

Abstract
Generating stable antibodies is an important goal in the development of antibody-based drugs. Often, thermal stability is assumed predictive of overall stability. To test this, we used different internally created antibodies and first studied changes in antibody structure as a function of pH, using the dye ANS. Comparison of the pH50 values, the midpoint of the transition from the high-pH to the low-pH conformation, allowed us for the first time to rank antibodies based on their pH stability. Next, thermal stability was probed by heating the protein in the presence of the dye Sypro Orange. A new data analysis method allowed extraction of all three antibody unfolding transitions and showed close correspondence to values obtained by differential scanning calorimetry. T1%, the temperature at which 1% of the protein is unfolded, was also determined. Importantly, no correlations could be found between thermal stability and pH50, suggesting that to accurately quantify antibody stability, different measures of protein stability are necessary. The experimental data were further analyzed using a machine-learning approach with a trained model that allowed the prediction of biophysical stability using primary sequence alone. The pH stability predictions proved most successful and were accurate to within pH ±0.2.
Keywords: biophysics, thermal stability, pH stability, machine learning, Sypro Orange, DSC
Introduction
Antibodies and antibody-based drugs are a relatively novel class of therapeutics gaining in popularity given their high specificity, long half-life, and the more recent “modular” approach to their design.1,2 During the development process, antibody-based drugs encounter a wide range of conditions: the crowded intracellular environment during expression; harsh measures to disrupt the cell membrane; flow through tubing and several solution conditions during purification; freeze-drying or formulation; and potentially less than optimal storage conditions. Upon administration, potentially after resolubilization, the drug is exposed to the crowded and hostile environment of subcutaneous tissue and/or blood. To ensure accurate dosage, highest effectiveness, and fewest side effects to the patient, it is critical that the drug is not modified by any of these conditions.3 At the research and development stage, it is impossible to anticipate all conditions that may affect the drug. By judiciously measuring and improving the biophysical stability of a candidate drug, however, it is hoped that its expression is maximized, purification and formulation are straightforward, patient administration is easier, and, most importantly, side effects or an immune response in the patient are minimized.
Thermal stability is a common method used to study protein stability, usually by measuring the midpoint of the unfolding transition, the melting temperature, Tm.4–9 Differential scanning calorimetry (DSC) is often used as it gives a quantitative view of the process and can be used to assign the multiple transitions often seen in the unfolding of antibodies.8,10 A plate-based fluorescence assay using Sypro Orange, a dye that binds to hydrophobic regions of a protein, is often used as it allows for higher throughput using less protein.5,9 A similar method can also be used to characterize the ability of different ligands in stabilizing the protein under investigation.11 The analysis of the unfolding curves, however, has not been well established, and generally only the first unfolding transition of an antibody is studied.5,9 Even then, the analysis was found to systematically underestimate the midpoint of this transition when compared with DSC.9
It is often thought that thermal stability is a good predictor of overall stability.12–14 In published experiments, proteins were incubated in different formulations at elevated temperatures. After several weeks, the level of aggregation was found to correlate well with the protein melting point, Tm.12–14 A close study of the unfolding curves, however, shows that the incubation temperatures were sufficiently high that the protein was starting to unfold. Consequently, a formulation-induced Tm shift of only a few degrees would change dramatically the extent of unfolding, influencing the amount of aggregation. It is therefore unclear if these experiments truly reveal a link between thermal stability and inherent long-term storage stability, or simply that more unfolded protein solutions will aggregate more. Indeed, a different study has shown that not the thermal stability per se, but the reversibility of unfolding may be the important factor,15 whereas other biophysical measurements suggest there may be a link to expression levels.16,17 Overall, therefore, the link between Tm and overall stability does not seem very well established.
This observation suggests that it is important to probe aspects of stability, for example, stability to denaturants,4 pH,7,18 salt or buffer components,7,19 and aggregation tendency.6,18–20 The effect of pH seems particularly relevant, since antibodies are affinity chromatography purified at low pH and low pH is used to inactive viruses. At low pH, antibodies adopt a non-native conformation that is quite stable and not a “molten globule” but may be more aggregation prone.4,18,21
Having measured stability, different approaches have been used to predict stability based on sequence alone.22,23 Data sets used to “train” different models often incorporated more than 100 clearly defined pairs of stable and less-stable molecules. Even so, simulations appear better at predicting trends than absolute values.22
In this article, we set out to test whether different stability measurements are correlated. We developed an assay to study the range of pH values at which the conformational change to the low pH structure occurred. We devised a more accurate analysis method to extract individual unfolding transitions from Sypro Orange-monitored thermal denaturation data and compared the data to that obtained from DSC experiments. We show that thermal and pH stabilities appear not to be strongly correlated, highlighting for the first time the importance of measuring many different aspects of stability. Finally, we developed a machine-learning model to predict the stability of antibodies based on their sequence alone. The results are encouraging, although further work is necessary, in particular for the thermal stability.
Results
The ANS fluorescence intensity in the pH stability assay was constant from pH 9 down to pH 4, at which point it started to increase. Below pH 2, no more change was observed [Fig. 1(A)]. Increasing protein concentration was found to increase the ratio of the fluorescence intensity at low pH to that at neutral pH, without affecting the pH50, the halfway point in the transition [Fig. 1(B), Table I). Increasing the ANS concentration had a similar effect (data not shown). The same protein incubated at different pH values was also followed by intrinsic fluorescence and CD. Overlaying the rescaled data sets, the signals could easily be compared and were found to overlay well [Fig. 1(C)]: the fitted pH50 values were 2.76 ± 0.07 (ANS), 2.67 ± 0.04 (intrinsic fluorescence), and 2.66 ± 0.22 (CD). The pH50 values from 53 antibodies were plotted as a histogram, showing a range from pH 1.8 to pH 3.2 [Fig. 1(D)].
Figure 1.
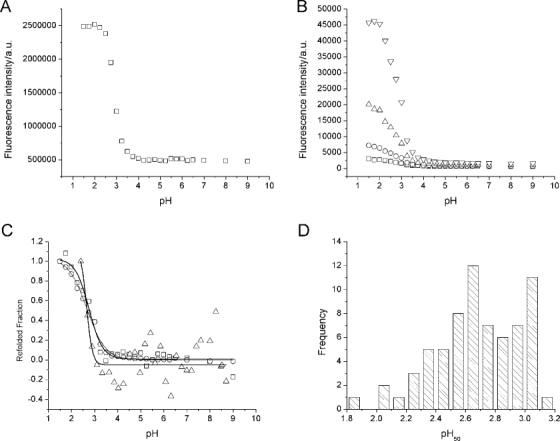
Antibody pH stability. A: A representative antibody, incubated at 0.1 mg mL−1 and different pH values overnight before adding 100-fold molar excess ANS over protein and reading the fluorescence, exciting at 360 nm and measuring emission at 500 nm. B: A second antibody, incubated at different concentrations: (□) 0.05 mg mL−1, (○) 0.1 mg mL−1, (▵) 0.2 mg mL−1, and (▿) 0.4 mg mL−1. At each concentration, ANS was present in 1:100 protein:ANS molar ratio. C: pH stability using ANS fluorescence (□), intrinsic fluorescence (○), and CD signal at 205 nm (▵). Lines are fitted sigmoidals with pH50 values of pH 2.76 ± 0.07 (ANS), pH 2.67 ± 0.04 (intrinsic fluorescence), and pH 2.66 ± 0.22 (CD). D: Histogram showing the frequency of all pH50 values measured.
Table I.
Relation Between Protein Concentration, Fluorescence at pH 2 and at pH 7, and the pH50 Obtained
| Concentration (mg mL−1) | Fluorescence ratio pH 2/pH 7 | pH50 |
|---|---|---|
| 0.05 | 5 | 2.75 ± 0.04 |
| 0.1 | 10 | 2.67 ± 0.04 |
| 0.2 | 17 | 2.67 ± 0.03 |
| 0.4 | 29 | 2.71 ± 0.04 |
As a function of protein concentration, the ratio of ANS fluorescence intensities at pH 2 and pH 7 increases, whereas the pH50 remains within error.
We next looked at the thermal stability of the antibodies using a modified fluorescence-based method with dye Sypro Orange. Plotting fluorescence intensity against temperature, a decrease in fluorescence was observed from 20°C to around 50°C [Fig. 2(A)]. At higher temperatures, this baseline decrease was less pronounced and easily corrected by subtracting a linear baseline. Above about 50°C, the protein starts to unfold, and an increase in fluorescence intensity was observed, reaching a maximum around 75°C and then decreasing [Fig. 2(A)].
Figure 2.
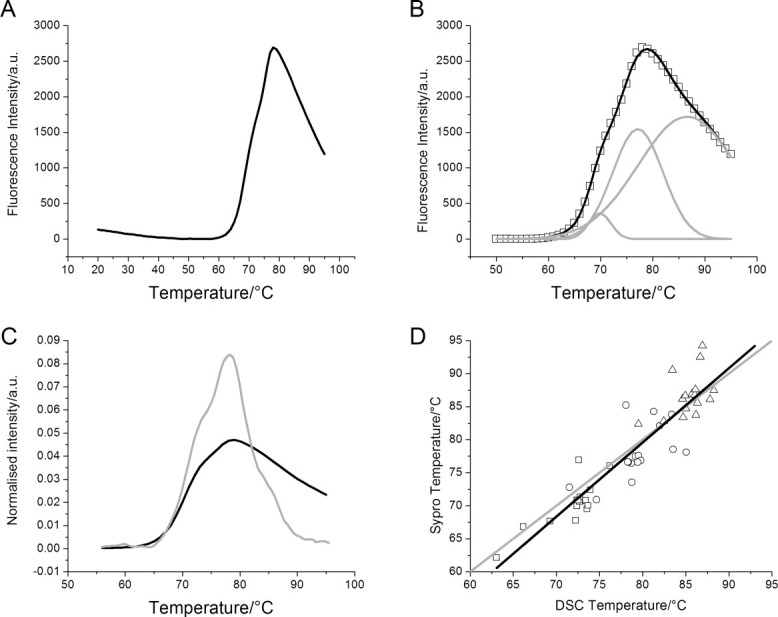
Sypro Orange thermal stability data. A: Representative data showing Sypro Orange fluorescence intensity as a function of temperature. The data have been baseline corrected. B: The same data fitted to three Gaussians. The original data are shown in symbols (□), the three Gaussians are shown as gray lines, and the sum of the Gaussians is the black line. C: Comparing Sypro Orange data with DSC data. Both curves have been normalized to an area under the curve of one. D: For 16 proteins, both Sypro Orange and DSC data were collected; the individual data for Tm1 (□), Tm2 (○), and Tm3 (▵) are shown here. The gray line shows the case where DSC and Sypro Orange data would be identical. The black line is a straight line fit to the actual data points. The slope is 1.12, R = 0.92, and R2 = 0.85.
The unfolding data were fitted to three Gaussians (see Materials and Methods for a detailed procedure) to extract the three unfolding transitions10 expected for an antibody [Fig. 2(B)]. Unfolding data for the same antibody were also collected by DSC [Fig. 2(C)]. The unfolding transitions from the Gaussian fits (Sypro Orange fluorescence data) and those from a thermodynamic analysis (DSC data) of 16 different proteins were plotted against each other [Fig. 2(D)]. A straight line fit through all the data had a slope of 1.12, with correlation coefficient R = 0.92 (P < 0.0001). The coefficient of determination, R2, which indicates how much variance in one variable can be predicted from the other, was high at 0.85. A further statistical analysis24 was also performed (see Supporting Information).
Similar to the pH50 values, histograms of the Tm values and the widths of the three thermal transitions were plotted [Fig. 3(A,B)]. A spread of about 16°C is observed for the Tm values [Fig. 3(A)]. The T1%, the temperature at which 1% of the protein is unfolded (see Materials and Methods), was calculated, and this is shown as a histogram in Figure 3(C).
Figure 3.
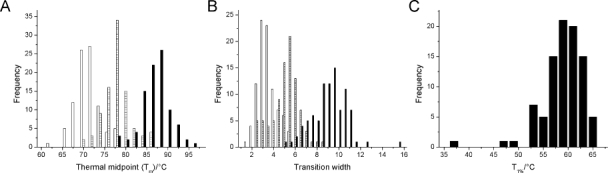
Histograms of thermal unfolding data. A: Histogram showing the distribution of the thermal transition midpoints, Tm1 (open bars), Tm2 (dashed bars), and Tm3 (solid bars). B: Histogram of the widths (standard deviations) of the Gaussians used to fit the Sypro Orange data, width for Tm1 (open bars), Tm2 (dashed bars), and Tm3 (solid bars). C: Histogram of T1% data, the temperature at which 1% of the protein is unfolded.
To look for correlations between the measured biophysical factors, the pH50 values were plotted against the different thermal indicators (Fig. 4). The correlation factor R and the coefficient of determination R2 were low, and the linear regression slopes were below 0.04 for all plots (Fig. 4, Table II).
Figure 4.
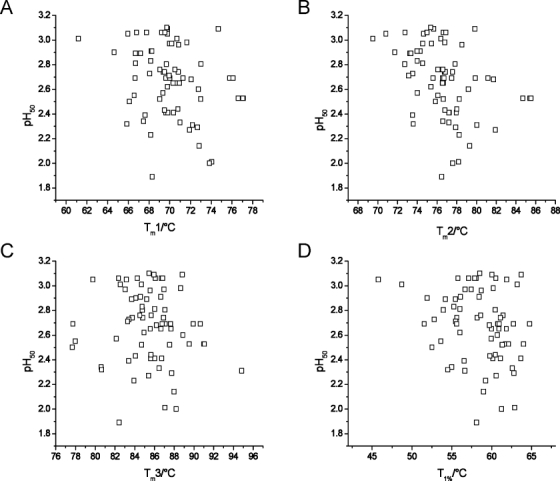
Thermal and pH stability plots. Shown are plots of pH50 against Tm1 (A), Tm2 (B), Tm3 (C), and T1% (D).
Table II.
pH50 and Thermal Transitions Correlations and Slopes
| pH50 plot against… | R factor | R2 factor | Slope |
|---|---|---|---|
| Tm1 | −0.260 | 0.068 | −0.026 |
| Tm2 | −0.389 | 0.151 | −0.038 |
| Tm3 | −0.087 | 0.076 | −0.008 |
| T1% | −0.248 | 0.061 | −0.019 |
Correlation coefficient R, coefficient of determination R2, and slope for linear fits to the plots shown in Figure 4, of pH50 against Tm1, Tm2, Tm3, and T1%.
As part of the model building for predicting stability based on sequence alone, cross validation was used in the model selection process. Figure 5 shows the performance of the pH50 models built in cross validation with the support vector machine (SVM) parameters listed in Table III, where for each number of selected features ranging from 1 to 100, the median of the 25 × 5 = 125 models is shown.
Figure 5.
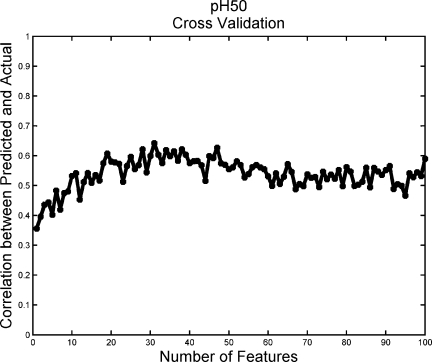
pH50 cross-validation curve for model selection. Model selection is performed across internal SVM parameters gamma, cost, and epsilon, as well as the number of features used to construct the model. Median SVM cross-validation performance with the selected parameter values is shown as a function of the number of features.
Table III.
Cross Validation and Test Set Performance, and Parameter Values of the Selected Models
| Cross validation | Test set | Selected parameters | ||||||
|---|---|---|---|---|---|---|---|---|
| Correlation, median | Correlation, MAD | Correlation | AUC | Number of features | Gamma | Cost | Epsilon | |
| Tm1 | 0.376 | 0.193 | 0.267 | 0.738 | 100 | 0.25 | 1 | 0.01 |
| Tm2 | 0.36 | 0.209 | 0.0741 | 0.786 | 45 | 0.0156 | 0.5 | 0.001 |
| Tm3 | 0.562 | 0.144 | 0.211 | 0.725 | 45 | 0.0625 | 4 | 0.01 |
| T1% | 0.471 | 0.13 | 0.0938 | 0.306 | 100 | 0.0625 | 1 | 0.001 |
| pH50 | 0.676 | 0.226 | 0.639 | 0.762 | 20 | 0.125 | 4 | 0.01 |
The cross-validation performance is given by the Pearson correlation coefficient between the predicted and actual values of the stability measures. Models yielding a high median and a low median absolute deviation (MAD) correlation coefficient are favored in the model selection. Performance on the test set is measured by the correlation of the predicted and actual stability measures, as well as the AUC (area under the receiver operating characteristic (ROC) curve) found by dichotomizing the stability measures into the two classes above and below the training set median.
Predictions for pH50 are shown in Figure 6. The accuracy of those predictions, as well as for the thermal measures, is shown in Table III. When the problem is converted to a two-class classification problem by dichotomizing the stability measures into those above and below the training set median, the accuracy of the predictions improves as indicated by the area under the curve (AUC) values being above 0.5 (fully random), except for the T1% (Fig. 6, Table III).
Figure 6.

Model predictions on the pH50 test set. A: Predicted pH50 plotted against actual pH50 for test set antibodies. Error bars show the variance in the predictions across leave-one-out training. B: ROC curve for two-class classification predictions of pH50s. The two classes are defined as high (above the training set median) and low pH50s.
Discussion
The antibodies used were taken from our internal database, representing different species (mouse, rat, and human), isotypes, and germlines. Raised against a variety of antigens, including clinically relevant and “dummy” targets, these antibodies' sequences differed significantly. A total of 77 antibodies were analyzed for thermal stability; of those 53 were also analyzed in the pH stability assay.
We first developed an assay to determine at which pH the structure of an antibody changed. To our knowledge, this is the first systematic study of the transition, rather than the low pH state per se. To measure the pH stability, protein A purification conditions were mimicked: mixtures of protein A loading and elution buffers were used to incubate antibodies at pH values from pH 9.0 to 1.5. ANS was used as an indicator for changes in conformation as it is known to bind specifically to partially folded protein conformations, resulting in increased and blue-shifted fluorescence emission.25,26 The observed increase in ANS fluorescence [Fig. 1(A)], suggested binding of the dye and, therefore, a loss of structure. Different protein and ANS concentrations were found to give rise to different fluorescence intensities [Fig. 1(B), Table I). Balancing the limited amount of protein available and the possibility that large excesses of ANS might induce conformational change, an antibody concentration of 0.1 mg mL−1 (or ∼0.67 μM) and a 100-fold molar excess of ANS over protein were chosen.
To confirm that the observed change in fluorescence corresponded to a change in structure, the same protein at the same concentration was studied by intrinsic protein fluorescence and circular dichroism (CD) spectroscopy [Fig. 1(C)]. The intrinsic fluorescence data were found to overlay the ANS curve well. When compared with far UV CD studies, the detailed shape of the CD curve was somewhat different to that observed for ANS binding, potentially due to far UV CD spectroscopy measuring secondary structure, rather than a combination of both secondary and tertiary structures. The relatively similar curves and closely similar fitted pH50 values suggested that the ANS assay accurately reflects the change in secondary and tertiary structures.
A histogram of pH50 values [Fig. 1(D)] showed a spread of values. It should be noted that these values are likely specific to the buffer system used. Lower pH50 values correspond to antibodies whose structures change at a lower pH values than those with higher pH50, suggesting they are more stable to pH-induced structural changes. Without significantly more data, it is neither possible to define the “best” pH50 nor possible to define a meaningful pH50 cutoff value above which antibodies become “too unstable” to be deemed successful drug candidates. The pH50 is an indicator of protein stability and may have a bearing on formulation studies, but by itself it is unlikely to be indicative of overall drug behavior. A more powerful approach would be to treat the pH50 as one data point to be considered alongside other stability measurements to come to decisions about which is the better candidate.
In light of this, the thermal stability of these antibodies was studied. To minimize protein usage and increase throughput, a Sypro Orange dye-based assay was developed based on previous work.5,9,27 The dye binds to unfolding proteins, resulting in increased fluorescence.5,9,27 The dye is delivered as a 5000× solution; structure and concentration are not given by the manufacturer. We found the best conditions to be 0.1 mg mL−1 protein and 2.5× dye.
The fluorescence intensity decrease seen from 20°C to about 50°C [Fig. 2(A)] is due to a temperature-induced lowering of the quantum yield of the dye.28 In the region where the antibody unfolded, above 50°C, the unfolding curve often contained a shoulder on either side of the maximum, suggesting the presence of more than one transition. We developed a method to extract the expected antibody unfolding transitions, corresponding to the CH2, Fab, and CH3 domains10 by postulating that Sypro Orange, similar to ANS, binds to hydrophobic pockets but has decreased affinity for folded or completely unfolded protein. Thus, low fluorescence intensity would be expected at low temperature, while the protein is in a native state; higher fluorescence at intermediate temperatures, where the protein is partially unfolded; and low fluorescence again at high temperatures when the protein is completely unfolded. The CH2, Fab, and CH3 domains in an antibody do not unfold at the same temperature,10 giving rise to more complicated curve profiles.
The thermal data were fitted to three Gaussian curves, one for each unfolding domain: CH2, Fab, and CH3. A Gaussian curve, and therefore each unfolding transition, is defined by the midpoint (the Tm), the width (the standard deviation), and the amplitude (fluorescence intensity in this case). No quantitative relationship exists between dye binding and intensity: a high intensity could be the result of many dye molecules bound weakly or a few molecules bound strongly. We therefore focused on the Tm values and widths of each transition in our analysis. To determine the accuracy of this fitting procedure, we compared our fluorescence data to data obtained from DSC experiments [Fig. 2(C)]: although the intensities varied, the main peak and its shoulders occurred at very similar temperatures, suggesting the data are very similar. In a plot of the Tm values from the fluorescence against those from DSC experiments [Fig. 2(D)], the R and R2 factors indicate a good correlation between the two data sets, but these factors do not indicate whether the two methods agreed and a further statistical analysis24 was performed. This showed that there is good overall agreement with little bias between the two techniques, but for individual data points the variation can be quite significant (Supporting Information Fig. S1). Therefore, the high throughput and small quantities of protein required make the Sypro Orange assay a powerful approach to screen the thermal stability of a large number of early drug candidates, but for a detailed characterization of promising late-stage drug candidates, DSC would be preferred.
Looking at the histogram of Tm values [Fig. 3(A)], even the lowest Tm1 was 62°C, a temperature an antibody is extremely unlikely to encounter during production, formulation, storage, or following administration. In the absence of evidence that Tm directly corresponds to overall stability, it is not clear that this antibody is necessarily “worse” than one with a Tm1 of 77°C. Furthermore, if two proteins have the same Tm, the protein with the wider, less cooperative unfolding transition will start to unfold earlier and, therefore, be effectively less stable. The onset of unfolding, therefore, might help better interpret this data. This requires consideration of the transition width, a factor not usually taken into account. A widespread of values was observed, especially for Tm3, the third transition [Fig. 3(B)]. In some cases, the width for Tm3 was such that the corresponding CH3 domain started to unfold at the same temperature as the CH2 or Fab domain [Fig. 2(B)]. Combining the Tm and width values, the T1%, temperature at which the protein was 1% unfolded or the onset of unfolding, was calculated (see Materials and Methods). The T1% values are distinctly lower than those for the midpoints of the transitions [Fig. 3(C)]. The lowest T1% observed, 36.7°C, was around physiological temperature; in other words, in vivo this protein would be 1% unfolded, with potentially deleterious consequences. The individual Tm transitions of this protein (70.5, 76.7, and 79.1°C) did not suggest that the T1% would be low. This example highlights that a careful consideration of thermal stability is crucial and that the usual citation of just the Tm values is not enough to characterize the thermal stability of an antibody.
To determine if the pH50 and Tm were correlated and, hence, whether one was predictive of the other, they were plotted against each other (Fig. 4). No obvious trends could be found, and these two measures of stability, therefore, are not well correlated. Even though the thermal stability could be measured for individual domains in the antibody (Fab, CH2, and CH3), while the pH stability was globally measured, a correlation between the pH stability of one domain and its thermal stability would be expected to be evident in the plots in Figure 4. Given the interplay between the domains in an antibody, it is not clear if measuring the pH stability on individual domains would yield results that were predictive of antibodies as a whole. Similarly, because of the limited sample set, subdividing the data set, for example, by species or isotype, would result in too few antibodies per class and so decrease the predictive power. The lack of a correlation could be expected because a thermal challenge is very different to a pH challenge. It also implies that there is no single measure of stability and to fully understand the stability of antibody candidate drugs, therefore, several measures of stability ought to be considered. We have highlighted two in this report but others are of interest as well, for example, the tendency to form aggregates, given their role in loss of effectiveness and increased immunogenicity,29 or protein dynamics.30,31
As all antibodies have nearly identical folds, we wondered if our sequences and stability data could be used to build an in silico model to predict stability based on sequence alone. The data were used to train epsilon regression support vector machines to predict the antibody thermal and acidic stabilities as continuous valued quantities using sequence data alone. It is possible to use a classifier to predict stability classes for the antibodies by dichotomizing the stability measurements, but the more difficult approach of predicting numerical values was chosen because it provides a means for predicting both the direction and magnitude of any stability changes due to induced mutations.
A novel approach was used to select the properties to describe individual amino acids: instead of principal component analysis,32 the different properties described in the AAindex database33 were clustered into 100 groups, and one representative property from each cluster was chosen (see Materials and Methods). The resulting number of features used to define each protein sequence was still relatively large when compared with the number of samples. This situation is often referred to as the “curse of dimensionality,” a phrase ascribed to Bellman34 referring to a situation where there are many variables but relatively few data points. To guard against overfitting, 25 times repeated fivefold cross validation in the model selection process was used. The performance of the pH50 models, shown in Figure 5, shows that although there is some noise in the curve, the general trend suggests that although the chosen model is probably not the global optimum, it is unlikely to suffer from severe overfitting. It may be that in the context of a modestly sized dataset, overfitting is most effectively avoided by models that favor more predictions that tend toward the mean. Models with this property would be likely to exhibit the relatively higher test set AUC than test set correlations as seen for the thermal transition endpoints (Table III).
Predictions for the pH50 values worked the best, with the average prediction being within 0.2 pH units of the measured values (Fig. 6). The accuracy of the prediction is significantly smaller than the range of pH50 values observed (from pH 1.8 to 3.2) and is comparable to the resolution in the pH experiment, increasing confidence that this model is appropriate for the predictions. The results presented in Table III show a range of predictive accuracies among the five endpoints, pH50, Tm1, Tm2, Tm3, and T1%. Converting the problem to a two-class, above/below the training set median prediction problem, the difference in performance between the pH50 predictions and the thermal transition predictions disappears, as can be seen by the test set AUC measures (Fig. 6, Table III).
For some endpoints, for example, Tm2, there is an apparent disparity between the two measures of test set prediction performance. For these endpoints, the low test set correlations (indeed for the Tm2 and the T1% endpoints, the test set correlations are only marginally above the chance value of zero) appear to be caused by the majority of the tests set stability measure predictions being tightly clustered around the mean. The relatively high value of the test set AUC (well above its chance value of 0.5) for these same endpoints indicates that the classification of the antibodies according to the dichotomized endpoints is relatively accurate. The combination of low correlation and high AUC is therefore indicative of accurate predictions of the direction of stability change relative to the training set mean, but frequent underestimation of the magnitude of the change. This may be the result of avoiding overfitting using models trained on a small data set.
Predicting protein stability is seen as a way of predicting which molecules are more likely to form better drugs. Ultimately, the goal of this work is to predict which molecule will behave best under large-scale GMP manufacturing conditions and in the clinic—not which molecule has the better biophysical properties such as pH50, Tm, aggregation propensity, and so forth. Given sufficiently more data, it may be possible to define threshold values. This may also make the computational prediction more tractable. Obtaining more data should be done ideally in an industry-wide manner. If a public library of anonimised sequences and stability measurements of antibodies and antibody-based drug candidates across the industry could be created, without legal and intellectual property implications, that could be an excellent way to determine these threshold stability values for the thermal, pH, and other stability measures. Not only would this help cut development costs by weeding out poor molecules earlier, it would greatly aid the development of better drugs for patients.
In conclusion, we have studied a selection of antibodies representing different species, isotypes, and germlines. By incubating the antibodies in buffers ranging from pH 9 to 1.5, we newly defined the pH50 as the pH at which half the protein has undergone a structural change; the pH50 ranged from pH 1.8 to 3.2. A high-throughput dye fluorescence thermal stability assay was developed with a new analysis method that allowed extraction of the three thermal transitions expected of an antibody as well as T1%, the value at which the protein is 1% unfolded. A range of values for the thermal transitions and T1% was found. One protein had a T1% that was within physiological range even though the unfolding midpoints did not suggest that the protein would start to unfold at such a low temperature.
No correlation could be found between the pH and thermal stability data, suggesting that different aspects of stability may not be correlated and that to truly characterize the stability of a biological drug candidate, several aspects of stability must be measured and considered together.
Machine-learning approaches could predict the pH50 to pH ±0.2, but the thermal stability could not be accurately predicted. Recasting the problem into whether a certain protein had a pH50 or thermal transition above or below the median for that measurement, predictions of all properties improved.
Materials and Methods
Antibodies
For these experiments, 77 antibodies chosen on material availability from an internal database had been expressed in mammalian cells in-house and purified on a protein A column followed by preparative size exclusion chromatography (SEC). Samples were 98% pure or better by SEC. Different glycosylation patterns due to the use of different cell lines were not found to significantly affect the properties measured. Most antibodies were in a phosphate-buffered saline (PBS) solution, comprised of 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, and 1.47 mM KH2PO4, pH 7.2, or in a His:sucrose buffer, consisting of 10 mM histidine and 5% sucrose, pH 6. Protein concentrations varied but were usually 1–5 mg mL−1.
pH stability solutions
By titrating a protein A loading buffer (650 mM sodium sulfate, 20 mM sodium citrate, 20 mM boric acid, and 20 mM sodium phosphate, pH 9) and protein A elution buffer (20 mM citric acid and 150 mM sodium chloride, pH 2.5), 24 solutions from pH 9 to 1.5 were prepared. For buffers with pH lower than 2.6, the protein A elution buffer was adjusted with 1 M HCl. For fluorescence experiments, 98 μL of each of the pH buffers was placed in black, clear-bottom 96-well plates (Corning, Lowell, MA). Antibody solutions were concentrated to 5 mg mL−1 where necessary, using MicroCon 30-kDa cutoff filters (Millipore, Billerica, MA), and 2 μL aliquots were added to the 96-well plate for a final protein concentration of 0.1 mg mL−1 (∼0.67 μM for an antibody). For CD experiments, samples were made up in Eppendorf tubes to a total volume of 200 μL (i.e., 196 μL buffer and 4 μL antibody solution). Otherwise, treatment was identical.
ANS fluorescence
Following sealing and storage at 4°C for 24 h, the plate with different pH solution and protein was equilibrated at room temperature for 30 min. Aliquots of 5 μL 8-anilinonaphthalene-1-sulfonic acid ammonium salt (ANS; Sigma-Aldrich, St. Louis, MO) in RODI water were added to a final ANS concentration of 67 μM, 100-fold molar excess over antibody. In this plate-based assay, 240 μg of protein was needed per experiment. Given the relatively limited amounts of protein available, 53 antibodies were analyzed in this assay. Fluorescence intensity was read immediately on an Infinite M1000 plate reader (Tecan Systems, San Jose, CA) exciting at 360 nm and reading the emission at 500 nm (20 nm bandwidth). The fluorescence signal of ANS in the various buffers was negligible and therefore not subtracted from the protein samples. Plotting fluorescence intensity against pH, a Boltzman sigmoidal curve was fitted to the data using Origin 7 SR2 (OriginLab, Northampton, MA). The pH50 value corresponded to the pH value where the fluorescence was at half maximum.
Attempts were made to find the onset of the change in fluorescence. One attempt looked for the intersection of straight lines fitted to the constant high pH region and to the range of maximal fluorescence change (pH 3–4). Given the steep change in fluorescence over a narrow pH range, this method was overly sensitive to the inclusion or exclusion of a single data point. In a different approach, the onset of change was defined as the first datapoint to fluoresce more brightly than median of the high pH region plus 1.5 times the noise in that region. This method was found to be overly sensitive to noisy signals. The sigmoidal analysis-based pH50 data were therefore regarded as superior.
Intrinsic fluorescence
A plate with the protein incubated at different pH was placed in the Infinite M1000 plate reader (Tecan Systems, San Jose, CA) and the intrinsic fluorescence measured, exciting at 280 nm (slit width 10 nm) and scanning the emission from 300 to 400 nm. The intensity at 340 nm was used for studying pH-induced conformational changes.
Circular dichroism spectroscopy
The centrifuge tubes with the buffered antibody solutions were taken and in turn, each sample was placed in a 1-mm path length quartz cuvette (Starna, Atascadero, CA). The CD spectra were recorded on a Jasco J-715 CD spectrometer (Jasco, Easton, MD), scanning from 250 to 196 nm in 0.5-nm steps, at 100 nm min−1 and averaging four scans. Spectra were smoothed (Savitzky-Golay, 9 points width), baseline and concentration corrected, and plotted. The signal was found to change most with pH at 205 nm, and this value, following rescaling from 0 (at pH 9) to 1 (at pH 1.5), was used to compare the data to intrinsic fluorescence and ANS binding data.
Sypro Orange experiment
Samples were tested in quadruplicate in a 96-well plate format, using a volume of 50 μL per well. Antibodies were diluted to 0.1 mg/mL in PBS, and Sypro Orange dye (5000× stock solution in DMSO; Invitrogen, Carlsbad, CA) was added to a final concentration of 2.5×. Only 20 μg protein was needed to run each protein in quadruplicate, and up to 24 samples could be run on one 96-well plate, thereby making this method remarkably more time and protein efficient than DSC, where typically 100 μg protein is needed per sample. The 96-well optical reaction plate (Applied Biosystems, Foster City, CA) was sealed with acetate plate sealers (Thermo Electron Corporation, Waltham, MA) and transferred to an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). Parameters included absolute quantification (standard curve), 96-well clear, detector name SYBR, and none for the passive reference selections. Following equilibration at 20°C, the temperature was raised to 22°C for 15 s and then decreased to 21°C for 45 s. This was necessary due to instrument and software limitations on the methods that could be written. The fluorescence measurement was collected at the end of this 45 s timeframe. This cycle of temperature increase by 1°C was repeated up to 95°C. The data were transferred to Excel for fitting.
Sypro Orange data analysis
The raw data from the rtPCR instrument (fluorescence intensity in arbitrary units against cycle number) were baseline corrected, and three Gaussian curves were fitted to the region of interest, from the onset of the unfolding transition to the highest recorded temperature. “Goodness of fit” was judged by minimizing the sum of the squared differences between each data point and each fitted point, akin to χ2. Subsequently, quality of the fit was judged visually and by assessing the residuals. Starting values for midpoint, width, and amplitude were not found to influence the final fit values. Based on the fluorescence data, we cannot determine which transition corresponds to the unfolding of which domain,. From low to high temperature, therefore, the three transitions were called Tm1, Tm2, and Tm3, with width 1, width 2, and width 3, respectively. At each melting temperature, the domain to which that Tm corresponds is half unfolded; therefore, given the Tm and width values, it was possible to find a numerical solution to the sum of these three Gaussians to determine at which temperature each protein was 1% unfolded. This temperature is referred to as the T1%. All calculations were performed in Excel 2003 using macros written in-house.
DSC
Protein samples were diluted to 250 μL at 0.3 mg mL−1. A PBS buffer blank was used for the reference sample. Sample and blank were thoroughly degassed using a MicroCal ThermoVac Sample Degassing and Thermostat (Microcal, Northampton, MA) at 8°C and dispensed into the appropriate cells of a MicroCal VP-DSC Capillary Cell MicroCalorimeter (MicroCal, Northampton, MA). Following equilibration for 4 min at 15°C, samples were scanned up to 100°C at a rate of 100°C h−1. A filtering period of 20 s was selected. Raw data were baseline and concentration corrected, and the data were fit to an MN2-State Model with three transitions using the Origin 7 RS2 (OriginLab Corporation, Northampton, MA) software package.
Machine learning
To train a support vector machine to predict protein properties from primary sequence information, all protein sequences must be represented as vectors of the same length, despite variations in sequence lengths. A sliding window representation accomplishes this by measuring the frequencies of short motifs in the sequences. Any amino acid property may be used to generate a set of sliding window motif frequencies. Over 500 amino acid physicochemical properties have been collected in the AAindex database,33 but many are highly correlated pairs (or triples, etc.). Including redundant properties increases the computational cost of training and may decrease predictive performance.35 Although principal component analysis can be used to generate nonredundant features,32 these features lack the direct interpretability of the physicochemical properties themselves. For this reason, we reduced the AAindex properties to 100 prototypes by clustering and choosing the most representative property from each cluster. Clusters were formed such that each member was correlated above a threshold value with at least one other cluster member. The threshold was fixed to produce exactly 100 clusters. Correlation coefficients were calculated between properties within each cluster, and the property with the highest average was taken as the prototype. Each prototypical property was normalized to a range of [0,1].
For each property, the 20 amino acids were ranked by the property value and assigned to one of three bins: the lowest seven, the middle six, or the highest seven. There are 34 = 81 distinct windows four residues wide for each property. Each window is slid along the antibody sequence, and the number of times four consecutive amino acids match the window is counted. The count is then normalized by the number of positions the window slid through (sequence length minus three), producing features in the range of [0,1]. Each sequence is thereby represented in 81 × 100 = 8100 dimensions, regardless of sequence length.
Epsilon regression support vector machines with radial basis function kernels (using the CRAN package e1071) were trained independently for each endpoint pH50, T1%, and thermal transitions Tm1, Tm2, and Tm3, using 10 times repeated random stratified fivefold cross-validation. The danger of overfitting is greatly reduced by nesting model selection within the cross-validation loop.36,37 Prediction quality was measured with the Pearson correlation coefficient (R) between the predicted and the true endpoint values for the antibodies in the 1/5 of the data used as the test set.
Features were selected within the cross-validation loop by calculating the absolute value of the correlation coefficient between each feature and the endpoint using the 4/5ths of the data selected as the training set for that loop. Models were generated using the top feature, the top two features, and so on to the top 100 features.
Model selection was used to find parameter values for the support vector machine: epsilon, gamma, and cost, as well as the number of features to be used for model construction. The models were selected to maximize the selection criterion f(m) = (Median(Rm) − 1.2·MAD(Rm)), where m is a collection of models with a specific number of features and fixed SVM parameter values, Rm is the collection of correlation coefficients between the predictions of m and the true endpoint values, and the MAD is the median absolute deviation: a robust alternative to the standard deviation. Inclusion of the term −1.2·MAD(Rm) in the model selection criterion penalizes models with highly variable performance, further reducing the risk of overfitting by favoring models that are both accurate and consistent.
Limited numbers of cross-validation repeats can cause the selection of suboptimal model parameters. Increasing the number of repeats improves the accuracy of the performance estimates and increases the likelihood of selecting a near optimal model, albeit at increased computational cost. By borrowing information across feature numbers, the variance in the performance estimates can be reduced without a large increase in computational cost. This was accomplished by smoothing the selection criterion f(m) curves (Fig. 5) over feature number through convolution with a discrete Gaussian kernel of width five.
After parameter value and feature number selection, models were constructed using the complete dataset. The performance of these models on the independent test set is shown in Table III.
Acknowledgments
MRHK acknowledges Peter Lapan's helpful suggestions and discussions on the further statistical analysis of the DSC and Sypro Orange fluorescence results.
Supplementary material
References
- 1.Demarest SJ, Glaser SM. Antibody therapeutics, antibody engineering, and the merits of protein stability. Curr Opin Drug Discov Dev. 2008;11:675–687. [PubMed] [Google Scholar]
- 2.Dimasi N, Gao C, Fleming R, Woods RM, Yao XT, Shirinian L, Kiener PA, Wu H. The design and characterization of oligospecific antibodies for simultaneous targeting of multiple disease mediators. J Mol Biol. 2009;393:672–692. doi: 10.1016/j.jmb.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 3.Mukovozov I, Sabljic T, Hortelano G, Ofosu FA. Factors that contribute to the immmunogenicity of therapeutic recombinant human proteins. Thromb Haemost. 2008;99:874–882. doi: 10.1160/TH07-11-0654. [DOI] [PubMed] [Google Scholar]
- 4.Welfle K, Misselwitz R, Hausdorf G, Hohne W, Welfle H. Conformation, pH-induced conformational changes, and thermal unfolding of anti-p24 (HIV-1) monoclonal antibody CB4-1 and its Fab and Fc fragments. Biochim Biophys Acta. 1999;1431:120–131. doi: 10.1016/s0167-4838(99)00046-1. [DOI] [PubMed] [Google Scholar]
- 5.Yeh AP, McMillan A, Stowell MH. Rapid and simple protein-stability screens: application to membrane proteins. Acta Crystallogr D Biol Crystallogr. 2006;62:451–457. doi: 10.1107/S0907444906005233. [DOI] [PubMed] [Google Scholar]
- 6.Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol. 2008;376:926–931. doi: 10.1016/j.jmb.2007.10.075. [DOI] [PubMed] [Google Scholar]
- 7.Garidel P, Hegyi M, Bassarab S, Weichel M. A rapid, sensitive and economical assessment of monoclonal antibody conformational stability by intrinsic tryptophan fluorescence spectroscopy. Biotechnol J. 2008;3:1201–1211. doi: 10.1002/biot.200800091. [DOI] [PubMed] [Google Scholar]
- 8.Ionescu RM, Vlasak J, Price C, Kirchmeier M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J Pharm Sci. 2008;97:1414–1426. doi: 10.1002/jps.21104. [DOI] [PubMed] [Google Scholar]
- 9.He F, Hogan S, Latypov RF, Narhi LO, Razinkov VI. High throughput thermostability screening of monoclonal antibody formulations. J Pharm Sci. 2009;99:1707–1720. doi: 10.1002/jps.21955. [DOI] [PubMed] [Google Scholar]
- 10.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355:751–757. doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 11.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez M, Murature DA, Fidelio GD. Thermal stability of human immunoglobulins with sorbitol. A critical evaluation. Vox Sang. 1995;68:1–4. doi: 10.1111/j.1423-0410.1995.tb02535.x. [DOI] [PubMed] [Google Scholar]
- 13.Remmele RL, Jr,, Nightlinger NS, Srinivasan S, Gombotz WR. Interleukin-1 receptor (IL-1R) liquid formulation development using differential scanning calorimetry. Pharm Res. 1998;15:200–208. doi: 10.1023/a:1011902215383. [DOI] [PubMed] [Google Scholar]
- 14.Kasraian K, Kuzniar A, Earley D, Kamicker BJ, Wilson G, Manion T, Hong J, Reiber C, Canning P. Sustained in vivo activity of recombinant bovine granulocyte colony stimulating factor (rbG-CSF) using HEPES buffer. Pharm Dev Technol. 2001;6:441–447. doi: 10.1081/pdt-100002252. [DOI] [PubMed] [Google Scholar]
- 15.Remmele RL, Jr,, Bhat SD, Phan DH, Gombotz WR. Minimization of recombinant human Flt3 ligand aggregation at the Tm plateau: a matter of thermal reversibility. Biochemistry. 1999;38:5241–5247. doi: 10.1021/bi982881g. [DOI] [PubMed] [Google Scholar]
- 16.Shusta EV, Kieke MC, Parke E, Kranz DM, Wittrup KD. Yeast polypeptide fusion surface display levels predict thermal stability and soluble secretion efficiency. J Mol Biol. 1999;292:949–956. doi: 10.1006/jmbi.1999.3130. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia GG, Pechmann S, Dobson CM, Vendruscolo M. Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem Sci. 2007;32:204–206. doi: 10.1016/j.tibs.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 18.Ejima D, Tsumoto K, Fukada H, Yumioka R, Nagase K, Arakawa T, Philo JS. Effects of acid exposure on the conformation, stability, and aggregation of monoclonal antibodies. Proteins. 2007;66:954–962. doi: 10.1002/prot.21243. [DOI] [PubMed] [Google Scholar]
- 19.Kameoka D, Masuzaki E, Ueda T, Imoto T. Effect of buffer species on the unfolding and the aggregation of humanized IgG. J Biochem. 2007;142:383–391. doi: 10.1093/jb/mvm145. [DOI] [PubMed] [Google Scholar]
- 20.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci USA. 2009;106:11937–11942. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiskoot W, Bloemendal M, van Haeringen B, van Grondelle R, Beuvery EC, Herron JN, Crommelin DJ. Non-random conformation of a mouse IgG2a monoclonal antibody at low pH. Eur J Biochem. 1991;201:223–232. doi: 10.1111/j.1432-1033.1991.tb16278.x. [DOI] [PubMed] [Google Scholar]
- 22.Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel. 2009;22:553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Middaugh CR, Fang J. A novel scoring function for discriminating hyperthermophilic and mesophilic proteins with application to predicting relative thermostability of protein mutants. BMC Bioinformatics. 2010;11:62. doi: 10.1186/1471-2105-11-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Int J Nurs Stud. 2010;47:931–936. [PubMed] [Google Scholar]
- 25.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 26.Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res. 2008;25:1487–1499. doi: 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 2004;332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 28.Lakowicz JR. Principles of fluorescence spectroscopy. New York: Springer; 2006. [Google Scholar]
- 29.Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006;8:E501–E507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumoulin M, Canet D, Last AM, Pardon E, Archer DB, Muyldermans S, Wyns L, Matagne A, Robinson CV, Redfield C, Dobson CM. Reduced global cooperativity is a common feature underlying the amyloidogenicity of pathogenic lysozyme mutations. J Mol Biol. 2005;346:773–788. doi: 10.1016/j.jmb.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Martinez JA, Sola RJ, Castillo B, Cintron-Colon HR, Rivera-Rivera I, Barletta G, Griebenow K. Stabilization of alpha-chymotrypsin upon PEGylation correlates with reduced structural dynamics. Biotechnol Bioeng. 2008;101:1142–1149. doi: 10.1002/bit.22014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rackovsky S. Sequence physical properties encode the global organization of protein structure space. Proc Natl Acad Sci USA. 2009;106:14345–14348. doi: 10.1073/pnas.0903433106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawashima S, Pokarowski P, Pokarowska M, Kolinski A, Katayama T, Kanehisa M. AAindex: amino acid index database, progress report 2008. Nucleic Acids Res. 2008;36:D202–D205. doi: 10.1093/nar/gkm998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellman RE. Adaptive control processes: a guided tour. Princeton, NJ: Princeton University Press; 1961. [Google Scholar]
- 35.Nicodemus KK, Malley JD. Predictor correlation impacts machine learning algorithms: implications for genomic studies. Bioinformatics. 2009;25:1884–1890. doi: 10.1093/bioinformatics/btp331. [DOI] [PubMed] [Google Scholar]
- 36.Kohavi R. Montreal, Quebec, Canada: Proceedings of the Fourteenth International Joint Conference on Artificial Intelligence; 1995. A study of cross-validation and bootstrap for accuracy estimation and model selection; pp. 1137–1143. [Google Scholar]
- 37.Hawkins DM, Subhash CB, Mills D. Assessing model fit by cross-validation. J Chem Inf Comput Sci. 2003;43:579–586. doi: 10.1021/ci025626i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.