

Abstract
Terpenoids include structurally diverse antibiotics, flavorings, and fragrances. Engineering terpene synthases for control over the synthesis of such compounds represents a long sought goal. We report computational design, selections, and assays of a thermostable mutant of tobacco 5-epi-aristolochene synthase (TEAS) for the catalysis of carbocation cyclization reactions at elevated temperatures. Selection for thermostability included proteolytic digestion followed by capture of intact proteins. Unlike the wild-type enzyme, the mutant TEAS retains enzymatic activity at 65°C. The thermostable terpene synthase variant denatures above 80°C, approximately twice the temperature of the wild-type enzyme.
Keywords: terpene synthases, protein-engineering, phage display, thermostability
Introduction
To date, terpenes comprise nearly half of the known natural products. The tremendous diversity of terpene structures ranges from simple, linear hydrocarbons to complex cyclic structures. Terpene synthases (also known as terpene cyclases) initiate the biosynthesis of over 20,000 different terpene products, all stemming from linear isoprenoid precursors.1 The class of synthases responsible for sesquiterpene (C15) synthesis have been extensively studied including the determination of X-ray crystal structures for at least five enzymes: Fusarium sporotrichioides trichodiene synthase (PDB accession code 1JFG, E.C. number 4.2.3.6), Streptomyces UC5319 pentalenene synthase (1PS1, E.C. 4.2.3.7), Penicillium roqueforti aristolochene synthase (1DI1, E.C. 4.2.3.9), Aspergillus terreus aristolochene synthase (2E4O, E.C. 4.2.3.9), and Nicotiana tabacum 5-epi-aristolochene synthase (5EAT, E.C. 4.2.3.9).
Sesquiterpene synthases use the linear precursor, farnesyl diphosphate (FPP, 1), to generate more than 300 distinct cyclic sesquiterpenes including 5-epi-aristolochene (2) and pentalenene (3) (Fig. 1).2 Catalyzing some of the most complex carbon–carbon bond forming reactions found in chemistry and biology,3 all known sesquiterpene synthases are soluble monomers or homodimers with molecular weights between 40 and 65 kDa. Each sesquiterpene synthase active site provides a shaped cavity for the binding and orientation of the flexible substrate. During the cyclization, the enzyme cleaves the diphosphate functionality from FPP, and then chaperones the reactive carbocation intermediates, thus guiding the formation of a single major product with striking regiochemical and stereochemical specificity.4 Despite a conserved mechanism for carbocation initiation and structural similarity, sesquiterpene synthases share only a minimal sequence identity of 6–15%. The structural basis for the precise control over product regiochemistry and stereochemistry remains poorly understood.4
Figure 1.
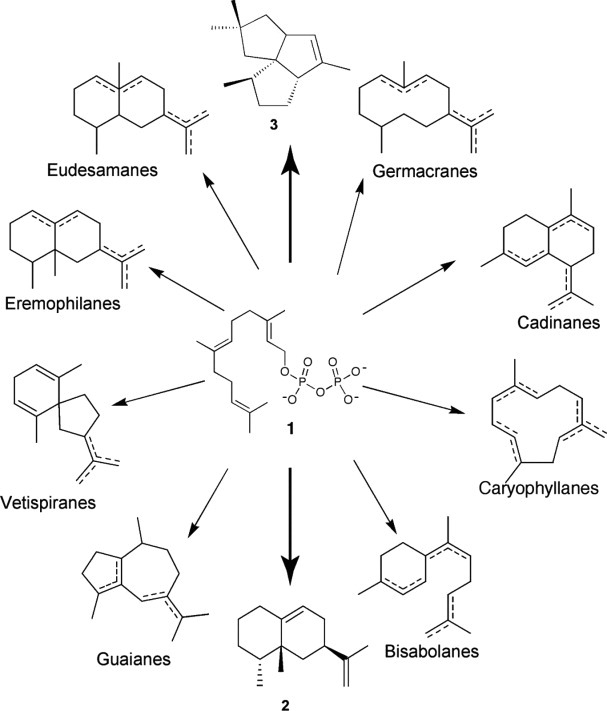
Biosynthetic diversity of sesquiterpenes generated from a single linear FPP (1) substrate. (2) = 5- epi-aristolochene, (3) = pentalenene. Figure adapted from Ref.48.
Control of regiospecific and stereospecific C–C bond forming reactions under mild, environmentally benign conditions represents an important goal in synthetic chemistry. Terpene synthases display virtuoso abilities to trigger and direct carbocation cyclizations, and therefore could fulfill this goal. However, the catalytic rates for these enzymes are far too low for practical applications. For example, under Vmax conditions, tobacco epi-aristolochene synthase (TEAS) has a catalytic efficiency (kcat/Km) of 2.1 × 104 (M−1 s−1).5 Additionally, as shown here, TEAS denatures at relatively low temperatures. Thus, enhanced thermostable terpene synthases offer an attractive approach to potentially increase the product yields obtained from these enzymes by enabling catalysis at higher temperatures.
Engineering enzymes to alter catalytic activity, protein folding, or thermostability is increasingly used and has been reviewed.6,7 In theory, a directed evolution approach could be performed on any enzyme without prior knowledge of protein structure or function. Although there are examples of the rational stabilization of enzymes,8 more typically, libraries of genes encoding the protein of interest are assembled by error prone polymerase chain reaction (PCR),9,10 recombination,11 or DNA shuffling10,12,13 to produce engineered enzymes for biosynthetic pathways,14 increased enantioselectivity,9,15 resistance to organic solvents,10 or increased thermostability.16
Here, we apply a statistical, computationally assisted design strategy (SCADS) to provide a model system for developing directed evolution methods for terpene synthases. SCADS identified mutations to potentially increase the thermostability of TEAS. Oligonucleotide-directed mutagenesis was then used to program the mutations into a phage-displayed protein, which provided the basis for developing selections to improve TEAS thermostability. After verifying proper folding by circular dichroism (CD), activity assays were carried out at elevated reaction temperatures to compare the wild-type and potentially thermostable TEAS. The experiments represent the first steps for efficient, in vitro selections of terpene synthases.
Results and Discussion
Although of particular interest due to the range of products generated, terpene synthases present substantial challenges to developing selection and screening methods to allow high throughput tailoring of their C–C bond forming activities. The product sesquiterpenes are nonessential for host survival, which obviates a conventional selection or screen for cell viability.17 For example, we unsuccessfully attempted to use growth of E. coli bearing terpene synthase expression vectors on phosphate deficient plates supplemented with FPP as a selection for terpene synthase activity. No selection for plasmids bearing the terpene synthase expression vector was observed. Secondly, the most common screening method for terpene synthase activity relies on gas chromatography–mass spectrometry (GC–MS);18 this relatively low throughput method requires over-expression and purification of individual protein variants. More recently, a medium thoroughput whole-cell system has been used to screen terpene synthase libraries of active-site mutants.19
This report details progress in developing selections for this fascinating class of enzymes. The reported in vitro selections use thermostability as a surrogate for protein activity. To develop the selection, two different proteins provide a model systems – wild-type TEAS and a computationally designed mutant TEAS.
Selection for TEAS thermostability
The SCADS algorithm was used to identify mutations of particular amino acids in different TEAS positions. This method has been successfully used to design a variety of protein systems,20,21 including a water-soluble version of the membrane-bound KcsA potassium channel22 and a de novo catalytically active monomeric, helical di-iron protein.23 The algorithm suggests mutations based on sidechain interactions consistent with the nearby protein backbone, neighboring sidechains, and the local environment. An “environmental energy score” has been developed to quantify the propensities of the amino acids for local densities of Cβ atoms and hence burial within a protein structure.24 The environmental energy of each residue was calculated, and the 12 residues having the highest such energy that are more than 12 Å from the substrate (so as to minimize impact on enzyme activity) were targeted for mutation (Table I). The 12 computationally predicted mutations leading to these stability gains include both surface-exposed and buried residues largely near the surface of the TEAS enzyme that could increase its solubility and thermostability (Fig. 2). The mutations were consistent with the wild-type structure. Many of the suggested mutations eliminate hydrophobic patches on the protein surface, and introduce surface salt bridges. The substitutions also resulted in the highest environmental energy scores, which correspond to more favorable interactions, as determined by the SCADS algorithm. Such changes might be expected to improve protein thermostability and solubility.
Table 1.
Computationally Suggested Mutations
 |
The SCADS algorithm computes environmental energy values with higher values corresponding to increased favorability of residues at the indicated position. The mutagenic oligonucleotides (SCAD 1-6) were used for the described mutagenesis. PDB residue numbers are based on the PDB accession file 5EAU. [Color table can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 2.
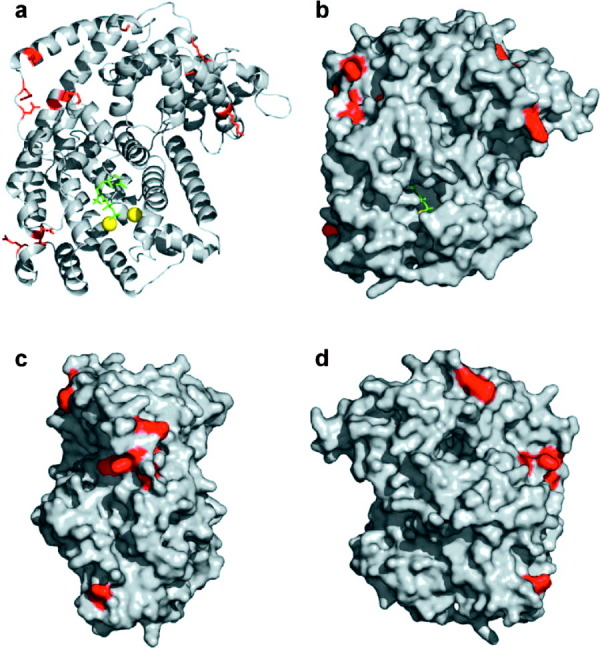
Location of computer-guided surface mutations on TEAS. (a) Cartoon representation of TEAS enzyme (grey) with FPP substrate (green) and two coordinated magnesium ions (yellow) bound in the active site. Site-directed mutations are depicted by the red colored sidechains. (b) Surface rendered front-view of TEAS enzyme active site and mutations. (c) Side-view (rotated 90° to the right) of TEAS surface mutations. (d) Back-view of TEAS surface mutations. Figure was drawn using atomic coordinates from the Protein Data Bank (entry 5EAU). Molecular graphics were made with PyMOL (http://pymol.sourceforge.net).
The thermostability of the expressed, computationally tailored, protein was confirmed by testing the susceptibility of the phage-displayed enzyme to proteolytic digestion. Resistance to proteolysis is a good indicator of protein stability at elevated temperatures.25 Such proteolytic susceptibility selection methods have previously been used in both mRNA26 and phage display systems.27–32 We use selection for the display of an epitope (c-myc peptide) on the N-terminus of TEAS as a marker for the intact protein. Thus, for the selection to work, protein unfolding at elevated temperatures must result in proteolysis, and loss of the c-myc tag.
A number of different proteases including proteinase K and trypsin were tested for viability in thermostability selections before chymotrypsin emerged as a protease with a slow rate of digestion at elevated temperatures. The choice of chymotrypsin reflects its specificity for aromatic residues and ability to remain active at elevated temperatures (in this case 37°C). The flexible, unstructured N-terminal extension of TEAS,33 located in the region near the epitope tag remained largely unaltered by mutagenesis, and presented a number of aromatic residues for protease digestion. Thus, a generic epitope tag for thermostability selection requires match with a protease specific for folding-sensitive motifs within the protein of interest.
To demonstrate selection for terpene synthase thermostability, a mixture of wild-type and mutant TEAS was selected for enrichment by intact display of a myc tag during proteolytic digestion at 37°C. Following chymotrypsin digestion, phage with the N-terminal myc tag still displayed were selected through binding to immobilized anti-myc antibody. After each round of selection, the ratio of wild-type to mutant TEAS was analyzed by PCR and digestion with an endonuclease. A NgoM IV site introduced during construction of the mutant TEAS, allowed differentiation between the wild-type and mutant DNA. As demonstrated by NgoM IV digestions, mutant TEAS was enriched substantially during four rounds of selection (Fig. 3).
Figure 3.

Phage display of TEAS and enrichment of thermo-TEAS DNA after four rounds of selection for thermostability. (a) Phage display of TEAS is demonstrated by ELISA as described previously.23 The negative control phage lack the ORF encoding TEAS (M13-KO7). (b) Lanes labeled “MW” and “Start” show a 1 kb DNA ladder and the naïve, initial mixture of wild-type and thermo-TEAS displayed on phage after PCR and a NgoM IV digestion, respectively. Lanes labeled 2, 3, and 4 represent the observed mixtures after the second, third, and fourth rounds of selection for thermostability, respectively. Clear enrichment for thermo-TEAS is observed. The “thermo-TEAS” lane provides a control. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The observed enrichment, through proteolysis resistance at a temperature causing denaturation for the wild-type enzyme, indicates the enhanced thermostability of the mutant TEAS. Addition of ionisable hydrophilic residues on the surface of TEAS likely expands a network of stabilizing salt-bridge interactions. Furthermore, replacement of a few buried hydrophilic sidechains with hydrophobic ones could also rigidify flexible regions of the protein, resulting in a tighter packing of the helices. Such tighter packing could in turn improve the proteolytic resistance by making surface helices and loops less accessible to protease digestion.
Secondary structure and melting point determination by circular dichroism
To investigate the effects of the computer-predicted mutations more thoroughly, we expressed and purified the wild-type and putatively thermostable mutant TEAS. Although the addition of charged residues on the surface of the enzyme could potentially improve solubility, the mutant TEAS, unlike wild-type, expressed only as an insoluble aggregate in inclusion bodies. Attempts to express thermo-TEAS at different temperatures and induction conditions proved unsuccessful, and the protein in the inclusion bodies was denatured before refolding and purification. Other thermal-stabilized proteins, such as Subtilisin BPN', also required protein refolding to yield the active enzyme.34 The folding of over-expressed, purified wild-type and mutant TEAS enzymes was examined by CD. Wild-type TEAS is a globular, largely α-helical protein lacking β-sheet secondary structure.33 On the basis of results from both CD and GC–MS activity assays, both enzymes appear to have retained an α-helical composition; furthermore, both wild-type and mutant TEAS likely folded into the correct protein structure, as demonstrated by retention of their catalytic activities [Fig. 4(a)].
Figure 4.
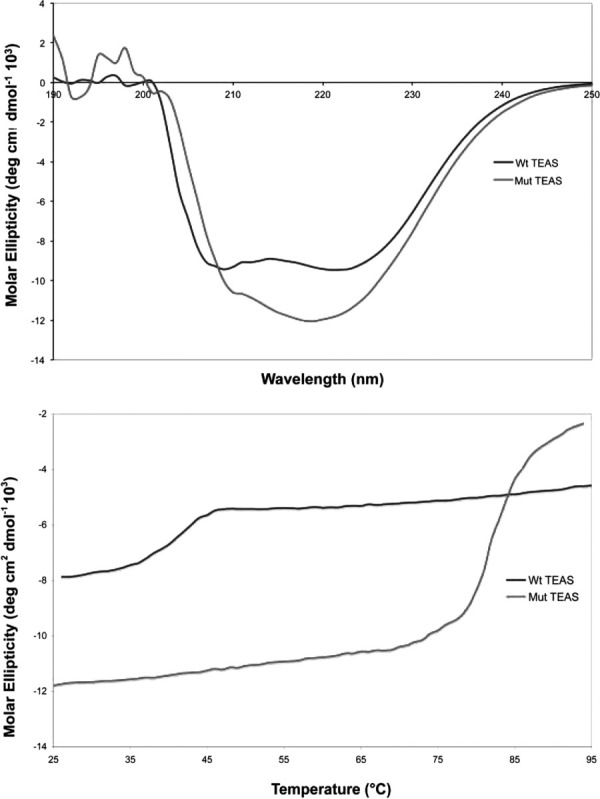
CD analysis of wild-type and refolded thermo-TEAS. (a) The far-UV spectra of wild-type and thermo-TEAS recorded at 20°C. (b) Temperature-induced unfolding of wild-type and thermo-TEAS, as determined by monitoring molar ellipticity at 222 nm. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The melting profile of wild-type and mutant TEAS was determined by monitoring the CD signal at 222 nm from 25 to 95°C. Under these conditions, wild-type TEAS denatured at 38°C [Fig. 4(b)]. The replacement of solvent-exposed hydrophobic residues and the addition of salt bridges on the surface of the mutant enzyme greatly enhanced the stability of the mutant TEAS. The mutant TEAS remains completely folded across the lower temperature range until denaturation occurs around 83°C [Fig. 4(b)]. The roughly doubled melting point of thermo-TEAS versus wild-type dramatically demonstrates the effectiveness of computational predictions made by the SCADS algorithm.
Thermostable TEAS activity at elevated temperatures
Next, we examined the catalytic performance of thermo-TEAS at elevated temperatures. At 25 and 37°C both enzymes were able to convert the natural substrate FPP to 5-epi-aristolochene with the products formed having identical retention times and fragmentation patterns by GC–MS (Fig. 5). Incubation of FPP under identical reaction conditions, without addition of enzyme, failed to generate product (data not shown). Thus, the mutations in the thermo-TEAS do not appear to have a major effect on the folded structure of the enzyme, which is required for the proper formation of the enzyme active site. However, compared with wild-type TEAS, thermo-TEAS had reduced levels of product forming activity at 37°C. The thermostability conferring mutations evidently decrease the net activity of the enzyme, perhaps by perturbing its conformational dynamics or through allosteric influence on the active site conformation.
Figure 5.
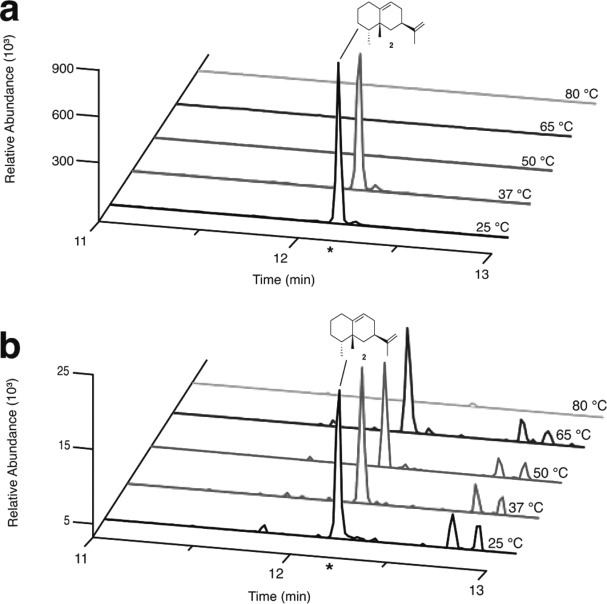
GC–SIM–MS of wild-type and thermo-TEAS reaction products at various temperatures. (a) Product formation of 5-epi-aristolochene by wild-type TEAS at the specified temperatures. (b) Product formation of 5-epi-aristolochene by thermo-TEAS. (*) Indicates the retention time expected for 5-epi-aristolochene (depicted), which was verified by the EI–MS fragmentation pattern. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
As expected from the CD-monitored denaturation experiment, temperatures above 40°C abolished wild-type TEAS activity [Fig. 5(a)]. Unlike the wild-type enzyme, thermo-TEAS could catalyze the cyclization of FPP to 5-epi-aristolochene at 50 and 65°C, but not 80°C [Fig. 5(b)]. Thus, both wild-type and mutant enzymes lose activity slightly below their melting temperatures. Unlike the wild-type TEAS, thermo-TEAS products include additional GC–MS peaks at all temperatures surveyed. For the additional products, the fragmentation patterns and MW (204 g/mol) are consistent with sesquiterpenes. Altered product specificity due to mutations distant from an enzyme active site has been observed previously, and likely results from modified active site dynamics and geometry.35,36 However, the strategy of introducing stabilizing mutations into TEAS resulted in a variant retaining activity at temperatures almost 30°C above the wild-type enzyme. Thus, thermo-TEAS provides the first example to our knowledge of a thermostable terpene synthase.
Although successful at increasing thermostability, the mutations decreased product yields at room temperature and 37°C, as assayed by GC–MS, possibly because of the slower product off-rates resulting from a more rigid protein fold. Release of the product is the rate-limiting step for wild-type TEAS,5,37,38 farnesyl diphosphate synthase and at least one other terpene synthase.39 However, reports of increased thermostability correlating with enhanced enzymatic activity largely involve enzymes catalyzing single step reactions. Thus, accommodating the different enzyme conformations required for the multistep catalysis and product release of TEAS could complicate the enhancement of catalysis through thermostability conferring mutagenesis.
Whereas this study focuses on one particularly thermostable mutant, the methods described could uncover thermostable mutants that display different dynamics with potentially higher productivity. The thermo-TEAS expressed well, but only as insoluble protein in inclusion bodies. Having established a viable selection for novel terpene synthase activities (catalysis at elevated temperatures), additional selections for protein solubility could also provide benefits.
Materials and Methods
Farnesyl diphosphate (FPP, 1) was from Sigma Aldrich. Oligonucleotide primers were purchased from MWG Biotech. The Gateway expression vector pHis9-GW, containing the wild-type TEAS gene, was kindly provided by Drs. Joseph P. Noel and Paul E. O'Maille of the Salk Institute (La Jolla, CA).
Computationally directed TEAS mutagenesis
The crystal structure of TEAS with a resolution of 2.15 Å was used for the design calculations (PDB accession code 5EAU).33 The inhibitor, trifluorofarnesyl diphosphate, and all crystallographic water molecules were removed, but two crystallographic Mg2+ ions were retained. Mutations consistent with the wild-type structure were identified using a SCADS, which allows variation of amino acid identity and side chain conformation (rotamer state).23,24 Up to 10 rotamer states where permitted for each residue in the protein,40 except for residues possessing nonrotamer conformations in the crystal structure and variation of sidechain conformation led to large increases in energy (>10 kcal/mol); such sites were constrained at the wild-type sidechain conformations. Amino acid identities were prepatterned at the mutated sites based on the number of Cβ atoms within 8 Å of the amino acid: for residues with 0–6 Cβ atoms, possible substitutions ADEGKNQRS were considered; for residues with 7–8 Cβ atoms, substitutions ADEFGIKLMNQRSVWY were considered; and for the most buried residues (10 or more Cβ atoms), substitutions AFGILMVW were permitted. Calculations were performed using a version of the Amber force field41 to quantify interatomic interactions, and the estimate of the total energy using such a potential averaged over sequences satisfying the desired constraints is denoted Ec. As described previously, the probabilities are obtained at a value of 1/Tc = 1.4, where 1/Tc is a Lagrange multiplier involving an effective temperature conjugate to Ec.23,24 At each mutated position, the most probable amino acid was selected.
Construction of mutant TEAS by oligonucleotide directed mutagenesis
A Nsi I-Afl II fragment encoding the wild-type TEAS gene was subcloned into a previously described phage display vector42 with TEAS-specific oligonucleotides TEAS-F and TEAS-R. A mutant TEAS gene with all 12 mutations suggested by the SCADS algorithm was constructed using an optimized version43 of the oligonucleotide-directed mutagenesis method developed by Kunkel.44 The mutations, encoded by six oligonucleotides, required two rounds of tri-oligonucleotide mutagenesis. A unique NgoM IV restriction site was introduced into the mutant TEAS gene during the mutagenesis to differentiate the mutant from the wild-type gene, an addition also resulting in a D333A mutation. The following oligonucleotides were used (mutation-encoding substitutions are underlined and restriction endonuclease sites are italicized in bold):
TEAS-F, 5′-GGCTATCGGATGCATCGCACCATCA CCATCACCATGCCTCAGCAGCAGTTGC-3′;
TEAS-R, 5′-TTTTTTCTTAAGAATTTTGATGGAGTCCACAAG-3′;
SCAD 1,5′-GGGGTGATCAGTTCCTTTCAGACTCCCGTGACAACCAGCGT
GCGGAAAAGTATATATATGCTC-3′;
SCAD 2,5′-GAACAAACGAGGAGTATGCTGCGTGCAACCGGAAGGAAATTGGC-3′;
SCAD 3,5′-GAGTCTCTTGCTAGTGATGTCAAAGGTCTGCTGATGTTGTATGAAGCTTCACATGTAAG-3′;
SCAD 4,5′-ACGCACTTGCTTTCTCCACTAAACATCTTGAATCTGCA GCTCC-3′;
SCAD 5,5′-CAAATTGGATTTCAACTTGCTCATGATGAAACACAAACA AGAACTTGCTCAAG-3′;
SCAD 6,5′-GATGCCATACAAAGATGGGATGAAAACGAAATCGACCGT CTG
CCGGCCTACATGAAAATCATCTTCAAAGCTATT CTAGATCTCTACAA-3′;
TEAS myc tag, 5′-GCCTCGGCTTATGCATCG GAACAGAAACTGATCTCCGAAGAA
GACCTGATGGCCTCAGCAGCAGTTG-3′.
Expression of phage-displayed TEAS
Colonies of E. coli strain XL1-Blue (Stratagene) harboring phagemids, encoding genes for myc-tagged wild-type, and mutant TEAS, were picked and grown in 10 mL of 2YT supplemented with tetracycline (5 μg/mL) for 8 h at 37°C. Cells were then coinfected with 10 μL of M13 KO7 helper phage (1010 pfu/mL, GE Healthcare), and further incubated for 20 min at 37°C. The cultures were then transferred to 50 mL of 2YT supplemented with carbenicillin (50 μg/mL), 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and grown overnight at 37°C. Phage displaying wild-type and mutant TEAS were purified by polyethylene glycol (PEG)–NaCl precipitation,49 resuspended in 0.5 mL of phosphate buffered saline (PBS), and verified for display by enzyme linked immunosorbent assay (ELISA).
Selections for increased thermostability
Myc-tagged wild-type and mutant TEAS phage (50 nM) were mixed in a 7:3 ratio, respectively. The protease chymotrypsin (1 μM) was added to each sample, and the temperature was increased to 37°C. After incubation for 7 min, the proteolytic digestion was quenched by adding 15 μL of stop solution (PBS, 0.05% Tween 20, 200 μM phenylmethylsulfonyl fluoride) for 10 min at room temperature. AMaxisorp immunoplate (96-well) was coated with anti-myc antibody (GeneTex, 1:2500 in 50 mM sodium carbonate buffer, pH 9.6) for 2 h at room temperature. The plate was then blocked for 30 min at room temperature with bovine serum albumin (BSA) (0.2%) in PBS, and washed three times with PT buffer (PBS, 0.05% Tween 20). The coated wells were incubated with the proteolyzed phage mixture (50 μL) for 1 h at room temperature, and washed 12 times with PT buffer. After washing, bound phage was eluted by the addition of 10 mM glycine (50 μL, pH 2.7) for 3 min and the acidified phage solution was removed. Eluted phage were used to infect 1 mL of log-phase XL1-Blue cells for 1 h at 37°C, before coinfecting with 1 μL of M13 KO7 helper phage (1010 pfu/mL) for 30 min at 37°C. The infected culture was transferred to 30 mL of 2YT supplemented with 50 μL of carbenicillin, and grown overnight for 16 h at 37°C.
After each of three rounds of selection, phage were isolated with PEG–NaCl precipitation, and resuspended with PBS as described above. The resulting phage was isolated, and the relative levels of wild-type and mutant genomes were analyzed by PCR amplification with TEAS-specific oligonucleotides. The mutant TEAS gene contained a unique NgoM IV restriction site used to monitor the enrichment of the mutant enzyme from the wild-type one. The relative intensities of wild-type and mutant DNA were quantified using Bio-Rad Quantity One 1D Analysis Software (version 4.5.0.046).
Wild-type TEAS expression and purification
Wild-type TEAS was expressed as described previously46 using the Gateway vector pHis9-GW transformed into E. coli BL21 (DE3) cells (Stratagene). Cells were grown in Terrific Broth supplemented with 40 μg/mL of kanamycin. When the OD600 reached 1.0, the cell culture was chilled on ice for 10 min, followed by IPTG induction (0.1 mM) for 4 h at 22°C. Cells were harvested by centrifugation, resuspended in a lysis buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 20 mM β-mercaptoethanol, 10% glycerol, and 1% Tween 20), and lysed with lysozyme and sonication.
Wild-type TEAS was purified to >95% purity by affinity chromatography with nickel-nitrilotriacetic acid (Ni2+–NTA) resin (Qiagen). After equilibrating the column with lysis buffer, the cell lysate was passed through the nickel resin, and washed with 10 column volumes of a wash buffer (50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 20 mM imidazole, 20 mM β-mercaptoethanol, and 10% glycerol). The protein was eluted with 10 column volumes of wash buffer supplemented with imidazole (250 mM). The expression level and purity of the protein were monitored by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For long-term storage, the eluted protein was concentrated by centrifugation using a 10 kDa MW cut-off concentrator (Millipore), dialyzed against storage buffer (50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM dithiothreitol (DTT)), before addition of glycerol (50% v/v final concentration), and stored at −20°C.
Mutant TEAS expression and purification
The open reading frame (ORF) encoding mutant TEAS was introduced into the Gateway expression vector pHis9-GW, and expressed under the same induction conditions as the wild-type enzyme. The mutant TEAS, however, formed inclusion bodies that required the following solubilization treatment with N-lauroylsarcosine.47
Inclusion bodies were resuspended in 100 mL of a wash buffer (20 mM Tris–HCl, pH 7.5, 10 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100, and 2% sodium deoxycholate), and centrifuged at room temperature for 15 min at 29,800g. The pellet was rewashed, and centrifuged again before resolubilization in 80 mL of N-lauroylsarcosine solution (50 mM N-cyclohexyl-3-aminopropanesulfonic acid, pH 11, 2% N-lauroylsarcosine, and 1 mM DTT). The solution was mixed gently for 1 h at 30°C, and centrifuged as described above. The soluble fraction was dialyzed against 20 mM Tris–HCl (pH 7.5) and glycerol was added to a final concentration of 10%. The protein sample was then concentrated to ≈2–3 mL by centrifugation using a 30 kDa MW cut-off concentrator (Millipore).
At 4°C, 35 μL of the resolubilized, concentrated fraction was added drop-wise to a beaker containing 100 mL of refolding buffer (200 mM arginine–HCl, 0.05% sodium azide, 1 mM EDTA, 100 mM Tris–HCl, 2 mM reduced glutathione, and 1 mM oxidized glutathione, pH 6.5), and mixed slowly by stirring. After 10 min, another 120 μL fraction was added drop-wise to the refolding buffer. Additional 120 μL drop-wise aliquots were added every 10 min until the entire resolubilized fraction was mixed into the refolding buffer. The beaker was then sealed with parafilm, and incubated at 4°C for 48–60 h with stirring.
After incubation, the refolded enzyme was concentrated to ≈20 mL by room temperature centrifugation at 3000g using a 30 kDa MW cut-off concentrator. The concentrated enzyme was dialyzed against 4 L of 500 mM NaCl, 50 mM NaH2PO4 (pH 8.0) overnight at 4°C before being purified by Ni2+–NTA affinity chromatography, concentrated, and stored as described above for wild-type TEAS.
TEAS circular dichroism
CD spectra were obtained on a Jasco Model 810 spectropolarimeter (equipped with a PFD-425S Peltier). The spectra were collected at 20°C over a range from 190 to 250 nm with a 2 s response time, 0.1 cm path length, and a 2 nm bandwidth. The scanning speed was 50 nm/min, and the data pitch was set at 1 nm. Protein unfolding was monitored by recording the CD signal at 222 nm over a temperature range from 25 to 95°C with a temperature rate increase of 40°C/h. The response time and bandwidth were 32 s and 2 nm, respectively. Before measurement, wild-type (0.5 mg/mL) and mutant TEAS (0.7 mg/mL) were dialyzed into 20 mM NaH2PO4 (pH 8.0) and 20 mM Tris–HCl (pH 8.0), respectively. The CD-monitored denaturation for wild-type TEAS was also measured in the Tris buffer, and the Tm remained identical (data not shown).
CD results were obtained in millidegrees (mdeg), which were then converted to molar ellipticity. The molar ellipticity in units of deg cm2 dmol−1 103 was derived from the equation: [θ]obs = mrw /10 ℓ C, where [θ]obs is the observed ellipticity in degrees, mrw is the mean molecular weight per residue (115 Da), ℓ is the cell length (0.1 cm), and C is the concentration of the enzyme samples in g/mL.
Terpene synthase activity assay
Terpene synthase activity for wild-type and mutant TEAS was confirmed by GC–MS as reported previously.18 In brief, reaction buffer (50 mM Tris–HCl, pH 7.5, and 20 mM MgCl2) was added to 3.5 μM terpene synthase and 50 μM of FPP in a 2 mL glass crimp-top vial (National Scientific) for a final reaction volume of 500 μL. The reactants were mixed, overlaid with 500 μL of ethyl acetate, capped, and incubated for 1 h at room temperature. Reactions were quenched by vortexing for 10 s, and the extracted hydrocarbon products analyzed by GC–MS. To test the thermostability attained for the mutant TEAS, activity assays were conducted in 1.5 mL screw-top vials as described above with 1 h incubation reactions at the following temperatures: 25, 37, 50, 65, and 80°C. No significant loss of solvent was observed at lower temperatures, but additional solvent was added to the 80°C sample to ensure the solvent level remained constant at the 500 μL mark.
GC–MS analysis of sequiterpenes
Extracted products were injected onto a Trace MS+ (Thermo Instruments) GC–MS system operated in electron ionization mode. The sesquiterpenes were separated on a DB-5 column (J&W Scientific) (30 m × 0.32 mm column × 0.25 μm film thickness). The GC oven temperature was ramped from 50 to 290°C at 10°C per minute. When necessary to increase the sensitivity of detection, the MS was operated in selected ion monitoring (SIM) mode, using m/z 105, 161, and 204, which are dominant, characteristic ions of the expected 5-epi-aristolochene products.
Acknowledgments
We thank Kyoto Pharmaceuticals (for a partial postdoctoral fellowship to K.K.), the American Chemical Society Division of Organic Chemistry Graduate Fellowship Award (awarded to J.E.D.), and Dr. Wytze van der Veer for expert technical assistance with the CD experiments.
Supplementary material
References
- 1.Greenhagen BT, Schoenbeck MA, Yeo Y-S, Chappell J. The chemical wizardry of isoprenoid metabolism in plants. Recent Adv Phytochem. 2003;37:231–251. [Google Scholar]
- 2.Croteau R, Cane DE. Monoterpene and sesquiterpene cyclases. Methods Enzymol. 1985;110:383–405. [Google Scholar]
- 3.Cane DE. Enzymatic formation of sesquiterpenes. Chem Rev. 1990;90:1089–1103. [Google Scholar]
- 4.Greenhagen B, Chappell J. Molecular scaffolds for chemical wizardry: learning nature's rules for terpene cyclases. Proc Natl Acad Sci USA. 2001;98:13479–13481. doi: 10.1073/pnas.261562898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rising KA, Starks CM, Noel JP, Chappell J. Demonstration of germacrene A as an intermediate in 5-epi-aristolochene synthase catalysis. J Am Chem Soc. 2000;122:1861–1866. [Google Scholar]
- 6.Bloom JD, Meyer MM, Meinhold P, Otey CR, MacMillan D, Arnold FH. Evolving strategies for enzyme engineering. Curr Opin Struct Biol. 2005;15:447–452. doi: 10.1016/j.sbi.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 7.Cherry JR, Fidantsef AL. Directed evolution of industrial enzymes: an update. Curr Opin Biotechnol. 2003;14:438–443. doi: 10.1016/s0958-1669(03)00099-5. [DOI] [PubMed] [Google Scholar]
- 8.Korkegian A, Black ME, Baker D, Stoddard BL. Computational thermostabilization of an enzyme. Science. 2005;308:857–860. doi: 10.1126/science.1107387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lipovsek D, Antipov E, Armstrong KA, Olsen MJ, Klibanov AM, Tidor B, Wittrup KD. Selection of horseradish peroxidase variants with enhanced enantioselectivity by yeast surface display. Chem Biol. 2007;14:1176–1185. doi: 10.1016/j.chembiol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 10.Moore JC, Arnold FH. Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nat Biotechnol. 1996;14:458–467. doi: 10.1038/nbt0496-458. [DOI] [PubMed] [Google Scholar]
- 11.Furukawa K, Suenaga H, Goto M. Biphenyl dioxygenases: functional versatilities and directed evolution. J Bacteriol. 2004;186:5189–5196. doi: 10.1128/JB.186.16.5189-5196.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aharoni A, Amitai G, Bernath K, Magdassi S, Tawfik DS. High-throughput screening of enzyme libraries: thiolactonases evolved by fluorescence-activated sorting of single cells in emulsion compartments. Chem Biol. 2005;12:1281–1289. doi: 10.1016/j.chembiol.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 13.Castle LA, Siehl DL, Gorton R, Patten PA, Chen YH, Bertain S, Cho HJ, Duck N, Wong J, Liu D, Lassner MW. Discovery and directed evolution of a glyphosate tolerance gene. Science. 2004;304:1151–1154. doi: 10.1126/science.1096770. [DOI] [PubMed] [Google Scholar]
- 14.Umeno D, Tobias AV, Arnold FH. Diversifying carotenoid biosynthetic pathways by directed evolution. Microbiol Mol Biol Rev. 2005;69:51–78. doi: 10.1128/MMBR.69.1.51-78.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reetz MT, Brunner B, Schneider T, Schulz F, Clouthier CM, Kayser MM. Directed evolution as a method to create enantioselective cyclohexanone monooxygenases for catalysis in Baeyer–Villiger reactions. Angew Chem Int Ed Engl. 2004;43:4075–4078. doi: 10.1002/anie.200460272. [DOI] [PubMed] [Google Scholar]
- 16.Giver L, Gershenson A, Freskgard PO, Arnold FH. Directed evolution of a thermostable esterase. Proc Natl Acad Sci USA. 1998;95:12809–12813. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hart EA, Hua L, Darr LB, Wilson WK, Pang J, Matsuda SPT. Directed evolution to investigate steric control of enzymatic oxidosqualene cyclization. An isoleucine-to-valine mutation in cycloartenol synthase allows lanosterol and parkeol biosynthesis. J Am Chem Soc. 1999;121:9887–9888. [Google Scholar]
- 18.O'Maille PE, Chappell J, Noel JP. A single-vial analytical and quantitative gas chromatography–mass spectrometry assay for terpene synthases. Anal Biochem. 2004;335:210–217. doi: 10.1016/j.ab.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 19.Yoshikuni Y, Ferrin TE, Keasling JD. Designed divergent evolution of enzyme function. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 20.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF. De novo design of a single-chain diphenylporphyrin metalloprotein. J Am Chem Soc. 2007;129:10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, Degrado WF. De novo design of a redox-active minimal rubredoxin mimic. J Am Chem Soc. 2005;127:5804–5805. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 22.Slovic AM, Kono H, Lear JD, Saven JG, DeGrado WF. Computational design of water-soluble analogues of the potassium channel KcsA. Proc Natl Acad Sci USA. 2004;101:1828–1833. doi: 10.1073/pnas.0306417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J Mol Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Kono H, Saven JG. Statistical theory for protein combinatorial libraries. Packing interactions, backbone flexibility, and the sequence variability of a main-chain structure. J Mol Biol. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 25.Parsell DA, Sauer RT. The structural stability of a protein is an important determinant of its proteolytic susceptibility in Escherichia coli. J Biol Chem. 1989;264:7590–7595. [PubMed] [Google Scholar]
- 26.Matsuura T, Pluckthun A. Selection based on the folding properties of proteins with ribosome display. FEBS Lett. 2003;539:24–28. doi: 10.1016/s0014-5793(03)00178-9. [DOI] [PubMed] [Google Scholar]
- 27.Distefano MD, Zhong A, Cochran AG. Quantifying beta-sheet stability by phage display. J Mol Biol. 2002;322:179–188. doi: 10.1016/s0022-2836(02)00738-6. [DOI] [PubMed] [Google Scholar]
- 28.Bai Y, Feng H. Selection of stably folded proteins by phage-display with proteolysis. Eur J Biochem. 2004;271:1609–1614. doi: 10.1111/j.1432-1033.2004.04074.x. [DOI] [PubMed] [Google Scholar]
- 29.Chu R, Takei J, Knowlton JR, Andrykovitch M, Pei W, Kajava AV, Steinbach PJ, Ji X, Bai Y. Redesign of a four-helix bundle protein by phage display coupled with proteolysis and structural characterization by NMR and X-ray crystallography. J Mol Biol. 2002;323:253–262. doi: 10.1016/s0022-2836(02)00884-7. [DOI] [PubMed] [Google Scholar]
- 30.Finucane MD, Tuna M, Lees JH, Woolfson DN. Core-directed protein design. I. An experimental method for selecting stable proteins from combinatorial libraries. Biochemistry. 1999;38:11604–11612. doi: 10.1021/bi990765n. [DOI] [PubMed] [Google Scholar]
- 31.Kristensen P, Winter G. Proteolytic selection for protein folding using filamentous bacteriophages. Fold Des. 1998;3:321–328. doi: 10.1016/S1359-0278(98)00044-3. [DOI] [PubMed] [Google Scholar]
- 32.Sieber V, Pluckthun A, Schmid FX. Selecting proteins with improved stability by a phage-based method. Nat Biotechnol. 1998;16:955–960. doi: 10.1038/nbt1098-955. [DOI] [PubMed] [Google Scholar]
- 33.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 34.Almog O, Gallagher T, Tordova M, Hoskins J, Bryan P, Gilliland GL. Crystal structure of calcium-independent subtilisin BPN' with restored thermal stability folded without the prodomain. Proteins. 1998;31:21–32. doi: 10.1002/(sici)1097-0134(19980401)31:1<21::aid-prot3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 35.Greenhagen BT, O'Maille PE, Noel JP, Chappell J. Identifying and manipulating structural determinates linking catalytic specificities in terpene synthases. Proc Natl Acad Sci USA. 2006;103:9826–9831. doi: 10.1073/pnas.0601605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiller B, Gershenson A, Arnold FH, Stevens RC. A structural view of evolutionary divergence. Proc Natl Acad Sci USA. 1999;96:12305–12310. doi: 10.1073/pnas.96.22.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cane DE, Chiu HT, Liang PH, Anderson KS. Pre-steady-state kinetic analysis of the trichodiene synthase reaction pathway. Biochemistry. 1997;36:8332–8339. doi: 10.1021/bi963018o. [DOI] [PubMed] [Google Scholar]
- 38.Mathis JR, Back K, Starks C, Noel J, Poulter CD, Chappell J. Pre-steady-state study of recombinant sesquiterpene cyclases. Biochemistry. 1997;36:8340–8348. doi: 10.1021/bi963019g. [DOI] [PubMed] [Google Scholar]
- 39.Laskovics FM, Poulter CD. Prenyltransferase; determination of the binding mechanism and individual kinetic constants for farnesylpyrophosphate synthetase by rapid quench and isotope partitioning experiments. Biochemistry. 1981;20:1893–1901. doi: 10.1021/bi00510a027. [DOI] [PubMed] [Google Scholar]
- 40.Bower MJ, Cohen FE, Dunbrack RL. Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: a new homology modeling tool. J Mol Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- 41.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta S, Weiner P. A new force-field for molecular mechanical simulation of nucleic-acids and proteins. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 42.Murase K, Morrison KL, Tam PY, Stafford RL, Jurnak F, Weiss GA. EF-Tu binding peptides identified, dissected, and affinity optimized by phage display. Chem Biol. 2003;10:161–168. doi: 10.1016/s1074-5521(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 43.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods Enzymol. 2000;328:333–363. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 44.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 45.Olszewski A, Sato K, Aron ZD, Cohen F, Harris A, McDougall BR, Robinson WE, Jr, Overman LE, Weiss GA. Guanidine alkaloid analogs as inhibitors of HIV-1 Nef interactions with p53, actin, and p56lck. Proc Natl Acad Sci USA. 2004;101:14079–14084. doi: 10.1073/pnas.0406040101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Maille PE, Chappell J, Noel JP. Biosynthetic potential of sesquiterpene synthases: alternative products of tobacco 5-epi-aristolochene synthase. Arch Biochem Biophys. 2006;448:73–82. doi: 10.1016/j.abb.2005.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgess RR. Purification of overproduced Escherichia coli RNA polymerase sigma factors by solubilizing inclusion bodies and refolding from Sarkosyl. Methods Enzymol. 1996;273:145–149. doi: 10.1016/s0076-6879(96)73014-8. [DOI] [PubMed] [Google Scholar]
- 48.Davis EM. Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes. In: Leeper FJ, Vederas JC, Croteau R, editors. Topics in Current Chemistry: Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids. Vol. 209. Berlin: Springer-Verlag; 2000. pp. 53–95. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.