

Abstract
β-Lactams are the most commonly prescribed class of antibiotics and have had an enormous impact on human health. Thus, it is disquieting that an enzyme called New Delhi metallo-β-lactamase-1 (NDM-1) can confer Enterobacteriaceae with nearly complete resistance to all β-lactam antibiotics including the carbapenams. We have determined the crystal structure of Klebsiella pneumoniae apo-NDM-1 to 2.1-Å resolution. From the structure, we see that NDM-1 has an expansive active site with a unique electrostatic profile, which we propose leads to a broader substrate specificity. In addition, NDM-1 undergoes important conformational changes upon substrate binding. These changes have not been previously observed in metallo-β-lactamase enzymes and may have a direct influence on substrate recognition and catalysis.
Keywords: β-lactams, NDM-1, metallo-β-lactamase, crystal structure
Introduction
β-Lactam antibiotics such as the cephalosporins and carbapenems remain the largest class of clinically used antibiotics worldwide.1 β-Lactams act by inhibiting cell wall biosynthesis, leading to membrane lysis and bacterial cell death. Specifically, they attenuate peptidoglycan biosynthesis by inhibiting the transpeptidase enzymes, involved in crosslinking adjacent peptidoglycan strands via their pentapeptide repeats.2 This strategy for antibiotic treatment has been very successful largely because of limited toxicity, excellent bioavailability, and broad activity.3
Unfortunately, bacteria have gained broad-spectrum resistance to β-lactam antibiotics, resulting in devastating treatment issues. An important cause of high-level bacterial resistance is the expression of enzymes called β-lactamases, which hydrolyze the four-member lactam ring of the antibiotic, yielding a product that is clinically inactive.4,5 β-Lactamases are found widely disseminated throughout Gram-positive and Gram-negative bacteria and are often encoded on mobile genetic elements, which facilitate their transmission.6
β-Lactamases have been grouped into four major classes (A–D) based on sequence homology.6 Classes A, C, and D use an active site serine as a nucleophile. However, the class B metallo-β-lactamase (MBL) enzymes use bound zinc atoms in the active site to help ionize and coordinate a nucleophilic hydroxide ion to mediate hydrolysis.7 β-Lactamase-induced resistance has led to two main therapeutic strategies: (i) introducing newly functionalized β-lactam antibiotics that are not recognized by β-lactamases; (ii) coadministration of β-lactam antibiotics with β-lactamase inhibitor compounds, active against the class A, C, and D enzymes. However, despite vast research efforts in this area, currently there are no effective inhibitors targeted against the emerging class B enzymes. MBL enzymes are now found widely disseminated throughout the world.6 Coupled with their broad-spectrum substrate profile, the clinical threat of MBLs is increasingly dire.
Recently, a novel resistance factor called the New Delhi metallo-β-lactamase (NDM-1) has been found to confer Enterobacteriaceae with nearly complete resistance to all β-lactam antibiotics including the carbapenems. This novel resistance gene was discovered in India and has rapidly spread throughout human populations on nearly every continent.8 NDM-1-positive Escherichia coli are now widespread in the environment and water supplies in India,9 likely a result of the fact that NDM-1 genes are typically located on readily transferable plasmids that are prone to rearrangement.10 Because of the selective advantage that NDM-1 confers and its propensity for plasmid-mediated horizontal gene transfer, it is feared that NDM-1 may herald the end of treatment with those drugs that are used clinically to fight Gram-negative infections.10,11
To gain a more complete understanding of the structural basis for the antibiotic resistance conferred by NDM-1, we have determined the crystal structure of the apo form of this enzyme to a resolution of 2.1 Å using molecular replacement phases generated from a previously determined MBL homolog VIM-4.12 Comparison of our apo-NDM-1 structure with the very recent product complex form of NDM-113 as well as other characterized class B MBLs provides important new insights into the catalytic mechanism within the active site as well as unique molecular features that may contribute to its broad-spectrum antibiotic specificity.
Results and Discussion
Structure solution
NDM-1 is a single-chain polypeptide, with a mature sequence from M27-R270 after cleavage of the signal peptide. The structure of apo-NDM-1 was solved by molecular replacement phasing and subsequently refined to a resolution of 2.1 Å (Supporting Information Table SI). The crystals are P1 with five protein chains in the asymmetric unit and a Matthews coefficient of 2.67, corresponding to a predicted solvent content of 53.6%, which roughly approximates the solvent content (44%) based on the model. In chains A, C, and D, there were additional residues at the C-terminus left over after thrombin cleavage of the HIS tag (Supporting Information Table SI). Because of missing density at the termini of the various chains, the following amino acids could be modeled: chain A (43–270 and K271-L272-V273-P274 of the tag), chain B (31–268), chain C (43-270 and K271-L272-V273-P274 of the tag), chain D (43–270 and K271-L272-V273 of the tag), and chain E (43–269). The final refined model has an R and Rfree of 21.7 and 25.1%, respectively, displays good stereochemistry as defined by PROCHECK,14 and contains 471 water and 10 Zn2+ ions within the asymmetric unit, all refined with an occupancy of 1.00. Each monomer had one residue (D90) in a disallowed region on the Ramachandran plot. This residue is a second-shell zinc coordinating ligand involved in extensive hydrogen-bonding interactions, which displays clear electron density, and has nearly identical phi/psi angles in the 1.3-Å product complex structure.13
Overall structure
NDM-1 displays the typical MBL αβ/βα fold, as observed in the previously characterized MBL enzymes VIM-2 and IMP-1 (sequence identity ∼32%), with an internal beta sandwich flanked by five solvent-exposed α-helices on its external face. Also in common, we observe the binuclear Zn-containing active site flanked by the L3 and L10 loops to locate near one edge of the β-sandwich in a shallow, broad groove [Fig. 1(A)]. However, when compared with the structure of the closely related enzyme VIM-2 (32% sequence identity and with a pair-wise RMSD of 1.22 Å on the 207 common CA atoms of the apo forms), two notable differences do exist. In particular, NDM-1 has an extra β-strand extension at the N-terminus of the enzyme as well as a β-turn with an elongated loop region (L8) in place of the strand β8 present in VIM-2 and other homologs [loop L8; Fig. 1(B)].
Figure 1.
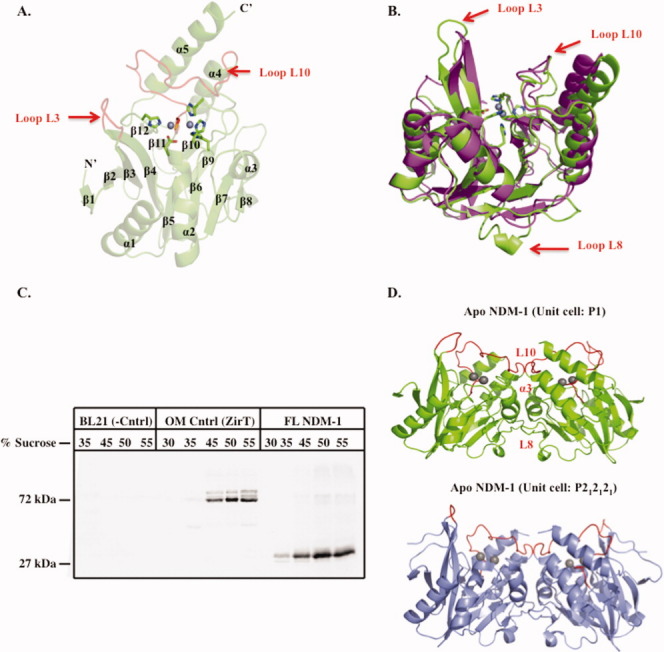
Overall structure of apo-NDM-1, NDM-1 localization, and crystallographic dimerization. A: Cartoon of apo-NDM-1 structure (green), active-site zinc ions (gray spheres), zinc ligands (stick, CPK coloring), and active-site loops L3 and L10 (red). Secondary structure designations are labeled. B: Superposition of NDM-1 on apo-VIM-2 (PDB ID 1K03)15 (magenta). Apo-NDM-1 zinc ligands are colored by atom, and the NDM-1 loops L3, L8, and L10 are highlighted with a red arrow. C: Western blot of a step sucrose gradient of membrane-associated 8xHIS FL-NDM-1. Negative control is E. coli BL21 membranes only. Positive control is outer membrane protein ZirT.16 D: NDM-1 crystallographic dimer interface for both apo (green) and ampicillinic acid-bound (gray, PDB ID: 3Q6X)13 forms. The L8, L10, and α3 helix constitute the dimer interface and are labeled on the apo structure (red).
Oligomerization state and membrane localization
Four of the five molecules within our P1 apo-NDM-1 unit cell and both chains in the P212121 product complex NDM-1 structure display the same dimer involving two protein chains rotated ∼180° relative to one another [Fig. 1(D)]. The fifth monomer in our apo structure sits on one face of the two dimer pairs in the asymmetric unit making crystal packing interactions with chains A, B, and C in the adjacent unit cell and is not involved in dimerization, likely reflecting the fact that NDM-1 can exist both as a monomer and dimer in solution (see below). The dimer interface has a buried surface area of ∼1900 A2 and is predicted to be stable in solution by the CCP4 program PISA.17 The loop L8 that is unique to NDM-1 forms key contacts involved in this dimer interface. These involve hydrophobic and van der Waals interactions between the α3 helix residues A143, L144, N146, and Q147 with residues; T162, F163, A165, and G167 in the L8 loop region. This suggests a functional role in dimerization for this unique insertion.
Supporting a potential physiological role for the dimer interface in our crystallographic data, we used size exclusion chromatography, dynamic light scattering, and chemical crosslinking to show that the M1-R270 NDM-1 (FL) enzyme exists both as a monomer and dimer in solution (Supporting Information Fig. S1). To our knowledge, the only other example of a class B MBL oligomer is the L1 functional tetramer.18,19 This dimerization of both purified and membrane-associated NDM-1 (unpublished data) may be an important feature for antibiotic resistance in a biological context.
Our biochemical analysis also suggests an unusual lipidation and outer membrane localization for NDM-1. Normally, MBL enzymes contain a Type I signal peptide. However, the LipoP server14 revealed probability scores of −0.39 and 1.06 for Type I (soluble) and lipidation signal peptides, respectively. As NDM-1 has a methionine rather than aspartic acid at position +2 from the lipidation signal cleavage site, it is expected to localize to the outer, rather than inner membrane.20,21 To probe the possibility of NDM-1 being a lipoprotein β-lactamase and study its potential localization, FL 8X HIS-tagged NDM-1 containing E. coli membrane samples were analyzed by a step sucrose gradient, followed by Western blot analysis using an anti-HIS antibody. Similar to our positive control (outer membrane porin ZirT),16 it was found that NDM-1 clearly predominated in the higher percentage sucrose layers (40–50% sucrose), suggesting that it preferentially localizes to the bacterial outer membrane [Fig. 1(C)].22 To our knowledge, there is only one other example of a lipoprotein β-lactamase enzyme (BRO-1),23 and the functional role of its membrane localization remains uncharacterized. However, the observation of a lipidation signal and outer membrane localization in a clinically important and broad-spectrum β-lactamase such as NDM-1 reopens this as a potentially important avenue of discovery in terms of antibiotic resistance and therapeutic development.
NDM-1 active site
Our 2.1-Å apo structure allows for a direct comparison between the apo and ampicillinic acid (product)-bound forms of NDM-1,13 revealing key conformational changes that occur around the active site, which further our understanding of MBL substrate binding. In particular, the flexible active-site loop L3 is elongated and disordered in the apo form; yet, upon ligand binding, β3 and β4 form an extended β-sheet interaction [Fig. 2(A)]. This zippering effect pulls the tip of the L3 loop further away from the zinc center, causing the side chain of M67 to reorient away from the zinc center by ∼4.9 Å, presumably to accommodate the substrate by interacting hydrophobically with the R1 phenyl group of ampicillin [Fig. 2(A)]. In contrast, L65 moves ∼2.1 Å closer to the zinc center and substrate upon binding. Together, residues L65, M67, and W93 form a hydrophobic face that tightly interacts with the R1 phenyl group of the substrate. The observed flexibility in the apoenzyme may be a mechanistic feature of MBL enzymes that provides the L3 loop with the plasticity required to form substrate-specific hydrophobic interactions. Therefore, altering the hydrophobicity of the R1 functional group may alter the affinity of β-lactam antibiotics for MBLs.
Figure 2.
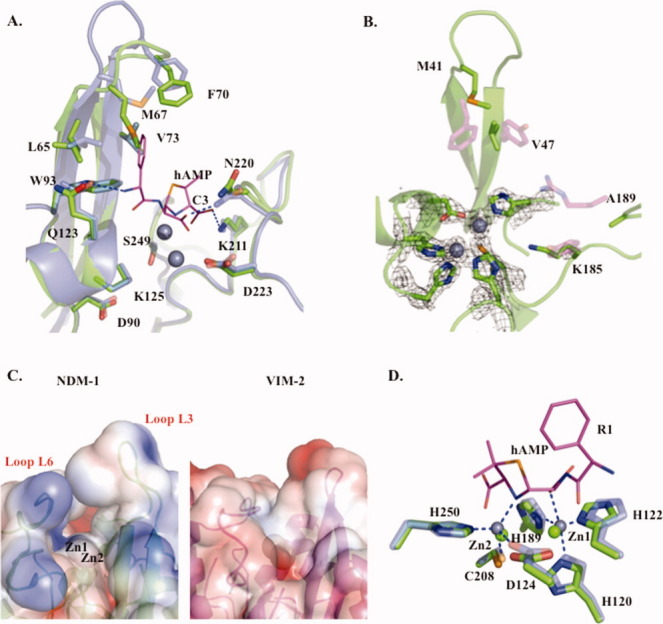
Apo-NDM-1/hydrolyzed ampicillin (hAMP)-bound NDM-1 and apo-NDM-1/apo-VIM-2 active-site comparisons. A: Apo-NDM-1/hAMP NDM-1 active-site overlay: apo-NDM-1 backbone (green), hydrolyzed ampicillin (hAMP) product complex (gray, PDB ID: 3Q6X)13 with selected active-site residues (sticks) labeled.13 B: Apo-NDM-1/apo-VIM-2 active-site overlay: apo-NDM-1 backbone (green), selected NDM-1 and VIM-2 active-site residues are shown as green and magenta sticks with cpk atom coloring, respectively. The 2Fo − Fc map at 2σ is shown for zinc ions and coordinating ligands. C: Electrostatic surface representation of apo-NDM-1 and apo-VIM-2 active-site clefts, calculated using APBS software, with a PARSE force field contoured at −3 and +3. D: Zinc coordination: zinc ions and ligands are in green and gray for apo-NDM-1 and hAMP-bound NDM-1. Coordination interactions are blue dashes, and hAMP is in cpk colored stick representation (carbons are magenta).
Upon ligand binding, the L10 loop also plays an essential role in interacting with the substrate. In particular, the N220 side-chain nitrogen is pulled ∼1 Å closer to the zinc center when comparing apo and ampicillinic acid-bound forms [Fig. 2(A)]. This shift is larger than the RMS differences in superposition of the 232 common CA atoms in the two models of ∼0.284 Å. This movement positions the highly conserved N220 for interaction with the lactam carbonyl group observed in the product complex structure.13 This conserved N220 residue together with Zn1 provide an oxy-anion hole, which helps to polarize the lactam carbonyl upon binding and facilitate nucleophilic attack by the adjacent hydroxide (see below).24 Taken together, the observed plasticity in the L3 and L10 loops provides NDM-1 with the ability to accommodate multiple substrates with differing molecular architectures.
Comparison of apo-NDM-1 and the apo-VIM-2 structures also indicates key differences in the shape and electrostatic characteristics of the active site, providing insight into possible features that govern the differences in specificity in these enzymes. In general, NDM-1 has a more extensive active-site cleft (NDM-1 SA = 520 Å2, VIM-2 SA = 450 Å2) and a more electrostatically neutral L3 loop than does VIM-2 [Fig. 2(C)]. It has been proposed that VIM-2 and VIM-4 are restricted in their substrate profile because of the presence of a narrow active-site cleft.12 The larger surface area of NDM-1 arises from the orientation of the active-site loops L3 and L10, which are positioned further away from the zinc active-site center (see also below) when compared with VIM-2 [Fig. 1(B)]. Another significant difference between the two species is that K211 of NDM-1, which orients the negatively charged carboxylate common to β-lactam substrates, is substituted by a tyrosine in VIM-2. However, an analogous electrostatic interaction with substrate arises from R228 on the L12 loop of VIM-2, which projects into the active site to interact with the β-lactam C3 carboxylate group.15 The protrusion of VIM-2 R228 into the active-site cleft reduces the size of the binding pocket, potentially limiting the VIM-2 substrate-binding profile. In addition, VIM-2 has several residues (including F61, D62, Y67, and R228) that project into the active-site cleft adding both charge and steric bulk. In equivalent positions, NDM-1 contains residues M67, P68, V73, and A215, which present more hydrophobicity in the L3 loop and less steric bulk in the active site [Fig. 2(B)]. We also observe that NDM-1 has an enhanced positively charged electrostatic profile around Zn1 compared with VIM-2 [Fig. 2(C)], which may serve to attract and orientate the negatively charged β-lactam carboxylate during hydrolysis. These active-site characteristics likely decrease steric interference with incoming substrates and facilitate a favorable electrostatic environment leading to a broad substrate profile. In this light, the design of mechanism-based inhibitors may be more effective than simple β-lactam functionalization as a therapeutic approach.
Catalytic mechanism
In our apo-NDM-1 crystal structure, all five of the NDM-1 monomers in the asymmetric unit have two zinc atoms in the active site as indicated by the refined electron density as well as accompanying anomalous diffraction peaks of data collected at the Zn edge using an appropriately tuned synchrotron X-ray source [Fig. 2(B), unpublished data]. In addition, the presence of zinc was confirmed by ICP mass spectrometry (Supporting Information Fig. S2). We therefore infer that NDM-1 uses a di-zinc-catalyzed mechanism for substrate hydrolysis much like other members of the class B1 and B3 MBLs. In this mechanism, Zn1 functions to orientate the substrate carbonyl bond, whereas Zn2 is required for interaction with the amide nitrogen of the substrate.7 An active-site hydroxide located between the two zinc ions, which is present in all five apo-NDM-1 monomers and hydrolyzed ampicillin-bound NDM-1,13 serves as nucleophile.25,26 It has been suggested that the nucleophilic hydroxide form is generated from bound water by the putative general base D120 (NDM-1 numbering). This hypothesis is supported by our structure, whereby D120 is in optimal position ∼2.8 Å away from the catalytic water.7 Following attack of the hydroxide on the carbonyl carbon, the peptide bond is broken with Zn2 acting as a Lewis acid to stabilize the charge on the nitrogen leaving group generated during this step.7
Generally, di-zinc MBLs such as NDM-1 have a lower Zn2 occupancy than Zn1.27 Our refined crystallographic data also suggest a consistent differential occupancy of the two zinc ions in each of the five active sites of the asymmetric unit, in that Zn1 refines with lower average temperature factors (B-factor 26.3 ± 3.7 Å2) than Zn2 (B-factor 33.7 ± 2.3 Å2). In our structures, Zn1 is tightly coordinated with tetrahedral geometry by three invariant histidine residues and a bridging water (H120, H122, H189, and W1), and the higher B-factor Zn2 is coordinated by the conserved D124, C208, H250, and water W1. Interestingly, ICP mass spectrometry revealed ∼1.5 mol of Zn per mole of enzyme (Supporting Information Fig. S2), suggesting that the Zn2 site is undergoing exchange with solvent. The observed zinc stoichiometry is lower than for members of the VIM or IMP MBLs,28,29 yet similar to the class B1 MBL SPM-1.27 For SPM-1, an increased Zn2 coordination distance by the conserved aspartic acid ligand was associated with the lower occupancy metal binding. In contrast, the NDM-1 Zn2 ligands (D124, C208, and H250) display relatively tight zinc coordination distances (∼2.1, 2.4, and 2.1 Å). However, NDM-1 displays substantial electropositive charge around the zinc center compared to that of VIM-2 [Fig. 2(C)]. In particular, K125, which immediately precedes the Zn2 ligand D124, presents a positive charge in close proximity to the Zn2-binding site [Fig. 2(A)]. Therefore, this charge may lead to a repulsion of Zn2.
Interestingly, the two zinc atoms display very similar Zn–Zn interatomic distances of ∼3.8 Å (±0.16 Å) in our ensemble of five apo-NDM-1 structures. However, in the recently determined product complex of NDM-1 with bound hydrolyzed substrate (ampicillinic acid), the Zn–Zn distance is substantially longer, ∼4.6 Å.13 Mutagenesis studies on the subclass B1 MBL (BcII) revealed a decrease in catalytic efficiency upon increasing the Zn–Zn distance.30 In theory, shortening the Zn–Zn distance should bring Zn2 closer to the β-lactam amide nitrogen, allowing for stronger coordination of the substrate and facilitating departure of the leaving group during catalysis. We suggest that the larger Zn–Zn distance observed in the hydrolyzed ampicillin form of NDM-1 represents a mechanism for product release. In our proposed model, the zinc ions are held closely together during substrate binding to facilitate close coordination of Zn2 with the amide nitrogen. However, the Zn–Zn distance increases following hydrolysis to weaken the interaction of the former amide nitrogen with Zn2, leading to departure of the product from the active site. This shift exemplifies the role of plasticity and active-site rearrangements in facilitating turnover in MBLs.
Evolutionary trends in antibiotic resistance
Among the most troubling attributes of MBL enzymes is their tendency of having a broad-spectrum substrate profile. Previously, eight residues in and around the active site of IMP-1 displayed a substrate-dependant sequence requirement, in that they were required for cleavage of some, but not all β-lactams tested.31 Despite the relatively low sequence identity between the two enzymes (∼32%), NDM-1 contains six of these residues (D90, K211, G219, N220, D223, and S249) [Fig. 2(A)]. Four of the six residues (K211, G219, N220, and D223) are located in the L10 loop and are thought to directly interact with β-lactam functional groups; however, residues D90 and S249 do not interact with substrate directly, but are second-shell Zn ligands, suggesting a substrate-dependant zinc requirement. Taken together, these amino acids point to an evolutionary trend in promoting broad-spectrum antibiotic resistance.
In summary, NDM-1 adopts the canonical MBL fold that facilitates a two zinc-catalyzed mechanism of β-lactam antibiotic hydrolysis. However, NDM-1 displays a larger active site and an electrostatic profile that can accommodate a wide variety of substrate molecules. In addition, NDM-1 displays important rearrangements around the active site upon substrate binding and product release. Taken together, the apo-NDM-1 crystal structure helps provide a molecular basis for broad-spectrum antibiotic resistance and represents a valuable step in the informed design of novel MBL inhibitors.
Materials and Methods
Protein expression and purification
Our sequence analysis suggests that the NDM-1 gene (NCBI code: FN396876) encodes an N-terminal Type II lipidation signal peptide, with a cleavage site located between C26 and M27 (as predicted by LipoP).32 The DNA sequence of the mature protein (M27 to R270) was synthesized by BioBasic and cloned into pUC57. This plasmid DNA was used as a template for PCR amplification of the NDM-1 gene (873 bp). Restriction free cloning produced a pET41 expression vector containing NDM-1 with an 8XHis tag on the N-terminus.33 NDM-1 protein was expressed in E. coli BL21 cells at 37°C until an ∼OD600 0.7 was reached, induced with 1 mM IPTG and grown at 25°C for 12–16 h. Harvested cells (∼20 g) were resuspended in 50 mL lysis buffer (50 mM Tris, pH 7, 150 mM NaCl, and one complete, EDTA-free protease inhibitor tablet from Roche) and lysed by French press at ∼12,000 p.s.i., and the lysate was centrifuged (45,000 rpm in a Beckman 60 Ti rotor) for 1 h. Supernatant was passed through a 1-mL Hi-Trap HP His column and pre-equilibrated in lysis buffer. Elution buffer (50 mM Tris, pH 7,. 5M NaCl, and 1M imidazole) was used to elute 8xHis NDM-1 with a gradient of imidazole from 0 to 0.5M in 40 min. NDM-1-enriched fractions were dialyzed into buffer A (20 mM HEPES, pH 8) and loaded onto a Mono Q 10/10 column equilibrated in buffer A. A continuous gradient of 10 column volumes (From A into A plus 2M NaCl) was used. NDM-1 was eluted at ∼400 mM NaCl. Fractions were passed over a Superdex 75 column using (20 mM HEPES, pH 6.8, 150 mM NaCl) as running buffer and concentrated to ∼68 mg/mL.
Crystallization, data collection, and structure determination
Crystals were grown by the sitting-drop method at 25°C using 0.2-μL protein solution and mixed with an equal volume of precipitant. Crystal conditions were 0.1M lithium sulfate, 0.63M ammonium sulfate, and 0.05M Tris at pH 8.5. Apo-NDM-1 crystals were soaked for ∼30 s in cryoprotectant solution (precipitant + 25% glycerol), followed by flash cooling in liquid nitrogen. Data were collected at beamline CMCF-2 of the Canadian Light Source and processed using HKL200034 and CCP4.35 Five percent of reflections were used for cross validation. The structure of NDM-1 was solved by molecular replacement with Phaser36 and the VIM-4 main-chain atoms (Protein Data Bank accession number 2WHG). Several cycles of manual rebuilding in Coot37 and refinement with strict NCS restraints using REFMAC (CCP4)38 were carried out. Simulated annealing was performed on the partially refined model using Phenix.39 In the final stages of refinement, TLS groups (residues 43–65, 71–183, and 255–269) were defined to allow for differences in loop regions between the five monomers. Zinc and waters ions were added manually by examination of Fo − Fc and 2Fo − Fc maps and refined at full occupancy. Coordinates and structure factors were deposited in PDB with entry code (3SPU). Figures were generated using PyMol,40 and calculation of the electrostatic surface was done using APBS.41
Sucrose density gradient centrifugation
E. coli BL21 cells expressing NDM-1 (M1-R270) were prepared and lysed as above. Lysate was centrifuged at 9000 rpm for 15 min followed by centrifugation (45,000 rpm in a Beckman 60 Ti rotor) for 1 h to isolate cell membranes. Membranes were resuspended in lysis buffer and layered atop a stepped sucrose gradient of 30, 35, 45, 50, and 55% sucrose in lysis buffer. Centrifugation was carried out at 39,000 rpm in a Beckman SW 41 Ti rotor for 16 h at 4°C, and sucrose layers were separated and analyzed by Western blot.
Acknowledgments
The authors gratefully acknowledge Liam Worrall, Susan Safadi, Robert Gruninger, and Gerd Prehna for their assistance. They thank personnel at CMCF-2 at the CLS synchrotron facility, Saskatoon, SK, for assistance with data collection.
Supplementary material
References
- 1.Dalhoff A, Thomson CJ. The art of fusion: from penams and cephems to penems. Chemotherapy. 2003;49:105–120. doi: 10.1159/000070616. [DOI] [PubMed] [Google Scholar]
- 2.Guignard B, Entenza JM, Moreillon P. Beta-lactams against methicillin-resistant Staphylococcus aureus. Curr Opin Pharmacol. 2005;5:479–489. doi: 10.1016/j.coph.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Triboulet S, Arthur M, Mainardi JL, Veckerlè C, Dubèe V, Nguekam-Nouri A, Gutmann L, Rice LB, Hugonnet E. Inactivation kinetics of a new target of {beta}-lactam antibiotics. J Biol Chem. 2011;286:22777–22784. doi: 10.1074/jbc.M111.239988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Livermore D. The zeitgeist of resistance. Antimicrob Agents Chemother. 2007;60(Supp.1):i59–i61. doi: 10.1093/jac/dkm160. [DOI] [PubMed] [Google Scholar]
- 5.Pitout JD, Laupland KB. Extended-spectrum beta-lactamase-producing Enterobacteriaceae: an emerging public-health concern. Lancet Infect Dis. 2008;8:159–166. doi: 10.1016/S1473-3099(08)70041-0. [DOI] [PubMed] [Google Scholar]
- 6.Bush K. Alarming β-lactamase-mediated resistance in multidrug-resistant Enterovacteriaceae. Curr Opin Microbiol. 2010;13:558–564. doi: 10.1016/j.mib.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Wang Z, Fast W, Valentine AM, Benkovic SJ. Metallo-beta-lactamase: structure and mechanism. Curr Opin Microbiol. 1999;3:614–622. doi: 10.1016/s1367-5931(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 8.Mollering RC. NDM-1—a cause for worldwide concern. N Engl J Med. 2010;363:2377–2379. doi: 10.1056/NEJMp1011715. [DOI] [PubMed] [Google Scholar]
- 9.Walsh TR, Weeks J, Livermore DM, Toleman MA. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet Infect Dis. 2011;11:334–335. doi: 10.1016/S1473-3099(11)70059-7. [DOI] [PubMed] [Google Scholar]
- 10.Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, Chaudhary U, Doumith M, Giske CG, Irfan S, Krishnan P, Kumar AV, Maharjan S, Mushtaq S, Noorie T, Paterson DL, Pearson A, Perry C, Pike R, Rao B, Ray U, Sarma JB, Sharma M, Sheridan E, Thirunarayan MA, Turton J, Upadhyay S, Warner M, Welfare W, Livermore DM, Woodford N. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis. 2010;10:597–602. doi: 10.1016/S1473-3099(10)70143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornaglia G, Giamarellou H, Rossolini GM. Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study. Lancet. 2011;11:381–393. doi: 10.1016/S1473-3099(11)70059-7. [DOI] [PubMed] [Google Scholar]
- 12.Lassaux P, Traorè DA, Loisel E, Favier A, Docquièr JD, Sohier JS, Laurent C, Bebrone C, Frère JM, Ferrer JL, Galleni M. Biochemical and structural characterization of the subclass B1 metallo-β-lactamase VIM-4. Antimicrob Agents Chemother. 2011;55:1248–1255. doi: 10.1128/AAC.01486-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, Hao Q. Crystal structure of NDM-1 reveals a common {beta}-lactam hydrolysis mechanism. FASEB J. 2011;2:1–9. doi: 10.1096/fj.11-184036. [DOI] [PubMed] [Google Scholar]
- 14.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 15.Garcia-Saez I, Docquir JD, Rossolini GM, Dideberg O. The three-dimensional structure of VIM-2, a Zn-beta-lactamase from Pseudomonas aeruginosa in its reduced and oxidized form. J Mol Biol. 2008;375:604–611. doi: 10.1016/j.jmb.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 16.Gal-Mor O, Gibson DL, Baluta D, Vallance BA, Finlay BB. A novel secretion pathway of Salmonella enterica acts as an antivirulence modulator during salmonellosis. PLoS Pathog. 2008;4:e1000036. doi: 10.1371/journal.ppat.1000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krissinel E, Henrick K. Inference of macromolecular assemblies from the crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 18.Ullah JH, Walsh TR, Taylor IA, Emery DC, Verma CS, Gamblin SJ, Spencer J. The crystal structure of the L1 metallo-beta-lactamase from Stenotrophomonas maltophilia at 1.7Å resolution. J Mol Biol. 1998;284:125–136. doi: 10.1006/jmbi.1998.2148. [DOI] [PubMed] [Google Scholar]
- 19.Simm AM, Higgins CS, Carenbauer AL, Crowder MW, Bateson JH, Bennett PM, Clarke AR, Halford SE, Walsh TR. Characterization of monomeric L1 metallo-beta-lactamase and the role of the N-terminal extension in negative cooperativity and antibiotic hydrolysis. J Mol Biol. 2002;277:24744–24752. doi: 10.1074/jbc.M201524200. [DOI] [PubMed] [Google Scholar]
- 20.Masuda K, Matsuyama S, Tokuda H. Elucidation of the function of lipoprotein-sorting signals that determine membrance localization. Proc Natl Acad Sci USA. 2002;99:7390–7395. doi: 10.1073/pnas.112085599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrandez Y, Condemine G. Novel mechanism of outer membrane targeting of proteins in Gram-negative bacteria. Mol Microbiol. 2008;69:1349–1357. doi: 10.1111/j.1365-2958.2008.06366.x. [DOI] [PubMed] [Google Scholar]
- 22.Viarre V, Cascales E, Ball G, Michel GPF, Filloux A, Voulhoux R. HxcQ liposecretin is self-piloted to the outer membrane by its N-terminal lipid anchor. J Mol Biol. 2009;284:33815–33823. doi: 10.1074/jbc.M109.065938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bootsma HJ, Aerts PC, Posthuma G, Harmsen T, Verhoef J, Dijk H, Mooi FR. Moraxella (Branhamella) catarrhalis BRO beta-lactamase: a lipoprotein of gram-positive origin? J Bacteriol. 1999;181:5090–5093. doi: 10.1128/jb.181.16.5090-5093.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Concha NO, Rasmussen BA, Bush K, Herzberg O. Crystal structure of the wide-spectrum binuclear zinc beta-lactamase from Bacteroides fragilis. Structure. 1996;4:823–836. doi: 10.1016/s0969-2126(96)00089-5. [DOI] [PubMed] [Google Scholar]
- 25.Page MI, Badarau A. The mechanisms of catalysis by metallo β-lactamases. Bioinorg Chem Appl. 2008:2008:576297. doi: 10.1155/2008/576297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crisp J, Conners R, Garrity JD, Carenbauer AL, Crowder MW, Spencer J. Structural basis for the role of Asp-120 in metallo-beta-lactamases. Biochemistry. 2007;18:10664–10674. doi: 10.1021/bi700707u. [DOI] [PubMed] [Google Scholar]
- 27.Murphy TA, Catto LE, Halford SE, Hadfield T, Minor W, Walsh TR, Spencer J. Crystal structure of Pseudomonas aeruginosa SPM-1 provides insights into variable zinc affinity of metallo-beta-lactamases. J Mol Biol. 2006;357:890–903. doi: 10.1016/j.jmb.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 28.Siemann S, Brewer D, Clarke AJ, Dmitrienko GI, Lajoie G, Viswanatha T. IMP-1 metallo-beta-lactamase: effect of chelators and assessment of metal requirement by electrospray mass spectrometry. Biochem Biophys Acta. 2002;1571:190–200. doi: 10.1016/s0304-4165(02)00258-1. [DOI] [PubMed] [Google Scholar]
- 29.Docquier JD, Lammotte-Brasseur J, Galleni M, Amicosante G, Frere JM, Rossolini GM. On functional and structural heterogeneity of VIM-type metallo-beta-lactamases. J Antimicrob Chemother. 2003;51:257–266. doi: 10.1093/jac/dkg067. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez JM, Martin M, Costello FJ, Tierney DL, Vila AJ. The Zn2 position in metallo-β-lactamases is critical for activity: a study on chimeric metal sites on a conserved protein scaffold. J Mol Biol. 2007;345:785–795. doi: 10.1016/j.jmb.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 31.Materon IC, Beharry Z, Huang W, Perez C, Palkill T. Analysis of the context dependant sequence requirements of active site residues in the metallo-beta-lactamase IMP-1. J Mol Biol. 2004;344:653–663. doi: 10.1016/j.jmb.2004.09.074. [DOI] [PubMed] [Google Scholar]
- 32.Juncker AS, Willenbrock H, Heijne G, Nielsen H, Brunak S, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van den Ent F, Löwe J. RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods. 2006;67:67–74. doi: 10.1016/j.jbbm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Winn MD. Overview of the CCP4 suite and current developments. Acta Cryst D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Winn M, Isupov M, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst D. 2000;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 39.Afonine PV, Grosse-Kunstleve RW, Chen VB, Headd JJ, Moriarty NW, Richardson JS, Richardson DC, Urzhumtsev A, Zwart PH, Adams PD. phenix. model_vs_data: a high-level tool for the calculation of crystallographic model and data statistics. J Appl Crystallogr. 2010;43:669–676. doi: 10.1107/S0021889810015608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLano WL. The Pymol molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 41.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.