

Abstract
The behavior of the amyloid β-peptide (Aβ) within a membrane environment is integral to its toxicity and the progression of Alzheimer's disease. Ganglioside GM1 has been shown to enhance the aggregation of Aβ, but the underlying mechanism is unknown. Using atomistic molecular dynamics simulations, we explored the interactions between the 40-residue alloform of Aβ (Aβ40) and several model membranes, including pure palmitoyloleoylphosphatidylcholine (POPC) and palmitoyloleoylphosphatidylserine (POPS), an equimolar mixture of POPC and palmitoyloleoylphosphatidylethanolamine (POPE), and lipid rafts, both with and without GM1, to understand the behavior of Aβ40 in various membrane microenvironments. Aβ40 remained inserted in POPC, POPS, POPC/POPE, and raft membranes, but in several instances exited the raft containing GM1. Aβ40 interacted with GM1 largely through hydrogen bonding, producing configurations containing β-strands with C-termini that, in some cases, exited the membrane and became exposed to solvent. These observations provide insight into the release of Aβ from the membrane, a previously uncharacterized process of the Aβ aggregation pathway.
Keywords: simulation, amyloid, molecular dynamics, membrane
Introduction
The “amyloid hypothesis” of Alzheimer's disease states that the aggregation and deposition of the amyloid β-peptide (Aβ) in neural tissue is central to the progression of the disease.1 Soluble oligomers of Aβ are believed to be the principal toxic species,2 and their toxicity is exerted through interactions with neuronal cell membranes.3 Aβ is produced by two sequential proteolytic cleavages of the amyloid precursor protein (APP) by β- and γ-secretases within the membrane.4,5 Thus, Aβ-membrane interactions are of critical importance in understanding the behavior of Aβ within the plasma membrane and the mechanism through which it exerts its toxic effects.
As the γ-secretase complex is localized in lipid rafts,6 which are liquid-ordered membrane microdomains enriched in cholesterol and sphingolipids, it stands to reason that it is this environment to which Aβ is exposed upon its production. The composition of these lipid rafts likely influences the structure and position of Aβ in the membrane, factors that may affect the release of Aβ from the membrane and the formation of toxic, low-molecular-weight oligomers. Although the facile release of Aβ from membranes is often depicted schematically in the literature, little attention has been paid to the actual mechanism by which Aβ enters the extracellular space. A thorough characterization of Aβ-membrane interactions is necessary, and understanding the behavior of Aβ in the membrane at the atomic level may provide insight into the earliest events of Alzheimer's disease.
To characterize Aβ-membrane interactions in a variety of systems, we explored a number of model membranes that contain many common membrane phospholipids, including systems containing single phospholipid components (POPC or POPS), a mixture of two lipid components (POPC/POPE), a model raft containing palmitoylsphingomyelin (PSM), cholesterol, and POPC, and a raft that included a physiologically relevant concentration of ganglioside GM1. Sphingomyelin (SM) and lipids containing phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS) headgroups are among the most common components of eukaryotic cell membranes.7,8 Thus, we examined the interactions of Aβ40 with these lipids and combinations thereof to establish a thorough biophysical characterization of Aβ dynamics in the plasma membrane and to understand if any of these lipids had unique characteristics that were relevant to the release of the peptide from the membrane.
Ganglioside GM1 (Fig. 1) is a component of lipid rafts and is enriched in the outer leaflet of the plasma membrane of cells in the central nervous system.9 It has been shown that Aβ-GM1 complexes may act as seeds for further Aβ aggregation,10–12 indicating that interactions between Aβ and GM1 are important to the progression of Alzheimer's disease. Aβ-GM1 complexes form most readily in lipid raft environments that are enriched with cholesterol.11,13 Further, GM1 has been found to enhance the formation of β-strand structures in Aβ, thus promoting aggregation.12,13 Although numerous studies have concluded that Aβ binds to the oligosaccharide headgroup of GM1,14–16 the influence of GM1 on the ability of Aβ to exit the membrane is thus far unexplored, and although numerous experiments have been conducted to characterize the interactions between Aβ and GM1, the existing results lack the detail that can be obtained from molecular dynamics (MD) simulations.
Figure 1.
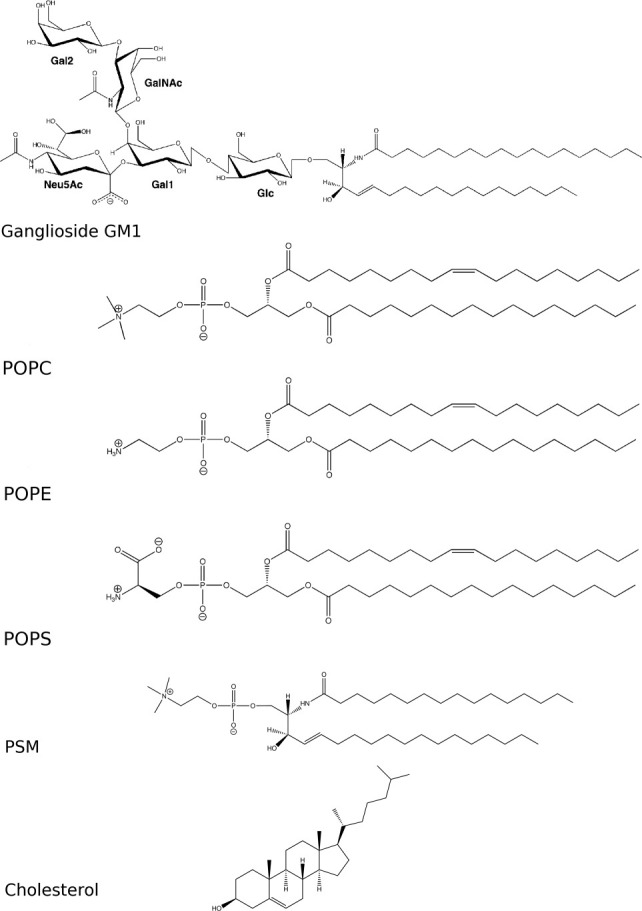
The structures of the lipids used in this work. For ganglioside GM1, the constituent sugar moieties are labeled as referred to in the main text: glucose (Glc), internal galactose (Gal1), N-acetylneuraminic acid (Neu5Ac), N-acetyl galactosamine (GalNAc), and terminal galactose (Gal2).
Results
The systems analyzed here describe the interactions of the Aβ40 peptide with a variety of model membranes with different biophysical and biochemical properties. The POPC membrane allows us to examine the interactions of Aβ with a neutral, zwitterionic lipid with mixed acyl chains. The POPC/POPE membrane is much like the POPC membrane, but includes ethanolamine headgroups, which provide hydrogen-bond donors by virtue of their primary amino groups. The POPS membrane contains headgroups with both hydrogen-bond donor (amino group) and acceptor (carboxylate and phosphate) groups. The raft systems allow us to examine the interactions of Aβ40 with a model of the environment in which it is produced, with a specific focus on the impact of ganglioside GM1 enrichment in this membrane microdomain.
Our results lead us to introduce the following mechanism for the release of Aβ from the membrane environment following γ-secretase cleavage. The N-terminal residues of Aβ interact with clusters of GM1 through hydrogen bonding and other electrostatic interactions, inducing the peptide to adopt β-hairpin configurations and allowing the C-terminal-embedded region to cross the membrane-water interface, aided by hydrogen bonding to nearby sugar moieties. These Aβ-GM1 interactions preclude other interfacial Aβ-lipid interactions involving the N-terminal residues that otherwise anchor the peptide to the membrane, which we observe in the simulations of Aβ40 in POPC, POPS, and POPC/POPE membranes. The stability of Aβ40 in POPC, POPS, and POPC/POPE indicates that Aβ may bind and insert into these membrane domains to exert its toxicity.
Throughout this section, simulations are categorized according to two factors, lipid type and Aβ40 model. There were two models, one wherein the C-terminal Val40 was protonated (model “CH”) and one wherein it was ionized (model “CI”). Lipids are identified according to headgroup (PC, PS, and PE). “Raft” refers to a lipid raft without GM1, whereas system identifiers with a “GM1” prefix indicate rafts that contain GM1. Individual simulations within each set are indicated by a numeral. For additional details, see Methods.
Secondary structure of Aβ40
Average secondary structure content for all simulations performed here is listed in Table I. The β-strand content reflects the sum of extended β-strands and isolated β-bridges, whereas “total helix” refers to the sum of α-, 310-, and π-helices.
Table I.
Average Secondary Structure Content of Aβ40 in All Model Membranes Over the Last 50 ns of All Trajectoriesa
| System | Coil | β-Strand | Bend | Turn | Total helix |
|---|---|---|---|---|---|
| PC-CH | 32 (4) | 2 (2) | 15 (8) | 11 (5) | 40 (16) |
| PC-CI | 38 (7) | 4 (5) | 22 (8) | 10 (4) | 27 (15) |
| PS-CH | 38 (3) | 2 (3) | 11 (2) | 14 (8) | 36 (6) |
| PS-CI | 35 (10) | 8 (8) | 14 (6) | 12 (3) | 30 (8) |
| PC/PE-CH | 40 (14) | 5 (6) | 21 (15) | 11 (8) | 22 (27) |
| PC/PE-CI | 31 (6) | 4 (3) | 12.3 (0.7) | 15 (6) | 38 (7) |
| Raft-CH | 35 (9) | 0.1 (0.1) | 12 (10) | 13 (6) | 34 (11) |
| Raft-CI | 48 (9) | 5 (8) | 21 (10) | 8 (2) | 7 (5) |
| GM1-CH | 33 (7) | 10 (8) | 25 (6) | 13 (10) | 20 (9) |
| GM1-CI | 45 (15) | 7 (7) | 21 (3) | 14 (7) | 15 (10) |
All values are expressed as percentages, with standard deviations given in parenthesis.
Aβ40 in POPC
PC-containing lipids comprise the most common types of phospholipids found on the extracellular surface of eukaryotic plasma membranes.7,8 Our previous work17,18 modeled Aβ40 in a fully saturated dipalmitoylphosphatidylcholine (DPPC) membrane, at elevated temperature (323 K) to maintain a fluid-phase membrane. POPC is a better model of a physiologically relevant membrane lipid, as it can be simulated at 310 K and remain fluid.
In POPC, the Aβ40 peptide tended to lose some of its initial α-helicity (45% at the beginning of the simulation) over time in favor of random coil structures. In sets PC-CH and PC-CI, Aβ40 retained 40% ± 16% and 27% ± 15% total helicity, respectively. The lower helical content in the PC-CI simulations relative to the PC-CH simulations was principally due to the deprotonation of the C-terminus, which caused Val40 and the other hydrophobic C-terminal residues to disorder and approach, but not cross, the membrane-water interface. β-Strand content in the Aβ40 peptides was low, 2% ± 2% for PC-CH simulations and 4% ± 5% for PC-CI simulations. These β-strands, which formed in four of the six simulations in this set, typically involved short stretches of amino acids (2–4 residues) at various positions within the N-terminal polar region. Snapshots from the end of the PC-CH and PC-CI simulations and secondary structure evolution are shown in Figures 1 and 2 of the Supporting Information.
Aβ40 in POPS
Although PS-containing phospholipids typically occur in the cytofacial leaflet of eukaryotic cell membranes,7,8 anionic lipids are frequently used in biophysical characterization of Aβ-membrane interactions.19–23 It has been suggested that cellular injury may expose anionic lipids, like POPS, to the extracellular space.20 Thus, Aβ-POPS interactions are important to characterize, because the dynamics and unfolding of the peptide may be related to such events.
The secondary structure content of Aβ40 in POPS was similar to the results of Aβ40 in POPC. The peptide retained 36% ± 6% and 30% ± 8% of its initial helicity, on average, in simulation sets PS-CH and PS-CI, respectively. The helical content is in good agreement with CD spectroscopy results of Aβ40 inserted in anionic lipids.19 Short β-strands developed in four of these six trajectories, with PS-CH simulations containing, on average, 2% ± 3% β-strand content and PS-CI, 8% ± 8%. As with simulations in POPC, N-terminal polar residues of Aβ40 bound to the membrane-water interface, associating with the charged lipid headgroups of POPS. Snapshots from the end of the PS-CH and PS-CI simulations and secondary structure evolution are shown in Figures 3 and 4 of the Supporting Information.
Aβ40 in POPC/POPE
Addition of POPE to a POPC membrane (which has only hydrogen-bond acceptor groups, phosphates) introduces many new hydrogen-bonding partners for Aβ, a fact that may affect the secondary structure of the peptide and its ability to bind the membrane. POPE, by virtue of its primary amine and phosphate groups, can engage in intermolecular (POPE-POPE, POPE-POPC, and POPE-Aβ) as well as intramolecular hydrogen bonding.
In the simulations of Aβ40 in POPC and POPS, model CH, with a protonated C-terminus, retained more of its initial helicity than model CI. In POPC-POPE membranes, however, this trend was reversed, with PC/PE-CH simulations averaging 22% ± 27% helicity and PC/PE-CI simulations, 38% ± 7%, although given the relatively wide standard deviations of these simulations, we cannot conclude that there is any significant difference between the different membranes with respect to helical content. In PC/PE-CH-1, Aβ40 lost all of its initial α-helicity, whereas in PC/PE-CH-2 and PC/PE-CH-3 and the entire PC/PE-CI set, at least some of the initial helicity was retained, as in the POPC and POPS simulations.
Aβ40 in POPC-POPE membranes developed similar β-strand content as the peptides in POPC and POPS systems, 5% ± 6% in the case of PC/PE-CH systems and 4% ± 3% in PC/PE-CI. Of note in this simulation set was the development of a short β-hairpin (V24GSNKG29) in Aβ40 in simulation PC/PE-CI-3. Ser26 and Asn27 formed the turn that connected the four-residue hairpin. This structure evolved because of the interactions of Asn27 and Lys28 side chains with POPE headgroups. Hydrogen bonding stabilized the turn, allowing β-strands to evolve over the last 10 ns of simulation PC/PE-CH-3. Snapshots from the end of the PC/PE-CH and PC/PE-CI simulations and secondary structure evolution are shown in Figures 5 and 6 of the Supporting Information.
Aβ40 in raft systems
The simulations of Aβ40 in POPC, POPS, and POPC/POPE presented here, as well as those of Aβ40 in DPPC published previously,17 indicate that the peptide possesses an intrinsic ability to form short β-strands and hairpins in a variety of membrane environments. Experiments have shown that ganglioside GM1 increases the level of β-strand content in Aβ.12,13 Toxic oligomeric species24 and mature Aβ fibrils25,26 are rich in β-strand content, indicating that the formation of β-strand elements is central to the aggregation cascade.
We found that the conversion of α-helix to β-strand was amplified by the presence of GM1, principally through the formation of β-hairpins, which we observed in 9 of 10 simulations of Aβ40 in GM1-containing rafts, all five GM1-CH trajectories and four GM1-CI trajectories (Figs. 7 and 8 of the Supporting Information). The β-hairpins occurred principally within the N-terminal 16 residues of Aβ40 and involved very specific residues, as discussed below in the context of hydrogen bonding. In one instance (simulation GM1-CI-5), a hairpin formed involving residues Val24-Gly25 and Lys28-Gly29, connected by a turn involving Ser26-Asn27. Formation of β-strand structures near glycines within residues 24–37 has previously been proposed as an important factor in the overall conversion of Aβ from α-helix to β-strand,27 but emergence of such structures has never before been observed in simulations of membrane-bound Aβ, indicating that this behavior is related to the presence of a suitable hydrogen-bond donor/acceptor environment, such as GM1 or POPE, as we observed above in the case of simulation PC/PE-CH-3. This phenomenon is discussed in greater detail below. Conversely, in simulations of raft systems lacking GM1, we found that β-strands were formed in only 6 of 10 total trajectories (two Raft-CH and four Raft-CI, Figs. 7 and 8 of the Supporting Information).
Destabilization of helical regions of Aβ40 occurred because of two principal factors in our simulations of Aβ40 in a raft environment: (i) hydrogen-bonding interactions between N-terminal residues and GM1 to form β-hairpins and (ii) exposure of C-terminal residues 29–40 to solvent to form random coil structures. The C-termini of Aβ40 in simulation sets GM1-CI and Raft-CI approached the membrane-water interface independently of the presence of GM1 (Figs. 9 and 10 of the Supporting Information) because of the ionized state of the C-terminal carboxylate of Val40, which snorkeled toward this polar environment. The result was a total helical (sum of α-, 310-, and π-helix) content of 7% ± 5% and 15% ± 10% for Raft-CI and GM1-CI simulation sets, respectively.
In the GM1-CH simulation set, interactions between Aβ40 and GM1 reduced the helical content of Aβ from 34% ± 11% in set Raft-CH to 20% ± 9% in set GM1-CH. GM1 caused Aβ40 to rise further out of the membrane (discussed below), resulting in closer proximity of the C-terminal residues to the polar interface than in the CH set, causing them to become disordered. In one of the simulations, GM1-CH-5, the total helical content was reduced to 9.7% as residues 29–40 were extracted from the membrane and exposed to solvent (discussed below), destabilizing the initial α-helix in favor of random coil structures. The total helical content of residues 29–40 as a function of their center-of-mass position relative to the interface is shown in Figure 2. In general, helical content dropped sharply when residues 29–40 were within 1 nm of the interface, an environment that is much more polar than the membrane core.
Figure 2.

Total helicity of C-terminal residues 29–40 as a function of distance relative to the interface, defined as the average phosphorus position over time. The total helicity is defined as the sum of α-, 310-, and π-helical structures, averaged over the last 50 ns of simulation time.
Several representative examples of β-hairpins that formed during our simulations are shown in Figure 3. The longest β-hairpins (8–10 residues) were formed at the interface of two or more GM1 molecules [Fig. 3(D,E)], an event that was independent of the protonation state of the C-terminus of Aβ40. These results provide insight into the mechanism underlying the experimental observations that Aβ binds to GM1 clusters, thereby seeding further aggregation.10–12 Gangliosides have been shown to dehydrate the membrane-water interface of phosphatidylcholine-containing lipid bilayers,28 and this phenomenon appears to play a functional role in terms of the unfolding of Aβ, facilitating the self-association of backbone amide groups that leads to β-strand hydrogen bonding. As a representative example, in simulation GM1-CH-4 [Fig. 3(A,B)], a β-hairpin formed 1.3 nm above the membrane-water interface, at which point the density of water is 14.6% lower than bulk solution, whereas in raft systems lacking GM1, the density of water at this same vertical position above the interface is only 3.9% below bulk [Fig. 3(A)]. In simulation Raft-CH-2 [Fig. 3(A,C), wherein the position of the β-hairpin was representative of those formed in the Raft-CH and Raft-CI sets], a β-hairpin was formed only 0.7 nm above the interface, a location where the water density is 9.4% lower than bulk.
Figure 3.
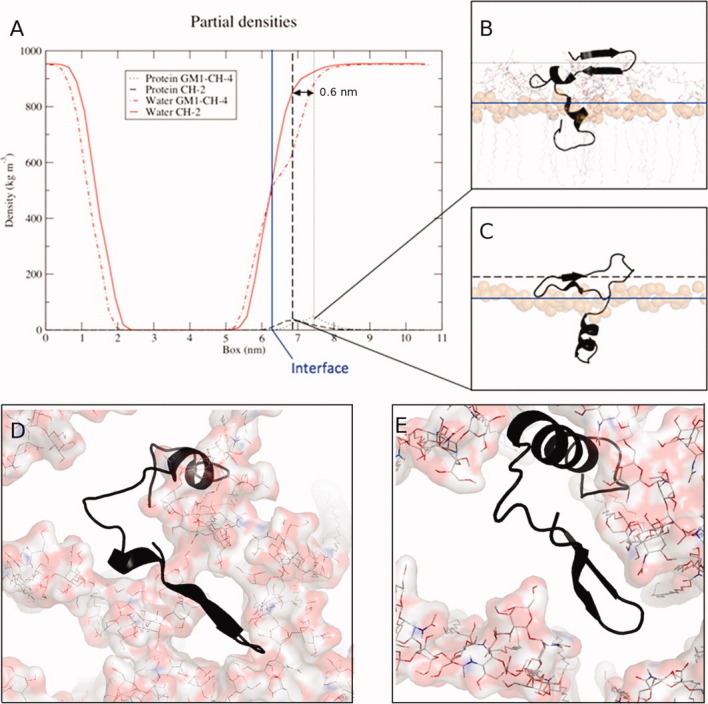
(A) Position of β-hairpin formation relative to the level of hydration along the z-dimension of the simulation box. The membrane-water interface is indicated as a solid blue vertical line, and the density maxima for the β-hairpins formed in simulations GM1-CH-4 and CH-2 are indicated by dotted and dashed lines, respectively. (B) Formation of an eight-residue β-hairpin in simulation GM1-CH-4 and (C) formation of a four-residue β-hairpin in simulation Raft-CH-2. Top-down views (along the z-axis) of two GM1-containing systems are showing the locations of β-hairpins at the interface of multiple GM1 molecules in simulations (D) GM1-CH-4 and (E) GM1-CH-1.
Hydrogen bonding between Aβ40 and lipids
Table II summarizes backbone and side-chain hydrogen bonding between Aβ40 and all phospholipids that were not part of raft membranes. In general, the side chains of Aβ40 formed more hydrogen bonds with surrounding lipids than did backbone groups, in agreement with experimental findings that Aβ-membrane interactions are largely mediated by nonspecific electrostatic interactions, such as those found between lipid headgroups and the charged and polar residues in the N-terminal region of Aβ.29
Table II.
Hydrogen Bonding Between Aβ40 and Lipids in All Nonraft Systemsa
| System | POPC | POPS | POPE | |
|---|---|---|---|---|
| PC-CH | Backbone | 4 (2) | — | — |
| Side chain | 9 (1) | — | — | |
| PC-CI | Backbone | 8 (3) | — | — |
| Side chain | 4.4 (0.8) | — | — | |
| PS-CH | Backbone | — | 6 (4) | — |
| Side chain | — | 19 (2) | — | |
| PS-CI | Backbone | — | 6 (3) | — |
| Side chain | — | 13 (1) | — | |
| PC/PE-CH | Backbone | 4 (2) | — | 2 (2) |
| Side chain | 4 (2) | — | 11 (1) | |
| PC/PE-CI | Backbone | 3 (1) | — | 4 (1) |
| Side chain | 3.5 (0.3) | — | 8 (4) | |
All values are expressed as the average number of hydrogen bonds, with standard deviations given in parenthesis.
Aβ40 in POPC
POPC lipids are only capable of serving as hydrogen-bond acceptors, having phosphate and ester groups that can interact with hydrogen-bond donors in Aβ40. Hydrogen bonds involving backbone amide groups were increased in the PC-CI set relative to PC-CH, principally because of the lower α-helical content of the Aβ40 peptide in these simulations relative to PC-CH. As described above, the ionized state of Val40 in the PC-CI series caused this residue to snorkel toward the membrane-water interface, destabilizing helicity in the C-terminal residues, as well as several other residues that reside at or below the interface. This destabilization led to exposed amide groups that interacted with POPC hydrogen-bond acceptors, particularly the ester functional groups connecting the glycerol backbone of POPC to the acyl chains. In PC-CI simulations, the number of hydrogen bonds to glycerol esters averaged 4 ± 2, whereas in PC-CH simulations, only approximately one hydrogen bond was formed with these groups. Hydrogen bonding to phosphate moieties was comparable between the two simulation sets, 3 ± 1 for PC-CH systems and 5 ± 2 for PC-CI.
Side-chain-POPC hydrogen bonds were decreased in PC-CI simulations relative to PC-CH, likely because of the backbone destabilization phenomena discussed above. In the PC-CI simulations, the backbone is more exposed to the surrounding lipids, causing their hydrogen-bonding acceptor groups to be occupied more frequently by exposed amide functional groups, leaving fewer open positions for other side-chain moieties that may be present.
Aβ40 in POPS
Hydrogen-bonding patterns in the PS-CH and PS-CI simulation sets were very similar, due largely to the fact that the Aβ40 peptide in these simulations adopted very similar secondary structures. Thus, the number of exposed backbone amides in each simulation was very similar. The slight increase in total helicity of Aβ40 in the PS-CI simulation set relative to those of PC-CI (Table I) caused the peptides in the PS-CI set to form fewer backbone-POPS hydrogen bonds than backbone-POPC hydrogen bonds in the PC-CI set. The embedded C-terminal residues were somewhat more helical in PS-CI than in PC-CI because of the repulsion between the negatively charged C-terminus and anionic interface. This electrostatic repulsion prevented the C-terminal residues from approaching the interface as closely as in the case of PC-CI simulations, leading to less destabilization of the helix and less overall hydrogen bonding involving backbone groups.
Numerous hydrogen bonds were formed between Aβ40 side-chain moieties and POPS lipids, principally involving headgroup functional groups. Unlike POPC, which contains only hydrogen-bond acceptors, POPS contains an amino group that serves as a hydrogen-bond donor and a carboxylate group that is an additional acceptor. These two amino acid-derived functional groups are the principal sites of hydrogen bonding between Aβ40 side chains and POPS lipids. Thus, although levels of backbone-lipid hydrogen bonding were comparable between the POPC and POPS systems, the presence of POPS lipids allowed for greater overall hydrogen-bonding capacity, particularly with the side chains of Aβ40. Thus, the N-terminal residues of Aβ40 are capable of extending along the interface, forming hydrogen bonds and other electrostatic interactions with several different moieties on the POPS lipids.
Aβ40 in POPC/POPE
A slightly smaller number of hydrogen bonds were formed between Aβ40 and POPC lipids in the equimolar POPC/POPE membrane than in the pure POPC membrane (Table II). This fact can be attributed to the presence of POPE lipids in the membrane. Although the backbone of Aβ40 participated in more hydrogen bonds with POPC than POPE, the side-chain moieties of Aβ40 formed significantly more hydrogen bonds to POPE than POPC, largely because of the presence of the primary amine in the POPE headgroup. Aβ40-amine hydrogen bonds accounted for 54.4% of the total Aβ40-POPE hydrogen bonds in the PC/PE-CH systems and 63.1% in PC/PE-CI.
Aβ40 in raft systems
Ganglioside GM1 contains an oligosaccharide headgroup that provides numerous sites for potential hydrogen bonding to Aβ (Fig. 1), substantially more per lipid than any of the other phospholipids analyzed above. In simulations of Aβ40 in GM1-containing rafts, Aβ40 formed hydrogen bonds with the GM1 molecules, most frequently with the Neu5Ac and Gal2 moieties (Table III). This behavior occurred independently of the protonation state of the C-terminus, because hydrogen bonds primarily involved the polar N-terminal residues. In simulations of Aβ40 in lipid rafts lacking GM1, the backbone and side chains of Aβ40 formed numerous hydrogen bonds to the phosphate moieties of the POPC and PSM lipids (Table IV). In GM1-CH systems, hydrogen bonds to POPC and PSM lipids were decreased by 68.0 and 43.4%, respectively, relative to Raft-CH systems. For GM1-CI models, hydrogen bonds to POPC and PSM were reduced by 58.8 and 30.7%, respectively. Hydrogen bonding to cholesterol and the sphingosine backbone groups of PSM was negligible (Table IV).
Table III.
Hydrogen Bonds Between Aβ and GM1 Sugar Moietiesa
| System | Glc | Gal1 | Neu5Ac | GalNAc | Gal2 | |
|---|---|---|---|---|---|---|
| GM1-CH | Backbone | 0.3 (0.6) | 0.3 (0.5) | 1.1 (0.7) | 1 (1) | 1.5 (0.9) |
| Side chain | 0.02 (0.03) | 0.1 (0.2) | 3 (2) | 1 (1) | 2 (1) | |
| GM1-CI | Backbone | 0.06 (0.05) | 0.0 (0.0) | 1 (1) | 0.8 (0.8) | 1.3 (0.9) |
| Side chain | 0.02 (0.04) | 0.3 (0.2) | 3 (2) | 0.6 (0.4) | 1.8 (0.7) | |
All values are expressed as the average number of hydrogen bonds, with standard deviations given in parenthesis.
Table IV.
Hydrogen Bonds Between Aβ and POPC, PSM, and Cholesterol for Raft-Only and GM1-Raft Systemsa
| System | POPC PO4 | POPC glycerol | PSM PO4 | PSM NH | PSM OH | Chol | |
|---|---|---|---|---|---|---|---|
| Raft-CH | Backbone | 3 (1) | 0.9 (0.7) | 3 (1) | 0.0 (0.0) | 0.5 (0.8) | 0.0 (0.1) |
| Side chain | 5 (1) | 2 (1) | 5 (2) | 0.0 (0.0) | 0.4 (0.4) | 0.3 (0.5) | |
| GM1-CH | Backbone | 0.9 (0.8) | 0.0 (0.0) | 0.9 (0.7) | 0.9 (0.6) | 0.0 (0.0) | 0.3 (0.7) |
| Side chain | 2.3 (0.9) | 0.0 (0.0) | 2.4 (0.9) | 0.3 (0.4) | 0.3 (0.4) | 0.0 (0.0) | |
| Raft-CI | Backbone | 4 (1) | 1.3 (0.9) | 3.4 (0.7) | 0.2 (0.4) | 0.1 (0.1) | 1 (2) |
| Side chain | 5 (1) | 1.3 (0.4) | 5 (2) | 0.0 (0.0) | 0.2 (0.2) | 0.0 (0.0) | |
| GM1-CI | Backbone | 2 (2) | 0.2 (0.5) | 2 (2) | 0.9 (0.8) | 0.1 (0.2) | 0.1 (0.2) |
| Side chain | 3 (1) | 0.2 (0.2) | 2.9 (0.9) | 0.4 (0.3) | 0.0 (0.0) | 0.1 (0.1) | |
All values are expressed as the average number of hydrogen bonds, with standard deviations given in parenthesis.
The presence of GM1 thus reduced the incidence of interfacial interactions between Aβ and the other lipids. Our previous work17,18 indicated that the N-terminus of Aβ40 associates strongly with the membrane-water interface in a DPPC model system, principally through electrostatic interactions with the zwitterionic lipid headgroups. In this study, analysis of three other nonraft phospholipid membranes indicates that Aβ associates strongly with these lipids, as well. Hydrogen bonding to the membrane-water interface is amplified in POPC/POPE and POPS systems relative to the pure POPC membrane because of the additional hydrogen-bonding capacity of the POPE and POPS lipids.
Previous work29 has concluded that Aβ associates with phospholipid headgroups through nonspecific electrostatic interactions, but interactions between Aβ and GM1 have been postulated to be based on specific interactions.30 Our findings indicate that specific, favorable interactions between Aβ and GM1 can effectively compete with nonspecific electrostatic interactions between Aβ and other nearby phospholipids that more often cause the N-terminal residues of Aβ to otherwise adopt coil or bend configurations (Figs. 7 and 8 of the Supporting Information).
Examination of the most persistent hydrogen bonds formed between Aβ and GM1 elucidates the roles of several important amino acids in these trajectories, most notably His6, Asp7, His13, and His14. Very recently, His13 and His14 have been identified as key residues for the binding of Aβ to GM1.31 The Neu5Ac moiety of GM1 has previously been proposed as an important binding site for Aβ, an effect that is enhanced by the presence of a terminal galactose residue (Gal2) in the oligosaccharide headgroup.30 In the five instances in which Asp7 interacted with either Neu5Ac or Gal2, a β-turn formed at this location, stabilizing the β-hairpin structure in nearby residues, a structure that has previously been proposed to contribute to neurotoxicity.32
Regarding the histidine residues, in 8 of 10 simulations of Aβ40 in GM1-containing rafts (four in each set, GM1-CH and GM1-CI), at least one of these histidine residues formed persistent hydrogen bonds (lasting continuously for 10 ns or more) with at least one sugar moiety on GM1, and in six of these instances, the sugar moiety involved was Neu5Ac. In eight of nine simulations that formed β-strands, at least one histidine residue was present in the β-strand, but in the absence of GM1, these residues were principally found in coil or bend configurations (Figs. 7 and 8 of the Supporting Information). The presence of carbohydrate groups and their roles as both hydrogen-bond donor and acceptor have been proposed to stabilize β-structures in the Aβ peptide, specifically in the vicinity of histidine residues at positions 6, 13, and 14 in the Aβ sequence.33–35 In addition to these hydrogen-bonding interactions, in simulation GM1-CI-5, the formation of β-strand and turn structures described above was promoted by hydrogen-bonding interactions formed by Gly29 with Neu5Ac, as well as Ser26 and Asn27 with Neu5Ac and GalNAc.
Position and orientation of Aβ40 in membranes
The initial orientation of Aβ40 in all of the systems modeled here was prepared such that hydrophobic residues 29–40 were embedded in the hydrophobic core of the lipid membrane, with the helix axis aligned with the membrane normal (Fig. 11 of the Supporting Information). The position and orientation of this peptide segment over time can be described by two principal factors that influence solvent exposure and thus aggregation propensity: (i) the location of residues 29–40 with respect to the membrane interface (defined as the average phosphorus plane of the extracellular leaflet of the bilayer) and (ii) the tilt angle of the principal axis of these same residues with respect to the membrane normal. The interactions of Aβ with the surrounding lipid matrix influence these two variables, which can be plotted as free-energy surfaces (Fig. 4). The free energy of a configuration with respect to these two variables, tilt (θ) and z-position (z), can be described by the following expression:
Figure 4.
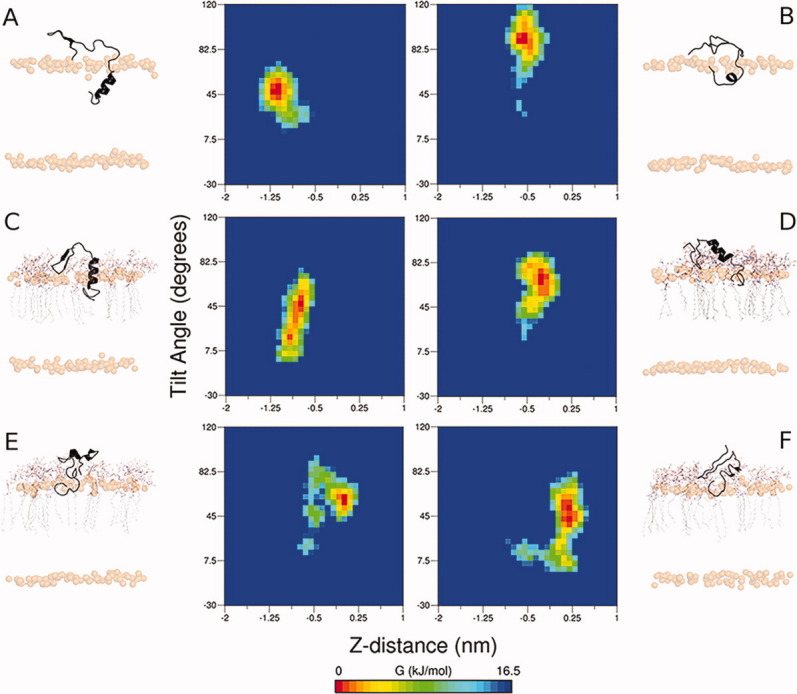
Free-energy surfaces as a function of both the C-terminal tilt angle (y) and vertical position of Aβ residues 29–40 relative to the average phosphate plane of the extracellular membrane leaflet (x). Representative images were chosen from each set for Aβ40 in 1:1:1 POPC:PSM:cholesterol raft systems: (A) Raft-CH-2 and (B) Raft-CI-4; GM1-containing raft systems: (C) GM1-CH-1 and (D) GM1-CI-1; and GM1-containing raft systems wherein the free-energy minimum was resided above the average phosphate plane: (E) GM1-CH-5 and (F) GM1-CI-5. Rendered images from each system correspond to a configuration taken from the energy minimum of each plot, with the Aβ40 peptide shown as a black ribbon, phosphorus atoms shown as transparent gold spheres, and GM1 molecules drawn as lines and colored by element.
The probability distribution is determined by binning the tilt and z-coordinate data from the MD trajectories. The maximum of the distribution (Pmax) is subtracted to give ΔG(z,θ) = 0 for the lowest point on the free-energy surface.
Table V summarizes the average values of the tilt angle and relative z-position for all membranes studied in this work.
Table V.
Average C-Terminal Tilt Angles and Vertical Positions of Aβ40 Residues 29–40 in All Model Membranes Over the Last 50 ns of All Trajectoriesa
| System | Tilt angle (°) | Relative position (nm) |
|---|---|---|
| PC-CH | 44 (27) | −1.3 (0.3) |
| PC-CI | 101 (10) | −0.8 (0.3) |
| PS-CH | 83.0 (0.4) | −1.4 (0.2) |
| PS-CI | 87 (30) | −1.13 (0.05) |
| PC/PE-CH | 37 (26) | −0.867 (0.009) |
| PC/PE-CI | 71 (5) | −1.1 (0.3) |
| Raft-CH | 36 (18) | −1.3 (0.4) |
| Raft-CI | 80 (22) | −0.6 (0.3) |
| GM1-CH | 48 (18) | −0.7 (0.5) |
| GM1-CI | 69 (11) | −0.3 (0.3) |
All values are expressed with standard deviations given in parenthesis.
Aβ40 in POPC
The work by Bokvist et al.19 indicated that Aβ40 can partially insert into zwitterionic membranes such as POPC, with its hydrophobic C-terminal residues oriented toward the hydrophobic interior of the membrane. Our results are in agreement with that finding. Although the embedded C-terminal residues of Aβ40 (residues 29–40) adopted a considerable tilt angle with respect to the bilayer normal, the center of mass of these residues remained well below the membrane-water interface (Table V). This vertical distance was −1.3 ± 0.3 nm in the case of the PC-CH simulations, and −0.8 ± 0.3 nm in the case of PC-CI. Thus, despite the fact that the C-terminal Val40 snorkeled toward the membrane-water interface (in both PC-CH and PC-CI, but to a greater extent in PC-CI wherein the C-terminus was ionized), the hydrophobic C-terminal residues did not exit the membrane (Figs. 1 and 2 of the Supporting Information). Aβ40 was stably inserted in this PC-containing membrane, as Bokvist et al.19 and others36,37 have predicted.
Aβ40 in POPS
In addition to proposing the partial insertion of Aβ40 into zwitterionic membranes, Bokvist et al. also demonstrated that Aβ40 could insert more deeply within anionic lipid membranes than those composed of neutral lipids. With respect to the C-terminal position relative to the interface, our simulations of Aβ40 in POPS showed that the Aβ40 peptide remained more deeply embedded in the membrane than in the case of the POPC systems, −1.4 ± 0.2 nm for PS-CH systems and −1.13 ± 0.05 nm for PS-CI systems. The significantly deeper insertion of the PS-CI systems relative to the PC-CI systems was derived from the negative charge on the C-terminal Val40 residue. Although it had a tendency to snorkel toward the polar membrane-water interface (Figs. 3 and 4 of the Supporting Information), it was ultimately repelled by the net negative charge of this environment and remained within the hydrophobic core of the membrane.
Aβ40 in POPC/POPE
In an equimolar mixture of POPC and POPE, Aβ40 also remained embedded in the membrane, regardless of the protonation state of Val40. The average relative z-position of residues 29–40 was −0.867 ± 0.009 in the case of PC/PE-CH and −1.1 ± 0.03 nm for PC/PE-CI. As with POPC and POPS systems, these residues tilted with respect to the bilayer normal, 37° ± 26° in the case of PC/PE-CH systems and 71° ± 5° in PC/PE-CI systems. These tilt angles are in good agreement with the findings of Ravault et al.,38 who studied the dynamics of a truncated form of Aβ, the peptide fragment from residues 29–42, in a 9:1 POPC:POPE membrane. To our knowledge, this is the only experimentally characterized system that is comparable to the one we have studied here. Although Ravault et al. explored a slightly different system over a longer timeframe (ms and higher) than is accessible to atomistic simulations (ns–μs), our results are in good qualitative agreement. In that study, and as shown by our results, the C-terminal residues of Aβ tended to localize just below the membrane-water interface, with the C-terminus tilted up to interact with this polar region (Supporting Information Figs. 5 and 6).
Aβ40 in raft systems
In lipid raft systems without GM1, the position of the center of mass of residues 29–40 remained well below the membrane-water interface, located at −1.3 ± 0.4 nm and −0.6 ± 0.3 nm for Raft-CH and Raft-CI systems, respectively, relative to the membrane-water interface (Fig. 4(A,B) and Supporting Information Figs. S9 and S10). Some tilting and disordering of residues 29–40 was intrinsic to Aβ in lipid rafts, as well, with Raft-CH systems tilting, on an average, 36° ± 18° and Raft-CI systems, 80° ± 22°. In the case of Raft-CI systems, the ionized state of Val40 caused the carboxylate moiety to snorkel toward the membrane interface in all five simulations, leading to greater tilt and proximity to the interface overall (Fig. 10 of the Supporting Information). However, based on the position of residues 29–40, Aβ40 clearly remained embedded in all raft systems in the absence of GM1. The rigidity of the membrane itself may also be a contributing factor in the approach of the Raft-CI peptides to the interface. Unlike the more fluid PC, PS, and PC/PE membranes, the cholesterol-rich lipid raft was more densely packed and thus likely limiting the dynamics of Aβ40. Our previous work18 demonstrated a reciprocal effect of Aβ40 on the surrounding membrane, such that nearby lipids tilted along with the peptide. In the case of an ordered raft domain, this tilting is disfavored.
In GM1-CH systems, the average position of residues 29–40 relative to the interface was at −0.7 ± 0.5 nm, an upward shift of +0.6 nm with respect to the Raft-CH systems. In GM1-CI systems, the average relative z-position was located at −0.3 ± 0.3 nm, a shift of +0.3 nm relative to the Raft-CI systems. Tilting of the C-terminal region occurred to a similar extent in GM1-containing systems as with the rafts lacking GM1, 48° ± 18° in GM1-CH systems and 69° ± 11° in GM1-CI systems [Fig. 4(C,D)]. What is most notable in the simulations of the GM1-containing rafts is that in one of the GM1-CH trajectories, residues 29–40 of the Aβ peptide exited the membrane, establishing a free-energy minimum at +0.06 nm above the membrane interface [Fig. 4(E)]. In two of the GM1-CI systems, Aβ also exited the membrane, finding free-energy minima at +0.25 nm [GM1-CI-5, Fig. 4(F)] and −0.03 nm (GM1-CI-4). The free-energy minimum of simulation GM1-CI-4 indicates that residues 29–40 of the peptide were effectively coincident with the membrane-water interface and spent substantial time above this interface during the trajectory, sampling configurations as high as +0.41 nm. As a result, the free-energy basin from simulation GM1-CI-4 is broader, such that configurations at +0.06 nm above the interface are separated from the free-energy minimum by only 1.38 kJ mol−1, accessible by thermal fluctuation at 310 K (RT = 2.58 kJ mol−1). Snapshots from the end of all simulations and corresponding free-energy surfaces are shown in Figures 9 and 10 of the Supporting Information.
The contours of the free-energy surfaces provide detail into the path along which Aβ40 proceeded to the free-energy minima in these trajectories. In the case of GM1-CH-5 [Fig. 4(E)], the presence of GM1 caused residues 29–40 of Aβ to tilt, cross the membrane interface, and reach equilibrium ∼+0.06 nm above the membrane-water interface. In the case of GM1-CI-5 [Fig. 4(F)], the Aβ40 peptide rose out of the membrane before tilting, establishing its free-energy minimum at +0.25 nm above the interface. In both GM1-CH-5 and GM1-CI-5, the backbone of hydrophobic amino acids in Aβ formed hydrogen bonds with nearby GM1 molecules. In GM1-CH-5, the backbone of Gly38 and Val39 interacted with Glc and the ceramide backbone of GM1. Upon forming these hydrogen bonds, the C-terminal residues tilted up toward the interface, drawing this region of the peptide out of the membrane. In GM1-CI-5, the backbone of G33LM35 hydrogen bonded with Glc and Neu5Ac, pulling the peptide up before residues V36GGVV40 tilted at the membrane-water interface. Despite subtle differences in the progression of these two trajectories, a common feature emerges: hydrogen-bonding groups, both donors and acceptors, on GM1 in the vicinity of the membrane-embedded residues of Aβ compete for native backbone hydrogen bonding and allow hydrophobic residues 29–40 to approach, and ultimately cross, the membrane-water interface. The oligosaccharide headgroup of GM1 sequesters the N-terminal residues of Aβ away from the membrane-water interface, blocking interactions (like those observed in the POPC, POPS, and POPC/POPE systems studied here) that otherwise anchor Aβ in the membrane. We note that in simulation PC/PE-CH-3, the presence of a hydrogen-bond donor group (a primary amine) induced the formation of a β-hairpin, but this group did not enable Aβ40 to escape the membrane. Thus, it appears that GM1 possesses a unique ability to facilitate this process.
Discussion
All previous MD studies of Aβ in membranes17,18,27,36,39–41 have consisted of only a single lipid type or an implicit model representing a membrane. In this work, however, we have explored numerous explicit model membranes, including the most complex lipid environments in which Aβ has been simulated to date, rafts that correspond very closely to the lipid matrix that Aβ encounters upon its production following γ-secretase cleavage of APP. These simulations provide insight into the behavior of Aβ during a time period for which experimental evidence is limited, but critical for understanding the early events in the development of Alzheimer's disease.
A previous MD study conducted on Aβ40 in a model DPPC bilayer showed that Aβ could spontaneously exit the membrane environment,27 but those findings are in direct contrast to experimental19,37 and theoretical17,36,39 demonstrations that Aβ40 remains partially inserted in such a lipid environment, with residues 29–40 embedded in the hydrophobic core of the bilayer. The discrepancy has been attributed to the use of better-quality force field models in the more recent studies.36 Using this improved force field model, we were able to reproduce experimental behavior of Aβ40 in DPPC.17,18 We have applied that same force field model in this study. It is important to note, however, that all force fields have some inherent limitations and biases that must be taken into account,42,43 and that studies on structurally dynamic proteins such as Aβ40 require great care in interpreting results. We note that, despite our efforts to comprehensively sample conformations that Aβ40 may adopt through relatively long simulations (100 ns) and several replicates, our data reflect the considerable heterogeneity intrinsic to the peptide. Thus, we focus on broader aspects of our observations while still considering the fact that the current limitations of atomistic simulations may prevent us from obtaining a complete understanding of structural transitions that occur over much longer time scales in vitro and in vivo.
In all of the simple membrane models explored here (POPC, POPS, and POPC/POPE), Aβ40 remained embedded within the membrane, with residues 29–40 well below the membrane-water interface. Ionization of C-terminal Val40 tended to cause some destabilization of the initial α-helical configuration of this segment of the peptide as Val40 snorkeled toward the interface. This behavior was observed regardless of the lipid composition of the surrounding membrane. In POPS, the ability of Val40 to approach the interface was diminished because of charge repulsion between the anionic lipids and the ionized C-terminus. Snorkeling contributed to the tilt of the embedded C-terminal region, a behavior that has been suggested to have implications for the toxicity of Aβ exerted through its interactions with membranes.18,23,38
The main secondary structural elements of Aβ40 in each of the simple model membranes were helices and random coils, though short, transient β-hairpins were observed, principally within its N-terminal polar region. The POPC, POPS, and POPC/POPE membranes did not significantly stabilize long β-strands, though it appears that the membrane environment may be conducive to the formation of such structures. Furthermore, these β-hairpin structures involved many different residues within the N-terminal sequence and were variable in terms of their location and spacing by intervening bends or turns. Thus, it appears that PC, PS, and PE lipids induce nonspecific β-strand formation.
In POPC, POPS, and POPC/POPE, Aβ40 anchored itself to the membrane through hydrogen bonding and other electrostatic interactions, in agreement with our previous findings regarding Aβ40 in DPPC.17,18 The increased hydrogen bonding between Aβ40 and POPS and POPC/POPE relative to POPC may explain why Aβ binds more tightly to these lipids than PC-containing lipids.19 In POPS and POPC/POPE, there are simply more functional groups available to form hydrogen bonds with Aβ40.
Clusters of ganglioside GM1 create a polar, dehydrated environment that extends more than 2 nm above the membrane-water interface. Experiments have shown that dehydrated environments facilitate amyloid aggregation by reducing the free-energy barrier of desolvating the membrane interface.44 The oligosaccharide headgroup of GM1 possesses hydrogen-bond donors and acceptors that compete for protein–protein hydrogen bonds and destabilize native α-helical structures, a behavior that accounts for the increased rate of β-strand formation observed experimentally.44 In our simulations, the formation of β-hairpins was stabilized by specific interactions between Aβ residues (His6, Asp7, His13, and His14) and GM1, most commonly Neu5Ac and Gal2, sugar moieties that have previously been predicted to be important in Aβ-GM1 binding.30 The sequestration of the polar N-terminus of Aβ away from the membrane-water interface, within the oligosaccharide headgroup of GM1 clusters, precluded nonspecific electrostatic interactions (like those of the POPC, POPS, and POPC/POPE systems) that otherwise anchor Aβ to the membrane.18 Our results indicate that GM1 clusters are required for this behavior. In simulations of Aβ40 in rafts containing only one GM1 molecule at random locations, Aβ40 bound to GM1, but the N-terminal region was not sequestered above the membrane-water interface. As a result, Aβ40 adopted positions, orientations, and secondary structure content that was indistinguishable from the results of raft-only simulations (data not shown).
The ability of GM1 to promote the release of Aβ40 from a lipid raft was a result of the presence of hydrogen-bonding groups near the membrane-water interface. Interactions between the peptide backbone of residues 29–40 and the Glc and Neu5Ac moieties of GM1 can lead to disordering of the peptide and transport out of the hydrophobic lipid matrix. The numerous hydrogen-bond donors and acceptors in the oligosaccharide headgroup of GM1 can effectively compete for the native intraprotein hydrogen bonding in residues 29–40, leading to destabilization of the helix and exit from the membrane. The phospholipid headgroups in rafts lacking GM1 have only hydrogen-bond acceptors (phosphate groups), which, as demonstrated here, interacted with Aβ40 in a manner that did not lead to efficient release of Aβ40 from the membrane. From this study and our previous work,17 it appears that the C-terminal residues of Aβ40 have some intrinsic capacity to approach, but not cross, the polar interface region, a process that was influenced to some degree by the protonation state of the C-terminal Val40. This effect may be enhanced in rigid, densely packed raft domains.
In general, model CI peptides approached the membrane-water interface more closely than did model CH peptides. Deprotonated peptides likely have a greater affinity for the polar interfacial environment, contributing to their stability in this region and eventual release from the membrane. Model CH peptides were capable of considerable C-terminal tilting in all membranes examined here, placing Val40 close to the interface, indicating that proton exchange between the peptide and any water molecules at the interface may be possible. Thus, a dynamic equilibrium between protonated and deprotonated C-termini may exist in vivo, but this is not a phenomenon that can directly be addressed using MD simulations, as bonds cannot break or form under classical mechanics assumptions.
The embedded C-terminal residues lost some of their initial α-helicity as they moved away from the hydrophobic core of the membrane and became more proximal to the polar interfacial environment. Our data indicate that GM1 facilitates this process for both CH and CI models of the Aβ40 peptide, enhancing the release of Aβ40 from its native lipid raft environment and promoting the exposure of hydrophobic, aggregation-prone residues to the extracellular environment.
Conclusion
There is an apparent paradox in the amyloid hypothesis. If Aβ exerts its toxicity by stably binding to, and inserting in lipid membranes, how does Aβ leave the membrane in the first place and gain access to the aqueous extracellular space? Our results provide insight into this mechanism and a very specific lipid that may resolve this issue. Aβ-GM1 interactions promote the structural conversion of Aβ from α-helix to β-strand, while simultaneously providing a scaffold that allows the C-terminal hydrophobic region of Aβ (residues 29–40) to unwind, tilt upward, and exit the membrane, a behavior that we observed in several simulations described here. Membrane exit was not observed in any of the simulations of Aβ40 in POPC, POPS, or POPC/POPE. Rather, the stability of Aβ in these environments may shed light on the ability of Aβ to reinsert into membranes and exert its toxic, membrane-destabilizing effects.
The behavior described here may provide insight into the earliest events in the Aβ aggregation cascade, the release of the Aβ peptide following its production in lipid rafts. It should be noted that our results do not preclude the possibility that traumatic brain injury or other neuronal damage might also contribute to the increased secretion of Aβ, as has been suggested previously.45,46 Prior damage to the plasma membrane may indeed release large amounts of Aβ and initiate the aggregation cascade in a different manner than that which is observed in otherwise healthy individuals. The mechanism we propose here may be useful for designing small-molecule inhibitors of Aβ-GM1 interactions, preventing the formation of aggregation-prone Aβ conformations and the release of Aβ from the plasma membrane during the earliest stages of Alzheimer's disease.
Methods
System construction
We conducted MD simulations of Aβ40 in model membranes consisting of pure POPC and POPS and an equimolar mixture of POPC and POPE. Coordinates and topologies for preequilibrated POPC47 and POPS48 membranes (128 lipids each) were obtained from D. P. Tieleman's site (http://moose.bio.ucalgary.ca) in the case of POPC and by personal communication in the case of POPS. Coordinates for the binary POPC-POPE system (288 total lipids, 144 each of POPC and POPE) were taken from the work by Leekumjorn et al.,49 with lipid topologies also from Tieleman's work.47,50 Lipid raft membranes contained POPC, PSM, and cholesterol, with and without 6.75 mol% GM1, which represents a physiologically relevant concentration of GM1.9 The coordinates and topologies for the model lipid rafts were taken from the work by Niemelä et al.51 Topologies for cholesterol and PSM were updated to replace GROMOS8752 atom types and charges with those of equivalent functional groups in the GROMOS96 53A6 force field (Supporting Information Tables I and II).53 The topology for GM1 was created using nonbonded parameters derived by Berger et al.54 for the acyl chain portion and those of the GROMOS96 53A6 force field for all other functional groups. The GROMOS96 53A6 force field was applied to the remaining components of the system. The topology for GM1 is given in Supporting Information Table III. From the initial raft configuration,51 a subset of lipids containing 222 lipids (74 each of POPC, PSM, and cholesterol) was extracted. Such a system is of sufficient size to accommodate the Aβ40 peptide and avoid spurious interactions across periodic boundaries during the MD simulations. To create a raft system containing ganglioside GM1, 15 PSM molecules in the extracellular leaflet of the raft were replaced with GM1 to give a total GM1 content of 6.75 mol% to reproduce the experimentally determined 6.59% ganglioside content in lipid rafts in vivo9 as accurately as possible. The rafts (both with and without GM1) were energy minimized and equilibrated for 20 ns before inserting the Aβ40 peptide. Residues 29–40 of Aβ40 were aligned with the membrane normal, with Lys28 coincident with the average phosphate plane, in agreement with experimental evidence of this location.55 Lipids were packed around the peptide using the InflateGRO method,56 modified in-house to accommodate multiple lipid types (in the case of raft and POPC/POPE systems). The remainder of the unit cell was filled with SPC water57 and ∼150 mM NaCl (including Na+ counterions).
The model of Aβ40 used here was taken from PDB entry 1BA4,55 using the same configuration from the NMR ensemble as in our earlier work.17,18 This structure was determined in the presence of sodium dodecyl micelles and, thus, is assumed to be a representative configuration for Aβ40 in the presence of a membrane. The transmembrane domain of APP and the polypeptide sequence that encompasses the Aβ sequence is believed to be principally helical with disordered solvent-exposed regions.58 For these reasons, we believe this starting structure to be applicable to modeling Aβ40 inserted in a membrane. For reasons described previously,17 both neutral (model “CH”) and ionized (model “CI”) C-termini were considered. Given that the γ-secretase complex has a solvent-accessible active site,5 both protonation states are plausible and potentially relevant to the dynamics of Aβ. Other titratable groups were assigned their typical protonation states at pH 7.4. The net charge on the CH peptide was −2, whereas that on the CI peptide was −3.
Molecular dynamics simulations
All simulations were performed with GROMACS, version 4.0.7.59 Periodic boundary conditions were applied in all directions. The real-space contribution to the Coulombic potential was truncated at 1.2 nm, and long-range electrostatics were calculated using the smooth particle-mesh Ewald method,60,61 using cubic-spline interpolation and a Fourier grid spacing of 0.12 nm. Van der Waals interactions were truncated at 1.2 nm, and dispersion correction was applied to the energy and pressure terms. All bond lengths were constrained using the P-LINCS algorithm,62 allowing an integration time step of 2 fs. Coordinates and energies were saved every 10 ps for analysis. Five independent simulations were produced for each raft system (Raft-CH, GM1-CH, Raft-CI, and GM1-CI) by assigning different random velocities from a Maxwell distribution at the outset of equilibration. Three independent simulations were generated for all other systems (PC-CH, PC-CI, PS-CH, PS-CI, PC/PE-CH, and PC/PE-CI).
Simulation systems were equilibrated in three phases, all of which used position restraints (kpr = 1000 kJ mol−1 nm−2) on all heavy atoms of the Aβ40 peptide. As the InflateGRO procedure removed all water molecules in the input configuration that needed to be added back later, the hydration of the equilibrated lipids was disrupted, requiring careful re-equilibration. The first phase used an isochoric-isothermal (NVT) ensemble, with temperature controlled by the Berendsen weak coupling algorithm.63 The NVT ensemble was applied for 100 ps, during which the temperature of the system was maintained at 100 K with a coupling constant of 0.1 ps. The protein, lipids, and solvent (including ions) were coupled separately. During NVT equilibration, position restraints were placed on lipid phosphorus atoms to restrict their motion to the x-y plane. Following NVT equilibration, the restraints on the lipids were removed, and simulated annealing was performed to heat the system linearly from 100 to 310 K over 500 ps under an isobaric-isothermal (NPT) ensemble. During annealing, the Berendsen weak coupling method63 was used to control both temperature and pressure (1 bar). Coupling constants for temperature and pressure were 0.1 and 2.0 ps, respectively, and pressure was coupled semiisotropically to allow independent deformations of the system in the x-y and z dimensions. Following annealing, 1 ns of NPT equilibration was performed, using the Nosé–Hoover thermostat64,65 and Parrinello–Rahman barostat.66,67 Coupling constants were the same as during the NPT equilibration phase. Production simulations were carried out for 100 ns under the same NPT ensemble in the absence of any restraints.
Analysis
All data analysis was conducted using tools present within the GROMACS distribution. Secondary structure assignments were determined using the DSSP algorithm.68 Images of Aβ40-membrane systems were generated with PyMOL.69 Statistical outliers were identified using a Q-test with 95% confidence. All values following ± are standard deviations, unless otherwise noted.
Acknowledgments
The authors thank Prof. Mikko Karttunen for providing lipid raft coordinates and topologies, Prof. D. Peter Tieleman for providing coordinates and topologies for the POPS membrane, Sukit Leekumjorn for providing coordinates and topologies for the POPC/POPE membrane, William J. Allen for critical reading of this manuscript, and Virginia Tech Advanced Research Computing for computing time on the SystemX supercomputer.
Supplementary material
References
- 1.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 3.Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 4.Cole SL, Vassar R. The role of amyloid precursor protein processing by BACE1, the β-secretase, in Alzheimer disease pathophysiology. J Biol Chem. 2008;283:29621–29625. doi: 10.1074/jbc.R800015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steiner H, Fluhrer R, Haass C. Intramembrane proteolysis by γ-secretase. J Biol Chem. 2008;283:29627–29631. doi: 10.1074/jbc.R800010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hur J-Y, Welander H, Behbahani H, Aoki M, Frånberg J, Winblad B, Frykman S, Tjernberg LO. Active γ-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 2010;275:1174–1187. doi: 10.1111/j.1742-4658.2008.06278.x. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda M, Kihara A, Igarashi Y. Lipid asymmetry of the eukaryotic plasma membrane: functions and related enzymes. Biol Pharm Bull. 2006;29:1542–1546. doi: 10.1248/bpb.29.1542. [DOI] [PubMed] [Google Scholar]
- 8.Devaux PF. Static and dynamic lipid asymmetry in cell membranes. Biochemistry. 1991;30:1163–1173. doi: 10.1021/bi00219a001. [DOI] [PubMed] [Google Scholar]
- 9.Sonnino S, Mauri L, Chigorno V, Prinetti A. Gangliosides as components of lipid membrane domains. Glycobiology. 2006;17:1R–13R. doi: 10.1093/glycob/cwl052. [DOI] [PubMed] [Google Scholar]
- 10.Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer's disease. Nat Med. 1995;1:1062–1066. doi: 10.1038/nm1095-1062. [DOI] [PubMed] [Google Scholar]
- 11.Kakio A, Nishimoto S-i, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem. 2001;276:24985–24990. doi: 10.1074/jbc.M100252200. [DOI] [PubMed] [Google Scholar]
- 12.Kakio A, Nishimoto S-i, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Interactions of amyloid β-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry. 2002;41:7385–7390. doi: 10.1021/bi0255874. [DOI] [PubMed] [Google Scholar]
- 13.Matsuzaki K, Horikiri C. Interactions of amyloid β-peptide (1-40) with ganglioside-containing membranes. Biochemistry. 1999;38:4137–4142. doi: 10.1021/bi982345o. [DOI] [PubMed] [Google Scholar]
- 14.Mikhalyov I, Olofsson A, Gröbner G, Johansson LB-Å. Designed fluorescent probes reveal interactions between amyloid-β(1-40) peptides and GM1 gangliosides in micelles and lipid vesicles. Biophys J. 2010;99:1510–1519. doi: 10.1016/j.bpj.2010.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Utsumi M, Yamaguchi Y, Sasakawa H, Yamamoto N, Yanagisawa K, Kato K. Up-and-down topological mode of amyloid β-peptide lying on hydrophilic/hydrophobic interface of ganglioside clusters. Glyconjugate J. 2009;26:999–1006. doi: 10.1007/s10719-008-9216-7. [DOI] [PubMed] [Google Scholar]
- 16.Yagi-Utsumi M, Kameda T, Yamaguchi Y, Kato K. NMR characterization of the interactions between lyso-GM1 aqueous micelles and amyloid β. FEBS Lett. 2010;584:831–836. doi: 10.1016/j.febslet.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Lemkul JA, Bevan DR. A comparative molecular dynamics analysis of the amyloid β-peptide in a lipid bilayer. Arch Biochem Biophys. 2008;470:54–63. doi: 10.1016/j.abb.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Lemkul JA, Bevan DR. Perturbation of membranes by the amyloid β-peptide—a molecular dynamics study. FEBS J. 2009;276:3060–3075. doi: 10.1111/j.1742-4658.2009.07024.x. [DOI] [PubMed] [Google Scholar]
- 19.Bokvist M, Lindström F, Watts A, Gröbner G. Two types of Alzheimer's β-amyloid (1-40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J Mol Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 20.Chi EY, Ege C, Winans A, Majewski J, Wu G, Kjaer K, Lee KYC. Lipid membrane templates the ordering and induces the fibrillogenesis of Alzheimer's disease amyloid-β peptide. Proteins. 2008;72:1–24. doi: 10.1002/prot.21887. [DOI] [PubMed] [Google Scholar]
- 21.Curtain CC, Ali FE, Smith DG, Bush AI, Masters CL, Barnham KJ. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer's disease amyloid-β peptide with membrane lipid. J Biol Chem. 2003;278:2977–2982. doi: 10.1074/jbc.M205455200. [DOI] [PubMed] [Google Scholar]
- 22.Ege C, Lee KYC. Insertion of Alzheimer's Aβ40 peptide into lipid monolayers. Biophys J. 2004;87:1732–1740. doi: 10.1529/biophysj.104.043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mingeot-Leclercq M-P, Lins L, Bensliman M, Van Bambeke F, Van Der Smissen P, Peuvot J, Schanck A, Brasseur R. Membrane destabilization induced by β-amyloid peptide 29-42: importance of the amino-terminus. Chem Phys Lipids. 2002;120:57–74. doi: 10.1016/s0009-3084(02)00108-1. [DOI] [PubMed] [Google Scholar]
- 24.Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET. Structural characterization of a soluble amyloid β-peptide oligomer. Biochemistry. 2009;48:1870–1877. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 25.Tycko R. Insights into the amyloid folding problem from solid-state NMR. Biochemistry. 2003;42:3151–3159. doi: 10.1021/bi027378p. [DOI] [PubMed] [Google Scholar]
- 26.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-β(1-42) fibrils. Proc Natl Acad Sci USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H. Conformational transition of amyloid β-peptide. Proc Natl Acad Sci USA. 2005;102:5403–5407. doi: 10.1073/pnas.0501218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müller E, Giehl A, Schwarzmann G, Sandhoff K, Blume A. Oriented 1,2-dimyristoyl-sn-glycerol-3-phosphorylcholine/ganglioside membranes: a Fourier transform infrared attenuated total reflection spectroscopic study. Band assignments; orientational, hydrational, and phase behavior; and effects of Ca2+ binding. Biophys J. 1996;71:1400–1421. doi: 10.1016/S0006-3495(96)79342-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terzi E, Hölzemann G, Seelig J. Self-association of β-amyloid peptide (1-40) in solution and binding to lipid membranes. J Mol Biol. 1995;252:633–642. doi: 10.1006/jmbi.1995.0525. [DOI] [PubMed] [Google Scholar]
- 30.Ariga T, Kobayashi K, Hasegawa A, Kiso M, Ishida H, Miyatake T. Characterization of high-affinity binding between gangliosides and amyloid β-protein. Arch Biochem Biophys. 2001;388:225–230. doi: 10.1006/abbi.2001.2304. [DOI] [PubMed] [Google Scholar]
- 31.Fantini J, Yahi N. Molecular basis for the glycosphingolipid-binding specificity of α-synuclekey role of tyrosine-39 in membrane insertion. J Mol Biol. 2011;408:654–669. doi: 10.1016/j.jmb.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Durell SR, Guy HR, Arispe N, Rojas E, Pollard HB. Theoretical models of the ion channel structure of amyloid β-protein. Biophys J. 1994;67:2137–2145. doi: 10.1016/S0006-3495(94)80717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaurin J, Chakrabartty A. Characterization of the interactions of Alzheimer β-amyloid peptides with phospholipid membranes. Eur J Biochem. 1997;245:355–363. doi: 10.1111/j.1432-1033.1997.t01-2-00355.x. [DOI] [PubMed] [Google Scholar]
- 34.Fraser PE, Nguyen JT, Chin DT, Kirschner DA. Effects of sulfate ions on Alzheimer β/A4 peptide assemblies: implications for amyloid fibril-proteoglycan interactions. J Neurochem. 1992;59:1531–1540. doi: 10.1111/j.1471-4159.1992.tb08470.x. [DOI] [PubMed] [Google Scholar]
- 35.Brunden KR, Richter-Cook NJ, Chaturvedi K, Frederickson RCA. pH-dependent binding of synthetic β-amyloid peptides to glycosaminoglycans. J Neurochem. 1993;61:2147–2154. doi: 10.1111/j.1471-4159.1993.tb07453.x. [DOI] [PubMed] [Google Scholar]
- 36.Reddy AS, Izmitli A, de Pablo JJ. Effect of trehalose on amyloid β (29-40)-membrane interaction. J Chem Phys. 2009;131:085101. doi: 10.1063/1.3193726. [DOI] [PubMed] [Google Scholar]
- 37.Yip CM, McLaurin J. Amyloid-β peptide assembly: a critical step in fibrillogenesis and membrane disruption. Biophys J. 2001;80:1359–1371. doi: 10.1016/S0006-3495(01)76109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ravault S, Soubias O, Saurel O, Thomas A, Brasseur R, Milon A. Fusogenic Alzheimer's peptide fragment Aβ (29-42) in interaction with lipid bilayers: secondary structure, dynamics, and specific interaction with phosphatidyl ethanolamine polar heads as revealed by solid-state NMR. Protein Sci. 2005;14:1181–1189. doi: 10.1110/ps.041291405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mobley DL, Cox DL, Singh RRP, Maddox MW, Longo ML. Modeling amyloid β-peptide insertion into lipid bilayers. Biophys J. 2004;86:3585–3597. doi: 10.1529/biophysj.103.032342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis CH, Berkowitz ML. Interaction between amyloid-β (1-42) peptide and phospholipid bilayers: a molecular dynamics study. Biophys J. 2009;96:785–797. doi: 10.1016/j.bpj.2008.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis CH, Berkowitz ML. Structure of the amyloid-β (1-42) monomer absorbed to model phospholipid bilayers: a molecular dynamics study. J Phys Chem B. 2009;113:14480–11486. doi: 10.1021/jp905889z. [DOI] [PubMed] [Google Scholar]
- 42.Best RB, Buchete N-V, Hummer G. Are current molecular dynamics force fields too helical? Biophys J. 2008;95:L07–L09. doi: 10.1529/biophysj.108.132696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matthes D, de Groot BL. Secondary structure propensities in peptide folding simulations: a systematic comparison of molecular mechanics interaction schemes. Biophys J. 2009;97:599–608. doi: 10.1016/j.bpj.2009.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukherjee S, Chowdhury P, Gai F. Effect of dehydration on the aggregation kinetics of two amyloid peptides. J Phys Chem B. 2009;113:531–535. doi: 10.1021/jp809817s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts GW, Gentleman SM, Lynch A, Graham DI. β A4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–1423. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 46.Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang S-H, Zhong Z, Shen Y, Simpkins JW, Tanzi RE. Depletion of GGA3 stabilizes BACE and enhances β-secretase activity. Neuron. 2007;54:721–737. doi: 10.1016/j.neuron.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tieleman DP, Sansom MSP, Berendsen HJC. Alamethicin helices in a bilayer and in solution: molecular dynamics simulations. Biophys J. 1999;76:40–49. doi: 10.1016/S0006-3495(99)77176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukhopadhyay P, Monticelli L, Tieleman DP. Molecular dynamics simulation of a palmitoyl-oleoyl phosphatidylserine bilayer with Na+ counterions and NaCl. Biophys J. 2004;86:1601–1609. doi: 10.1016/S0006-3495(04)74227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leekumjorn S, Wu Y, Sum AK, Chan C. Experimental and computational studies investigating trehalose protection of HepG2 cells from palmitate-induced toxicity. Biophys J. 2008;94:2869–2883. doi: 10.1529/biophysj.107.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tieleman DP, Forrest LR, Sansom MSP, Berendsen HJC. Lipid properties and the orientation of aromatic residues in OmpF, influenza M2, and alamethicin systems: molecular dynamics simulations. Biochemistry. 1998;37:17554–17561. doi: 10.1021/bi981802y. [DOI] [PubMed] [Google Scholar]
- 51.Niemelä PS, Ollila S, Hyvönen MT, Karttunen M, Vattulainen I. Assessing the nature of lipid raft membranes. PLoS Comp Biol. 2007;3:e34. doi: 10.1371/journal.pcbi.0030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Gunsteren WF, Berendsen HJC. Groningen molecular simulation (GROMOS) library manual, BIOMOS b.v. Groningen: University of Groningen; 1987. [Google Scholar]
- 53.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 54.Berger O, Edholm O, Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ. Solution structure of amyloid β-peptide(1-40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry. 1998;37:11064–11077. doi: 10.1021/bi972979f. [DOI] [PubMed] [Google Scholar]
- 56.Kandt C, Ash WL, Tieleman DP. Setting up and running molecular dynamics simulations of membrane proteins. Methods. 2007;41:475–488. doi: 10.1016/j.ymeth.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B, editor. Intermolecular forces. Dordrecht: Reidel; 1981. p. 331. [Google Scholar]
- 58.Beel AJ, Mobley CK, Kim HJ, Tian F, Hadziselimovic A, Jap B, Prestegard JH, Sanders CR. Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): does APP function as a cholesterol sensor? Biochemistry. 2008;47:9428–9446. doi: 10.1021/bi800993c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 60.Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 61.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 62.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J Chem Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 63.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 64.Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81:511–519. [Google Scholar]
- 65.Hoover WG. Canonical dynamics: equilibrium phase-space distributions. Phys Rev A: At Mol Opt Phys. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 66.Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. [Google Scholar]
- 67.Nosé S, Klein ML. Constant pressure molecular dynamics for molecular systems. Mol Phys. 1983;50:1055–1076. [Google Scholar]
- 68.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 69.DeLano WL. The PyMOL molecular graphics system. Palo Alto, CA: DeLano Scientific LLC; 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.