for gift of Ser2 phospho RNAPII antibody, R. Dhar and N. Krogan for strains, J. Dhakshnamoorthy, N. Komissarova and S. Mehta for helpful contributions, and members of the Grewal laboratory for discussions. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
METHODS
Yeast strains
Constructions of deletion or epitope tagged strains were done by standard PCR based methods. The tfs1Δ was a gift from N. Krogan (UCSF). Genetic crosses were used to construct double mutants. To test the requirement of RNAi in establishment of H3K9me in mlo3Δ strain, clr4+ was subcloned into pWH5 (pclr4+) and expressed under its endogenous promoter. Plasmids pREP3 (empty vector control) and pclr4+ were introduced by transformation in the indicated strains. Most strains used contained mat1-Msmt0 mating-type allele because these cells showed more pronounced suppression of RNAi mutants by mlo3Δ and tfs1Δ.
Chromatin immunoprecipitation (ChIP) and ChIP-chip
ChIP and ChIP-chip experiments were performed as previously described3. To test the requirement of RNAi in initiation of H3K9me at centromeres in mlo3Δ strains, cells were grown in EMM-Leu media. For the remaining ChIPs, cells were grown in rich media (YEA). Antibodies used were anti-Ser2phospho RNAPII (3E10) (ref. 58), anti-H3K9me (Abcam 1220), anti-Swi6 (ref. 3), anti-Flag (M2)-conjugated agarose (Sigma), anti-HA (12CA5, Covance), and anti-myc (A14, Santa Cruz).
RT-PCR and Northern blot analysis
For RT-PCR, total RNA was extracted from cells using the Master Pure™ Yeast RNA purification kit (Epicentre). One-hundred nanograms of RNA was amplified using the One-step RT-PCR kit (Qiagen) and strand specific primers. Northern blots were performed as described previously59
RNA immunoprecipitation (RIP)
RIP was performed as described previously60 with following modifications. The final ethanol precipitated RNA pellets were resuspended in 100 μl of 1X DNaseI buffer (Amplification grade, Invitrogen) and treated with 3 units DNaseI (Amplification Grade) at 37°C for one hour. The treated RNA samples were phenol/chloroform extracted, ethanol precipitated, and resuspended in 20 μl DEPC treated H2O. 1μl from each sample was subjected to RT-PCR analysis. RT-PCR was performed using the one-step RT-PCR kit from Qiagen with centromeric dh or fbp1 primers.
References
- 1.Grewal SI, Elgin SC. Transcription and RNA interference in the formation of heterochromatin. Nature. 2007;447:399–406. doi: 10.1038/nature05914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ekwall K. Epigenetic control of centromere behavior. Ann. Rev. Genet. 2007;41:63–81. doi: 10.1146/annurev.genet.41.110306.130127. [DOI] [PubMed] [Google Scholar]
- 3.Cam HP, et al. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 2005;37:809–819. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
- 4.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 5.Noma K, et al. RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat. Genet. 2004;36:1174–1180. doi: 10.1038/ng1452. [DOI] [PubMed] [Google Scholar]
- 6.Schalch T, et al. High-affinity binding of Chp1 chromodomain to K9 methylated histone H3 is required to establish centromeric heterochromatin. Mol. Cell. 2009;34:36–46. doi: 10.1016/j.molcel.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayne EH, et al. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell. 2010;140:666–677. doi: 10.1016/j.cell.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang K, Mosch K, Fischle W, Grewal SI. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat. Struct. Mol. Biol. 2008;15:381–388. doi: 10.1038/nsmb.1406. [DOI] [PubMed] [Google Scholar]
- 9.Sugiyama T, Cam H, Verdel A, Moazed D, Grewal SI. RNA-dependent RNA polymerase is an essential component of a self-enforcing loop coupling heterochromatin assembly to siRNA production. Proc. Natl. Acad. Sci. USA. 2005;102:152–157. doi: 10.1073/pnas.0407641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer T, et al. Diverse roles of HP1 proteins in heterochromatin assembly and functions in fission yeast. Proc. Natl. Acad. Sci. USA. 2009;106:8998–9003. doi: 10.1073/pnas.0813063106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugiyama T, et al. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell. 2007;128:491–504. doi: 10.1016/j.cell.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 12.Motamedi MR, et al. HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol. Cell. 2008;32:778–790. doi: 10.1016/j.molcel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamane K, et al. Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol. Cell. 2011;41:56–66. doi: 10.1016/j.molcel.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen ES, et al. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–737. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- 15.Kloc A, Zaratiegui M, Nora E, Martienssen R. RNA interference guides histone modification during the S phase of chromosomal replication. Curr. Biol. 2008;18:490–495. doi: 10.1016/j.cub.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Djupedal I, et al. RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev. 2005;19:2301–2306. doi: 10.1101/gad.344205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kato H, et al. RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science. 2005;309:467–469. doi: 10.1126/science.1114955. [DOI] [PubMed] [Google Scholar]
- 18.Bayne EH, et al. Splicing factors facilitate RNAi-directed silencing in fission yeast. Science. 2008;322:602–606. doi: 10.1126/science.1164029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith E, Shilatifard A. The chromatin signaling pathway: diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell. 2010;40:689–701. doi: 10.1016/j.molcel.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huertas P, Aguilera A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell. 2003;12:711–721. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 21.Strasser K, et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417:304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- 22.Houseley J, LaCava J, Tollervey D. RNA-quality control by the exosome. Nat. Rev. Mol. Cell. Biol. 2006;7:529–539. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- 23.Wang SW, Stevenson AL, Kearsey SE, Watt S, Bahler J. Global role for polyadenylation-assisted nuclear RNA degradation in posttranscriptional gene silencing. Mol. Cell. Biol. 2008;28:656–665. doi: 10.1128/MCB.01531-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buhler M, Haas W, Gygi SP, Moazed D. RNAi-dependent and -independent RNA turnover mechanisms contribute to heterochromatic gene silencing. Cell. 2007;129:707–721. doi: 10.1016/j.cell.2007.03.038. [DOI] [PubMed] [Google Scholar]
- 25.Halic M, Moazed D. Dicer-independent primal RNAs trigger RNAi and heterochromatin formation. Cell. 2010;140:504–516. doi: 10.1016/j.cell.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thakurta AG, Gopal G, Yoon JH, Kozak L, Dhar R. Homolog of BRCA2-interacting Dss1p and Uap56p link Mlo3p and Rae1p for mRNA export in fission yeast. EMBO J. 2005;24:2512–2523. doi: 10.1038/sj.emboj.7600713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernard P, Allshire R. Centromeres become unstuck without heterochromatin. Trends Cell Biol. 2002;12:419–424. doi: 10.1016/s0962-8924(02)02344-9. [DOI] [PubMed] [Google Scholar]
- 28.Hall IM, Noma K, Grewal SI. RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc. Natl. Acad. Sci. USA. 2003;100:193–198. doi: 10.1073/pnas.232688099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Provost P, et al. Dicer is required for chromosome segregation and gene silencing in fission yeast cells. Proc. Natl. Acad. Sci. USA. 2002;99:16648–16653. doi: 10.1073/pnas.212633199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang K, et al. Clr4/Suv39 and RNA quality control factors cooperate to trigger RNAi and suppress antisense RNA. Science. 2011;331:1624–1627. doi: 10.1126/science.1198712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamada T, Fischle W, Sugiyama T, Allis CD, Grewal SI. The nucleation and maintenance of heterochromatin by a histone deacetylase in fission yeast. Mol. Cell. 2005;20:173–185. doi: 10.1016/j.molcel.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Kulish D, Struhl K. TFIIS enhances transcriptional elongation through an artificial arrest site in vivo. Mol. Cell. Biol. 2001;21:4162–4168. doi: 10.1128/MCB.21.13.4162-4168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams LA, Kane CM. Isolation and characterization of the Schizosaccharomyces pombe gene encoding transcript elongation factor TFIIS. Yeast. 1996;12:227–236. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C227::AID-YEA905%3E3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 34.Kvint K, et al. Reversal of RNA polymerase II ubiquitylation by the ubiquitin protease Ubp3. Mol. Cell. 2008;30:498–506. doi: 10.1016/j.molcel.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 35.Iida T, Nakayama J, Moazed D. siRNA-mediated heterochromatin establishment requires HP1 and is associated with antisense transcription. Mol. Cell. 2008;31:178–189. doi: 10.1016/j.molcel.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simmer F, et al. Hairpin RNA induces secondary small interfering RNA synthesis and silencing in trans in fission yeast. EMBO Rep. 2010;11:112–118. doi: 10.1038/embor.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hilleren P, McCarthy T, Rosbash M, Parker R, Jensen TH. Quality control of mRNA 3'-end processing is linked to the nuclear exosome. Nature. 2001;413:538–542. doi: 10.1038/35097110. [DOI] [PubMed] [Google Scholar]
- 38.Libri D, et al. Interactions between mRNA export commitment, 3'-end quality control, and nuclear degradation. Mol. Cell. Biol. 2002;22:8254–8266. doi: 10.1128/MCB.22.23.8254-8266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grewal SI, Klar AJ. A recombinationally repressed region between mat2 and mat3 loci shares homology to centromeric repeats and regulates directionality of mating-type switching in fission yeast. Genetics. 1997;146:1221–1238. doi: 10.1093/genetics/146.4.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall IM, et al. Establishment and maintenance of a heterochromatin domain. Science. 2002;297:2232–2237. doi: 10.1126/science.1076466. [DOI] [PubMed] [Google Scholar]
- 41.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616. doi: 10.1126/science.1191078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadaie M, Iida T, Urano T, Nakayama J. A chromodomain protein, Chp1, is required for the establishment of heterochromatin in fission yeast. EMBO J. 2004;23:3825–3835. doi: 10.1038/sj.emboj.7600401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shanker S, et al. Continuous requirement for the Clr4 complex but not RNAi for centromeric heterochromatin assembly in fission yeast harboring a disrupted RITS complex. PLoS Genet. 2010;6:e1001174. doi: 10.1371/journal.pgen.1001174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moshkovich N, Lei EP. HP1 recruitment in the absence of argonaute proteins in Drosophila. PLoS Genet. 2010;6:e1000880. doi: 10.1371/journal.pgen.1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freitag M, et al. DNA methylation is independent of RNA interference in Neurospora. Science. 2004;304:1939. doi: 10.1126/science.1099709. [DOI] [PubMed] [Google Scholar]
- 47.Henderson IR, Jacobsen SE. Epigenetic inheritance in plants. Nature. 2007;447:418–424. doi: 10.1038/nature05917. [DOI] [PubMed] [Google Scholar]
- 48.Kavi HH, Birchler JA. Interaction of RNA polymerase II and the small RNA machinery affects heterochromatic silencing in Drosophila. Epigenetics Chromatin. 2009;2:15. doi: 10.1186/1756-8935-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guang S, et al. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010;465:1097–1101. doi: 10.1038/nature09095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orphanides G, Reinberg D. RNA polymerase II elongation through chromatin. Nature. 2000;407:471–475. doi: 10.1038/35035000. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Manley JL. Cotranscriptional processes and their influence on genome stability. Genes Dev. 2006;20:1838–1847. doi: 10.1101/gad.1438306. [DOI] [PubMed] [Google Scholar]
- 52.Pal-Bhadra M, Bhadra U, Birchler JA. RNAi related mechanisms affect both transcriptional and posttranscriptional transgene silencing in Drosophila. Mol. Cell. 2002;9:315–327. doi: 10.1016/s1097-2765(02)00440-9. [DOI] [PubMed] [Google Scholar]
- 53.Buhler M, Verdel A, Moazed D. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell. 2006;125:873–886. doi: 10.1016/j.cell.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 54.Lee JT. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–1842. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharp PA. The centrality of RNA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 56.Matzke MA, Birchler JA. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 2005;6:24–35. doi: 10.1038/nrg1500. [DOI] [PubMed] [Google Scholar]
- 57.Maison C, et al. SUMOylation promotes de novo targeting of HP1alpha to pericentric heterochromatin. Nat. Genet. 2011;43:220–227. doi: 10.1038/ng.765. [DOI] [PubMed] [Google Scholar]
- 58.Chapman RD, et al. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 59.Zofall M, et al. Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature. 2009;461:419–422. doi: 10.1038/nature08321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gilbert C, Svejstrup JQ. RNA immunoprecipitation for determining RNA-protein associations in vivo. Curr. Protoc. Mol. Biol. 2006 doi: 10.1002/0471142727.mb2704s75. Chapter 27, Unit 27 24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.