

Abstract
There is growing interest in the development of protein switches, which are proteins whose function, such as binding a target molecule, can be modulated through environmental triggers. Efforts to engineer highly pH sensitive protein–protein interactions typically rely on the rational introduction of ionizable groups in the protein interface. Such experiments are typically time intensive and often sacrifice the protein's affinity at the permissive pH. The underlying thermodynamics of proton-linkage dictate that the presence of multiple ionizable groups, which undergo a pKa change on protein binding, are necessary to result in highly pH-dependent binding. To test this hypothesis, a novel combinatorial histidine library was developed where every possible combination of histidine and wild-type residue is sampled throughout the interface of a model anti-RNase A single domain VHH antibody. Antibodies were coselected for high-affinity binding and pH-sensitivity using an in vitro, dual-function selection strategy. The resulting antibodies retained near wild-type affinity yet became highly sensitive to small decreases in pH, drastically decreasing their binding affinity, due to the incorporation of multiple histidine groups. Several trends were observed, such as histidine “hot-spots,” which will help enhance the development of pH switch proteins as well as increase our understanding of the role of ionizable residues in protein interfaces. Overall, the combinatorial approach is rapid, general, and robust and should be capable of producing highly pH-sensitive protein affinity reagents for a number of different applications.
Keywords: single domain antibody, camelid VHH, protein engineering, pH switch, combinatorial histidine scanning, phage display, dual-function, pKa, histidine
Introduction
Protein reagents, in particular antibodies, play increasing roles in fundamental research, such as imaging and affinity reagents, as well as numerous life science applications such as therapeutics, diagnostics, and biosensors. The development of library-based in vitro screening methods has enhanced antibody engineering by providing new routes to improve antibody binding affinity and stability. These methods, particularly synthetic-based libraries,1 provide protein engineering opportunities that are simply not possible using traditional immunization techniques and may play a significant role in developing custom-made designer antibodies suitable for any application.
In general, the nature of protein affinity reagents in most life science applications may be simplified as a one-way binding event, where antibody binding cannot be easily controlled (or turned off) without the addition of harsh solution conditions (e.g., pH extremes, denaturants, temperature, etc.). The use of such conditions frequently irreversibly damages the protein reagent and/or the target molecule of interest or is simply not compatible with the reaction/assays of interest. Consequently, the development of protein molecular switches,2,3 which are proteins whose function (e.g., binding) may be modulated (or linked) through an environmental trigger (e.g., pH or ligand) or conformational change, are desired in many applications.
The underlying physical basis for “protein switches” originates from coupled (or linked) equilibria. This is perhaps not very surprising; such coupled-equilibria provide powerful routes to biological regulation. Biology commonly uses coupled equilibria to generate pH-dependent binding events, where protonation of an ionizable residue(s) on a macromolecule is coupled to the protein binding equilibrium. Proton-linked binding events play an important role in biological regulation, which includes well-known examples such as the Bohr effect in hemoglobin4 and the pH-dependent binding of serine protease inhibitors for their cognate serine proteases.5,6 One of the most sensitive pH-dependent binding events involves human prolactin (hPRL), which is reported to display a large (500-fold) decrease in binding affinity for its receptor, hPRLR, over a relatively modest pH drop from pH 8 to 6.7 The high pH sensitivity of hPRL/hPRLR binding is likely to play a physiological role in function, such as terminating the signaling event on internalization into endosomes or aiding hormone recycling.7 Thermodynamically, this pH dependence is believed to be linked to the presence of several ionizable histidines within the hRPL/hPRLR interface.8 Ultimately, the sensitivity (or efficiency) of such pH switches is determined by the balance between the electrostatic (i.e., pH dependent) and nonelectrostatic components of the binding equilibrium.
Here, we address the hypothesis that a highly pH-dependent binding event requires multiple ionizable residues (e.g., histidine) and that these may be introduced successfully without interfering with affinity under the permissive pH conditions. Using a model antibody/antigen complex, anti-RNase A VHH/RNase A,9,10 a novel, synthetic, combinatorial library was developed where all 22 VHH interface positions sample histidine and the original wild-type residue. In doing so, all 4 × 106 unique histidine/wild-type combinations were generated and subjected to in vitro screening to isolate pH-sensitive clones. The resulting antibodies possessed significant histidine incorporation and displayed drastically reduced affinity with minor decreases in pH while retaining near wild-type affinity at physiological pH. Detailed binding studies, using isothermal titration calorimetry (ITC), were performed on two pH-sensitive clones to validate the engineered pH dependence. These results were consistent with several histidine groups participating in the proton-linked binding events. Overall, the library-based histidine scanning approach is both general and robust, and is capable of producing numerous highly pH-sensitive antibody variants that would not be generated using traditional design methods. In addition, these findings provide new insights for pH-dependent biological interactions and protein design efforts.
Results
Chemical basis for pH-dependent binding
Engineering protein switches into macromolecules is a considerable challenge. Much of these efforts have focused on modifications of existing biologically relevant switches.11,12 Only a handful of examples exist that introduce novel molecular control into existing interactions. Introduction of pH-dependent binding is an example where some success has been achieved. This includes selective covalent modification of interface tyrosines,13,14 as well as introducing interface histidine residues through structure-based modeling.15–20 Mechanistically, the engineering of pH sensitive binding requires the inserted residue (e.g., histidine) or functional group to undergo a change in pKa on binding [Fig. 1(A)]. This change in pKa value stems from the ionizable group's sensitivity to a change in microenvironments (e.g., those present in bound and unbound states or folded and unfolded states). For example, protein stability studies by Garcia-Moreno and coworkers have revealed a surprising range of shifts in pKa values for ionizable groups that become buried in dehydrated environments.21–23 Perhaps, most importantly, their work reveals that ionizable groups can tolerate seemingly unexpected environments, which may highlight their use in pH-dependent biological events.
Figure 1.
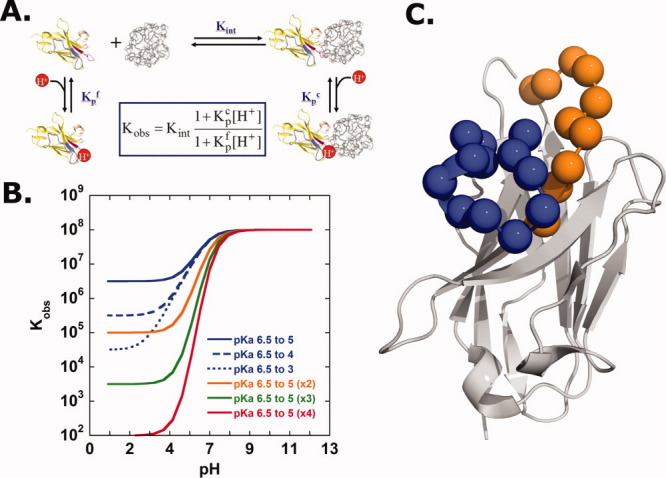
(A) Thermodynamic model for a single proton-linked protein binding event. (B) Simulation illustrating the pH dependence of the observed binding constant, Kobs, which is dependent on both the magnitude of the ionizable residue's pKa change, as well as the number of ionizable groups undergoing a pKa change upon binding. (C) Ribbon representation of the single domain anti-RNase A VHH antibody. The alpha-carbons of the 22 His/Wt interface residues are shown in spheres. CDR1: orange; CDR3: blue.
Although each of the examples of engineered pH-dependent binding, discussed above, did indeed introduce pH sensitivity, the results are quite modest considering what is thermodynamically possible. To illustrate this point, Figure 1(B) displays a plot of simulated binding data for a protein interaction exhibiting different extents of linked proton binding. The extent of pKa perturbation and, more importantly, the total number of ionizable groups undergoing pKa changes will significantly impact the degree of pH sensitivity. For example, a single, well-placed histidine group that experiences a significant pKa drop of 3.5 units on binding only exhibits limited pH sensitivity over the pH range of 7–4. The full thermodynamic effect of such linkage is only apparent at very low pH values where other ionization event (such as those involved with protein stability) will likely begin to dominate. On the other hand, when multiple histidines are present, with each experiencing modest 1.5 unit drops in pKa on binding, a dramatic enhancement of pH sensitivity is observed, where the maximum value of the slope of dlogK/dpH equals the number of ionizable groups involved (i.e., 2, 3, or 4). It is worth mentioning that ionizable groups besides histidine (e.g., Arg, Lys, Asp, and Glu) are likely not to be as useful for physiological pH switches because the expected pKa value shifts for these groups into the physiological range would require a significant cost in free energy, resulting in protein destabilization.21–23
In practice, engineering pH-sensitive binding through the introduction of one, two, or more ionizable groups into a protein interface is a difficult task as simple correlations between a histidine's pKa and its structural environment are not obvious.24 This is a problem that cannot be solved through prediction or simulation as we cannot predict shifts in pKa with sufficient accuracy. Furthermore, the inserted ionizable residue must not only undergo a pKa perturbation on binding but also not dramatically interfere with high-affinity binding at the permissive pH. Previous approaches to insert histidine residues within a protein interface were often guided by structural knowledge of the protein complex; however, such approaches have limitations as only a small subset of the structural and energetic space is ultimately sampled. These techniques also tend to be time and cost intensive. Finally, although knowledge of the protein complex structure may certainly be useful toward engineering pH sensitivity, many, if not most, antibody affinity reagents are limited to knowledge of only the amino acid sequence.
Design and selection of pH-dependent linkage
Here, we examine whether highly pH-sensitive binding could be engineered into a model antibody/antigen system, anti-RNase A VHH/RNase A,9 which was selected due to its high affinity (Kd ∼20 nM) and lack of appreciable pH dependence to the binding affinity (down to at least pH 3.5). To introduce significant pH dependence within the anti-RNase A VHH interface, a novel, combinatorial scanning phage display strategy was developed. When compared with traditional interface alanine-scans25 or shotgun alanine scans,26 where the loss or retention of function (e.g., binding, enzymatic activity) on side chain removal is revealed, here, a histidine interface scan probes locations that introduce new function (i.e., proton binding sites). This approach exploits the frequently observed plastic nature of protein interfaces, whereby a large percentage of interface residues can tolerate substitution without dramatic loss in binding affinity.27,28 Using oligonucleotide-directed mutagenesis stratagies,29 an M13-phage library was generated so that each residue position within the anti-RNase A VHH binding interface, which includes a total of 22 residues, was allowed to sample both histidine and the original wild-type residue Fig. 1(C). The resulting library (with a theoretical diversity of 1010) simultaneously ensures complete histidine saturation (i.e., every possible combination of histidine at each position is sampled within the interface), while allowing retention of wild-type residues that are necessary for high-affinity binding at physiological pH.
Phage selection included two key steps for dual-function screening: binding and elution. Initially, RNase A-binding library members were subjected to solution capture at physiological pH (7.4) using biotinylated RNase A and streptavidin-coated magnetic beads. Phage particles retained on the magnetic beads were subjected to several wash steps to remove low-affinity variants. The second step involved phage elution where the pH-sensitive variants were released using moderately acidic conditions (pH 4.0–5.5). These clones were then reamplified and subjected to additional rounds of pH 7.4 binding and acidic pH elution. After three rounds of selection, immunoassays were used to screen for clones possessing pH-dependent binding. These assays demonstrated that, unlike wild-type anti-RNase A VHH, complete loss of binding was observed for all 24 clones tested when the pH was lowered from pH 7.4 to 4.0 (data not shown). Furthermore, binding was reversible, as binding was recovered on increasing the pH back to physiological conditions.
The location and extent of histidine incorporation was determined through DNA sequencing. The sequences of the resulting 26 unique clones (of 48 sequenced) are displayed in Figure 2(A). Of the 22 residue positions sampled in the His/Wt library, 10 different positions within the CDR1 and CDR3 binding loops tolerated histidine incorporation. This relates to 45% incorporation coverage, suggesting histidines are very well tolerated within the small VHH interface. Spatially, histidines are present in both interface loops, CDR1 and CDR3, with a slight preference for CDR1. Figure 2(B,C) presents a histidine insertion “hot-spot” map of the VHH interface. There is a clear preference for histidine substitutions within the interface periphery. Conversely, interface residues that do not tolerate histidine substitutions (i.e., the cold-spots) are located centrally, some of which protrude into the VHH interior, where they participate in hydrophobic intramolecular interactions. The number of simultaneous histidine incorporations is perhaps the most striking observation. VHH variants possessed a minimum of two and up to a maximum of five histidines within the binding interface. Notably, approximately one-third of the unique clones had “paired” histidines, where two histidines are located adjacent to each other in sequence or space [Fig. 2(A)].
Figure 2.

(A) Amino acid sequences of pH sensitive VHH variants. (B) Top and (C) side views of the VHH interface side chain residues color coded by histidine hot-spot incorporation frequency. Color map provided in legend. RNase A-white sticks.
Detailed analysis of the pH dependence
To address the central question whether significant pH dependence can be introduced with multiple, well-placed histidine residues, a detailed analysis of the observed binding constant, Kobs, was necessary. ITC, which allows the determination of binding constants, Kb, with high accuracy, was used to measure the protein–protein binding constant over a range of pH values. Although ITC also provides information regarding the underlying binding enthalpy and entropy (Table 1, Supporting Information), the presence of multiple linked-protonation events occurring over the pH range of interest will make interpretation of enthalpy/entropy components nontrivial. This complexity originates from an observed binding enthalpy that will possess contributions from not only from the intrinsic protein–protein binding but also the underlying protonation enthalpies (in the free and complex state), as well as buffer contributions.30 Consequently, our analysis here is limited to Kobs values. Two prototypical VHH variants were selected to determine the binding properties over a range of pH values. This included VHH#10, which is the “consensus clone” possessing the three “hot-spot” histidine substitution sites, Y29, Y33, and Y101 and VHH#24, which possessed five interface histidines [see Fig. 2(A)]. Figure 3(A,B) displays the binding constant profiles from pH 4.5 to 8.0. At the permissive pH, 7.4, both variants VHH#10 and VHH#24, possessed binding affinities within twofold and fivefold of the wild-type value, respectively. This corresponds to dissociation constants, Kd, of 35 nM (VHH#10) and 91 nM (VHH#24). On decreasing the pH, the observed binding constants decreased by ∼104 over a range of ∼2 pH units. Determination of binding constants below pH 4.5 was not feasible, due to low binding signal; however, the pH profile displays no indication of a lower plateau near pH 4.5, suggesting the binding constant would continue to decline as pH is decreased further. This trend would correspond to a binding constant that is no greater than a value of 100 (Kd = 10 mM), at pH 4.0.
Figure 3.
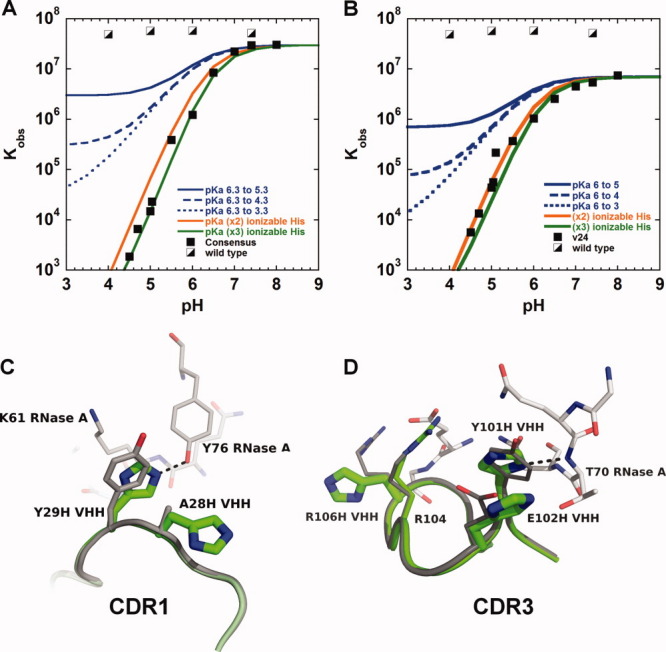
The pH dependence of the observed binding constant, Kobs, for the consensus VHH#10 (A) and VHH#24 (B) variants. For reference, the wild-type VHH/RNase A binding data is presented, as well as simulated curves for a single ionizable group undergoing a range of pKa changes. (C and D) Structural overlay of the interface loops CDR1 and CDR3, respectively, from the VHH-wt (grey; PDB ID: 2P49) and VHH#24 (light green) RNase A complexes. RNase A residues displayed in white. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To rule out simple pH-based unfolding as the cause of the decrease in binding affinity, VHH#10, VHH#24, and VHH-wt were analyzed by far-UV circular dichroism (Fig. 4). Overall, both pH-sensitive VHH variants exhibit no major change in structure down to pH 4.0, which is below the lowest pH performed using ITC (pH 4.5). The pH profiles of both variants are quite similar to that of the parent VHH-wt. In addition, thermal denaturation experiments were performed using circular dichroism (CD). At physiological pH, the wild-type, VHH#10, and VHH#24 variants all possessed comparable Tm values, 68.0 ± 0.3, 64.3 ± 0.1, and 66.4 ± 0.1°C, respectively [Fig. 5]. When the pH was lowered to pH 4.0, the Tm values were 61.2 ± 0.2, 53.4 ± 0.2, and 49.7 ± 0.2°C, for the wild-type, VHH#10, and VHH#24 variants, respectively [Fig. 5]. Despite a larger drop in Tm (relative to VHH-wt) when going from pH 7.4 to 4.0, both pH-sensitive VHH variants remained 100% folded up to ∼45°C at pH 4.0, well above the temperature used in binding analysis (10–25°C).
Figure 4.
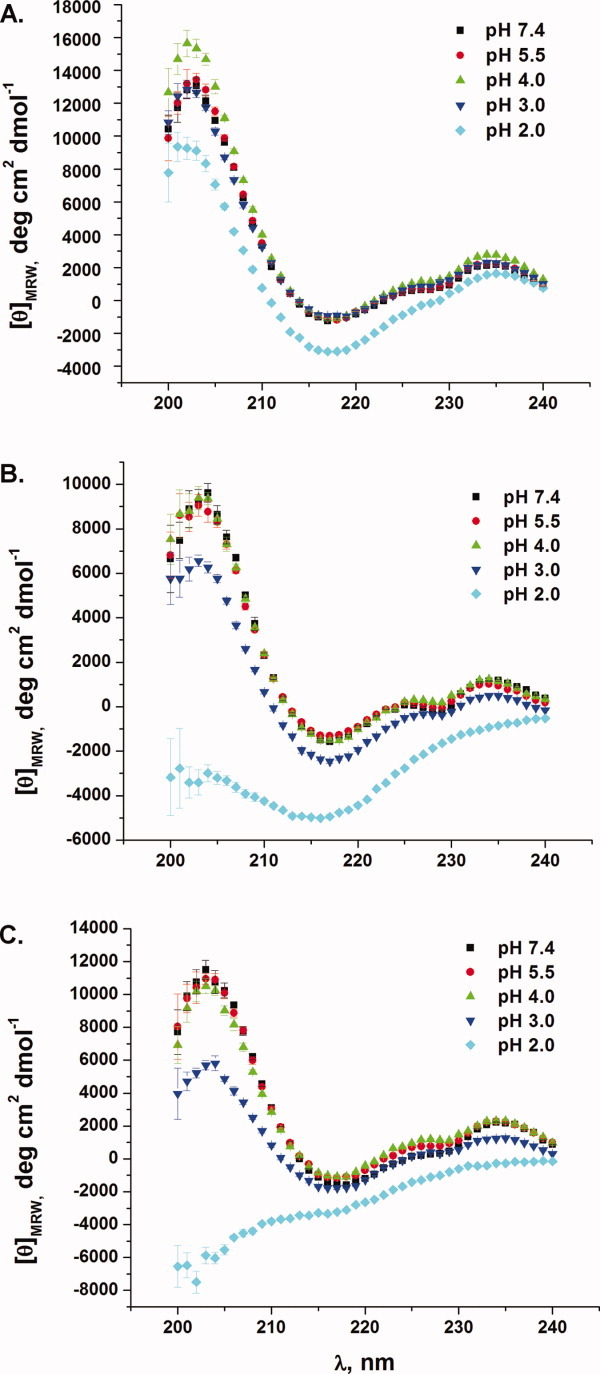
CD spectra as a function of pH for VHH-wt (A), VHH#10 (B) and VHH#24 (C). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.
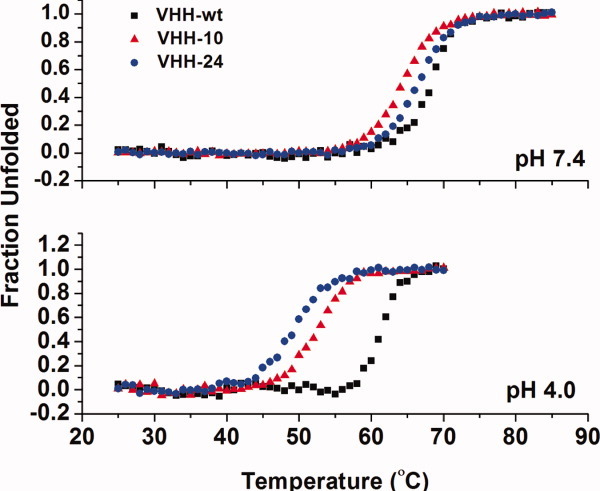
Temperature unfolding curves of the wild-type VHH, VHH#10, and VHH#24 at pH 7.4 and 4.0. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
When multiple ionizable groups undergo pKa changes on binding, detailed information of individual pKa values (or protonation thermodynamics) cannot be easily determined from simply fitting proton linkage models to Kobs versus pH data (or calorimetric data such as that described by Baker and Murphy30), due to a high correlation between the protonation thermodynamic parameters (e.g., pKa, ΔHprotonation, etc.). Despite this limitation, manual fits are capable of revealing insight into the extent of histidine participation, which is the major focus of this work. The simulated fits are presented in Figure 3(A,B). Data were analyzed using models that included one, two, or three, ionizable histidines (see Materials and Methods section for parameters). A simple assumption was made that each protonation equilibrium behaves independently. Although neither a single or double histidine model could adequately fit the VHH#10 data, a double histidine model could represent the VHH#24 data rather well; however, a significant pKa drop of at least four units was required for each histidine. On the other hand, a three histidine model matched the observed data using moderate ΔpKa values (within a range of 0.5–3). Although care must be taken not to overfit the data, by including a larger than necessary number of pKa parameters, the binding data for both variants, VHH#10 and VHH#24, appear best fit with at least three contributing histidine residues. Simple analysis of dlog Kb/dpH at low pH also suggests the slope reaches a value of 3.0, which is consistent with the simulated fits and that at least three histidines are present in both interfaces. Finally, an estimate of the number of protons released on forming the VHH/RNase A complex, due to a drop in histidine pKa values, may be examined by determining the slope of a linear fit to a plot of the observed enthalpy versus buffer ionization enthalpy (Fig. 1, Supporting Information). The VHH#10-RNase A binding event at pH 5.0 (10°C) possessed 1.78 ± 0.03 protons released on complex formation. Consequently, this analysis also indicates that multiple histidine groups must be responsible for the observed pH-dependent binding.
Structural analysis of the pH-dependent variant VHH#24
To understand the structural consequences of introducing a large number of histidines within the VHH interface, the X-ray structure of the VHH#24/RNase A complex was determined. Despite the introduction of five interface histidines (CDR1 residues: A28H and Y29H and CDR3 residues: Y101H, E102H, and R106H), no major structural changes are observed when compared to the original wt-VHH/RNase A complex beyond slight adjustments in side chain conformations [see Fig. 3(C,D)]. All five histidine substitutions are positioned in the interface periphery. Overall, VHH#24, buries slightly less surface area (540 Å2) versus VHH-wt (600 Å2), which is almost entirely due to loss of contact area from the R106H substitution.
With five potential histidines that may contribute to the observed pH sensitivity, straight-forward structural interpretations of VHH#24's pH-sensitive binding are not possible; however, the VHH#24/RNase A structure provides a model to make several predictions. In general, perturbations of an ionizable group's pKa value on protein–protein complex formation originate from changes in the residue's microenvironment. In view of this, the structural data reveals two of the five histidine residues bury significant side chain surface area in the VHH#24 complex, including Y29H and Y101H, which bury 82 and 61 Å2 of solvent accessible surface area (ΔASA) on forming the VHH#24/RNase A complex, respectively. As a comparison, the corresponding wild-type residue side chains bury 65 and 64 Å2, respectively. Both histidine side chains make hydrogen bonds with RNase A residues. Nɛ of Y29H forms a hydrogen bond with the side chain of Y77 from RNase A [Fig. 3(C)], while Nδ of Y101H forms a hydrogen bond with the main chain of T70 from RNase A [Fig. 3(D)]. Perhaps most relevant, both Y29H and Y101H possess a side chain nitrogen(s) that buries significant surface area. Y29H buries 13 and 29 Å2 for Nδ and Nɛ atoms, respectively, while Y101H's Nδ buries 22 Å2.
In addition to surface area burial, neighboring residues may also influence pKa values. For instance, both Y29H and Y101H substitutions possess adjacent histidine groups, A28H and E102H, respectively [see Fig. 3(C,D)] that may play a contributing role. The “dual-histidines” are observed to be in close proximity within the VHH#24/RNase A complex, which may result in reduced pKa values. Of course, the dual histidine environment would need to be different between the free and bound states for them to contribute toward pH-dependent binding. Such paired histidine groupings are observed in several of the sequenced variants [Fig. 2(A)], which may suggest they are involved in the pH-dependent binding mechanism. The Y101H/E102H “stacking” observed in the VHH#24 complex [Fig. 3(D)] is particularly interesting. Although E102H does not exhibit a change in ASA based on a simple rigid-body model of the complex, it is likely to be more exposed in the unbound state and therefore buries intramolecular surface in the bound state, which could influence (drop) E102H's pKa value. The VHH#24/RNase A structure reveals both Y29H and Y101H groups are also in the vicinity of residues likely to be positively charged. K61 of RNase A is within close proximity to Y29H [Fig. 3(C)] and R104 of VHH is near Y101H. The positively charged environment may also influence (reduce) pKa values of the histidines in the bound state. The remaining histidine, R106H, does not appear to make appreciable contact with RNase A and is not predicted to play a role in VHH#24's pH-dependent binding [Fig. 3(D)]. Overall, the crystal structure reveals at least two histidines clearly experience new environments on formation of the protein–protein complex with likely contributions from at least another histidine (e.g., E102H). This evaluation is in agreement with the pH-dependence data, which suggested contributions from as few as two or three histidines for VHH#24. Of course, these surface area calculations and interpretations should be used with caution as only knowledge of the complex state structure was used, while both free and complex structures contribute to the observed changes in pKa values. Ultimately, additional structure and thermodynamic investigations are necessary to dissect the individual roles each histidine plays in the observed pH-sensitive binding.
Discussion
Although previous efforts to engineer pH sensitive protein–protein interactions have resulted in pH-dependent binding, these efforts have generally resulted in modest sensitivity, often at the expense of high-affinity binding at the permissive pH. Typically, this limited pH sensitivity originates from the introduction of only a single ionizable group which is not capable of introducing a large enough change in ΔG to drive a pH switch. A pH switch, which must undergo a significant change in binding affinity over a short range in pH, can only be generated through the insertion of multiple ionizable groups that undergo a change in pKa on binding. In fact, the thermodynamic principles which govern a pH-binding switch are analogous to those which drive acid-induced protein unfolding, where multiple ionizable groups contribute to the loss of protein's stability. Consequently, the generation of a pH switch (whether binding or folding) requires an intimate balance of pH-dependent and pH-independent contributions. Here, a combinatorial histidine/wild-type library was developed to reveal antibodies which possessed this balance of pH-dependent and pH-independent terms. This combinatorial approach produced numerous, unique antibodies with highly pH-sensitive binding through the introduction of multiple interface histidine residues. Two main features likely contributed to the success of the method. First, the combinatorial library is robust; every possible combination of histidine and wild-type residue is sampled, thus providing an opportunity to coselect engineered variants that possess both high-affinity binding and pH sensitivity. Second, the approach exploits the frequently observed plasticity of protein interfaces,31,28 a quality which is clearly apparent from the results, as approximately half the interface could tolerate histidine incorporation without loss of function.
A significant advantage of the combinatorial approach is that detailed structural knowledge of the antibody (or other protein of interest) is not required, only knowledge of the interface residues. For instance, the only structural information that was used in development of the anti-RNase A VHH histidine library was knowledge that CDR1 and CDR3 are involved in binding the RNase A target. Histidines were broadly sampled throughout CDR1 and CDR3, including both surface exposed and scaffolding residues. The approach could be easily modified to explore additional interface or scaffolding residues or leave specific residues unchanged. In fact, the method is completely scalable up to larger interfaces (e.g., conventional antibodies with interfaces possessing both heavy (VH) and light (VL) variable domains) where as many as 30–35 residues could be sampled by combining modern phage29 or mRNA32 display technologies with trinucleotide (trimer) phosphoramidite-based degenerate oligonucleotides.1
Knowledge of the anti-RNase A VHH/RNase A complex structure9,10 allows a retrospective analysis into histidine “host” sites. Overall, an impressive amount of histidine incorporation was observed across the VHH interface; however, a distinct, centrally located group of 11 positions did not accommodate histidines [Fig. 2(B)]. Nine of these residues (Y31, M34, G35, G99, G100, L103, T107, Y108, and G109) are predicted to be significantly buried in the absence of RNase A and contribute only 40 Å2 of total surface area burial in the wild-type VHH/RNase A complex. The simplest explanation is that they cannot structurally tolerate a histidine substitution, due to their involvement in specific packing interactions which would be disrupted on histidine insertion. Similarly, the three glycine residues serve as “flexibility” points at the N- and C-terminal portions of the CDR3 loop, where insertion of a bulky histidine side chain would have detrimental effects on the conformation of the CDR3 loop for productive RNase A binding. Of the remaining two residues, D105 participates in an internal salt bridge with R45 that would be disrupted on histidine insertion, and T30 is likely located too far away from the RNase A interface (at least 8 Å).
In general, residues within the interface interior are poor histidine hosts, while the periphery residues appear well suited for histidine substitution. In particular, three histidine “hot-spots” [Fig. 2(B,C)] were identified by sequence analysis. VHH#10 possessed all three hot-spot histidines and displayed near wild-type affinity at pH 7.4 and an extremely sensitive pH binding profile [Fig. 3(A)]. Such sequence analysis could be particularly helpful in isolating pH sensitive clones. In general, histidine substitutions resulted in only minor structural changes in the VHH/RNase A complex as observed with the five histidine VHH#24/RNase A complex. The structure reveals that histidines appear to serve as good “place keepers” for many types of residues, in particular tyrosine residues, which have comparable side chain size. This preference for tyrosine residues is significant for pH-dependent antibody engineering, as examinations of naturally derived antibodies reveal tyrosine residues play a prominent role in antibody binding sites by participating in ∼25% of the antigen contacts.33 Consequently, tyrosine/histidine compatibility could easily be exploited for both screening- and nonscreening-based strategies to introduce new pH sensitivity.
Although a wild-type to histidine substitution inserts a protonation site, it is not necessarily sufficient to introduce pH dependence as the histidine must exhibit a pKa change on forming the protein–protein complex. There is no single structural “recipe” for such a change in proton affinity. A common finding is that the residue will experience a change in ΔASA between the bound and unbound states. In fact, the VHH interface sites that tolerate histidine substitutions are all located in regions where significant solvent surface area would be available in the unbound state. Many of these residues would be predicted to experience significant change on binding RNase A, including surface area burial, loss of conformational freedom, as well as new electrostatic environments. The structure of the VHH#24/RNase A complex reveals that three of the five histidine residues Y29H, Y101H, and E102H are predicted to experience a change in environment based on surface burial; however, their specific contribution to pH sensitivity cannot be determined from structure alone. In addition, VHH#24's A28H/Y29H and Y101H/E102H paired interactions may also play a significant role, as histidine “pairs” are observed across many of the pH sensitive clones. One possibility is that a histidine pair may contribute to pH sensitivity through nonindependent interactions, where on forming the complex their close proximity would reduce the apparent pKa values due to negative cooperativity. This observation is particularly interesting in light of the close structural proximity of histidines believed to be involved in the pH dependence of the hPRL hormone/receptor binding.8
Up to now, combinatorial approaches, including shotgun alanine scans of protein interfaces26,28,34 as well as methionine scanning of protein interior residues,35 have been quite effective in revealing the malleable nature of proteins. Here, a combinatorial histidine scan exploits this plasticity of VHH antibodies and takes new directions in dual function antibodies.36,37 Through coselection of both high-affinity antigen binding and binding sensitive protonation sites, this approach was extremely effective in generating a number of highly pH-dependent single-chain VHH antibodies. Most importantly, these pH sensitive switches were made possible by the incorporation of many histidines with perturbed pKa values. It is important to note that first principles alone could not likely be used to identify several suitable histidines insertion sites. Therefore, the protein complexes described here will help serve as useful models to better understand the role of electrostatics in protein–protein recognition. This general approach should perform well with other protein-affinity reagents. In fact, it is very likely that the number of histidine protonation sites could be further increased with larger interfaces, such as those found in conventional IgG, Fab, and scFv antibody fragments, which posses two variable domains (i.e., VH and VL). As antibodies play a vital role in numerous applications, including diagnostics, therapeutics, and protein reagents, the ability to rapidly introduce pH-dependent control of binding holds exciting promise. Perhaps, even more interesting will be the potential of new applications where the binding of complex mixtures of antibodies, with differing pH profiles, can be controlled through slight modifications in pH.
Materials and Methods
Simulations
The pH dependence of the observed binding constant, Kobs, for a proton linked, protein–protein binding event was simulated using the following general relationship:
where Kint is the protein–protein binding constant for the unprotonated species,  and
and 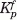 are the protonation binding constants (Kp = 10pKa) for the ionizable group(s) in the complex and free states, respectively, and n is the number of ionizable histidine groups. Overall, this relationship assumes independent, noninteracting, protonation events.
are the protonation binding constants (Kp = 10pKa) for the ionizable group(s) in the complex and free states, respectively, and n is the number of ionizable histidine groups. Overall, this relationship assumes independent, noninteracting, protonation events.
Simulated fits to the VHH#10 and VHH#24 include the following parameters. VHH#10 two histidine model:  ,
, 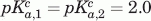 ,
, 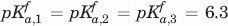 ,
, 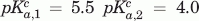 ,
, 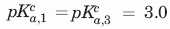 , VHH#24 two-histidine model:
, VHH#24 two-histidine model: 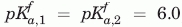 , , VHH#24 three histidine model:
, , VHH#24 three histidine model: 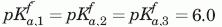 ,
, 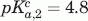 , .
, .
Histidine scanning library generation
Starting with a phagemid vector containing the anti-RNase A VHH antibody fragment (cAb-RN05) upstream of the M13 gene 3,10 Kunkel mutagenesis38 was used to create a binding incompetent VHH vector (phgmd-VHH-CDR13-stop) through the introduction of repeating stop codons throughout the CDR1 and CDR3 loops. Degenerate oligonucleotides (Sigma Genosys) were designed to encode both histidine and the original wild-type residue at each position within the CDR1 and CDR3 (CDR1: 5′-CGT CTG AGC TGC GCA GCA AGC SRT YAT SMT YAT MMT YAT MWT YAT MWK SRT TGG TTC CGT CAA GCA CCA GG-3′ and CDR3: 5′-CC TAC TAC TGC GCA GCA SRT SRT YAT SAW CWT CRT SAT CRT MMT YAT SRT CAW TGG GGT CAA GGC ACC C-3′, where S = G/C, R = A/G, Y = C/T, K = G/T, M = A/C, W = A/T). For many residue positions, the degenerate codon library involved toggling a single pair of nucleotides, while some positions required varying more than one codon position to allow sampling of both wild-type and histidine side chains. Consequently, the corresponding residue position can sample nonwild-type or histidine side chains; however, their inclusion in the library will not affect selection. Kunkel mutagenesis38 was used to generate the M13 phage library using the two histidine scanning oligonucleotides and single stranded deurycilated (dU-ssDNA) phgmd-VHH-CDR13-stop template using methods described previously.29 The histidine scanning library was verified through DNA sequencing.
Selection of pH sensitive VHH clones
Preparation of RNase A target
Bovine RNase A (Sigma-Aldrich) was biotinylated using EZ-Link Sulfo-NHS-SS-Biotin (Pierce, Rockford) according to the manufacturer's protocol (Promega, Madison WI).
Phage panning and selection
The initial round of phage selection included incubation of the phage library (1012 phage particles) with 2-μM biotinylated RNase A in TBS buffer (50 mM tris(hydroxymethyl) aminomethane (TRIS), 150 mM NaCl) pH 7.4. Phage that displayed binding competent VHH variants were then captured using magnetic streptavidin-coated beads (Promega, Madison WI) using a Kingfisher (Thermo Scientific) magnetic bead handler. Four wash steps were performed with TBS-Tween-20 buffer pH 7.4 to help eliminate nonspecific interactions and weakly bound VHH variants. pH-Sensitive VHH variants were selected by incubating the phage-bound beads in a low pH buffer (either pH 4.0 or 5.5). The eluted phage, which included the pH-sensitive VHH variants, were amplified following established protocols.29 Precipitated phage particles were resuspended in TBS, pH 7.4 for the next round of selection. In effort to increase selection stringency for both high affinity pH 7.4 binding and high pH sensitivity, the concentration of the RNase A target decreased from 2 μM down to 20 nM, while the pH elution started at pH 4.0 and increased to pH 5.5 (50 mM sodium acetate/HCl, 150 mM NaCl) over the four rounds of selection.
VHH expression and purification
To express the pH-sensitive VHH variants independently without the Gene-3 fusion, the VHH gene of interest was subcloned out of the phagemid vector and into a pET-21a expression plasmid (Novagen) using the primers (5′-GAGATATACATATGCATCATCATCATCATCATGAAAACCTGTACTTCCAGGGATCCCAAGTACAACTGGTAG-3′) and (5′-TAGACTCGAGGAATTCCTATTAGCTGCTTACGGTTACTTGG-3′), which introduce NdeI and EcoRI restriction sites along with an N-terminal hexa-His-TEV tag. Ligated clones were verified by DNA sequencing.
A 5-mL (LB/ampicillin) culture of E. coli BL21(DE3) containing pET-21a-HisTevVHH was grown overnight at 37°C. This culture was used to start a 50-mL subculture which, upon reaching mid-log phase, was used to inoculate a 1 L (LB/ampicillin) culture. VHH expression was induced with 1.0 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at optical density (OD)600 ∼ 0.5–0.8 and incubated at 20°C overnight. The cells were isolated by centrifugation (17,700g; 15 min) and subsequently frozen (−20°C) overnight. The frozen pellet was then resuspended in 10 mM of TRIS pH 8.0 and sonicated (21 Watts) for three cycles of 2 min on/2 min off with a Model 60 Sonic Dismembrator (Fischer Scientific). As the VHH clones were expressed as insoluble inclusion bodies, the lysed cells were centrifuged (22,700g; 20 min) to isolate the inclusion bodies/cell debris fraction. The pellet was washed with Tris-DE buffer [2% deoxycholic acid, 50 mM TRIS, 5 mM ethylenediaminetetraacetic acid (EDTA) pH 8.0], ddH2O and twice with 10 mM TRIS pH 8.0. The cells were centrifuged (22,700g; 20 min) after each wash step and the resulting supernatant discarded. The final washed inclusion body pellet was resuspended in unfolding buffer (6M guanidine HCl, 50 mM NaPi/HCl, 300 mM NaCl, 2.0-mM reduced glutathione (GSH), 0.2-mM oxidized glutathione (GSSG), pH 7.4) to solubilize the inclusion body and centrifuged (25,900g; 20 min) to remove any insoluble material. Next, the unfolded VHH was refolded by dropwise addition to refolding buffer (20 mM TRIS-HCl, 2.0 mM GSH, 0.2 mM GSSG, pH 7.4) at 4°C with moderate stirring. The final refolded solution corresponded to a 10× dilution of the unfolded protein. Refolded protein was centrifuged (25,900g; 15 min) to remove insoluble precipitate. The refolded VHH protein was subsequently purified as described previously.39
Isothermal titration calorimetry
All experiments were run with a VP-ITC titration calorimeter (MicroCal). Buffer matching was ensured by dialyzing the VHH variant and bovine RNase A (Sigma-Aldrich) overnight in 4 L of buffer at 4°C. Buffer conditions depended on the pH of the experimental run. All buffers contained 150 mM NaCl and 20 mM buffer. Buffers and their pH ranges included: phosphate (pH 6.0–7.4), TRIS (pH 8.0–9.0), acetate (pH 3.0–5.5), and piperazine (pH 4.3–6.3). Protein concentrations were determined by UV absorbance using a UV–visible spectrometer (Hewlett Packard). Extinction coefficients (280 nm) were 9,440 M−1 cm−1 (RNase A), 22,091 M−1 cm−1 (VHH#24), and 21,555 M−1 cm−1 (VHH#10). Extinction coefficient values were determined using methods described by Pace et al.40 For high-pH/high-affinity experiments, titrations were performed with VHH as the titrant at concentrations over a range of 40–200 μM. RNase A concentrations were typically one-tenth the respective concentration of VHH. For low-pH/low-affinity experiments, the methods of Turnbull and Daranas were followed.41 Generally, RNase A was used as the titrant at a concentration of 1 mM, and VHH concentration was 100-fold lower. To find conditions that gave sufficient heats of binding, a range of temperatures and buffers were used. Experiments were run at 25°C from pH 6 to 8 and 10°C from pH 5.5 to 4.5 with VHH as the titrant. With RNase A as the titrant experiments were run at 25°C from pH 4.7 to 4.5. Dilution experiments (titrant into buffer) were performed to account for heats of dilution. Data were analyzed using Origin with the Microcal ITC add-on available from the manufacturer.
Circular dichroism
All experiments were run with an Aviv Instruments circular dichroism spectrometer model 215. Wavelength scans were carried out at 25°C from 240 to 200 nm in phosphate buffer saline (PBS) (10 mM sodium phosphate, 150 mM sodium chloride, pH 7.4, pH 3.0, or pH 2.0) and acetate (10 mM sodium acetate, 150 mM sodium chloride pH 4.0 or pH 5.5). A quartz cuvette was used with a 1-mm path length. Temperature denaturation experiments were carried out at 207 nm from 25 to 85°C at 1° increments. The Tm values were then determined with Origin Labs.
Structure determination of the anti-RNase A VHH#24/RNase A complex
About 20 mg mL−1 of anti-RNase A VHH#24 TBS (10 mM Tris, 300 mM NaCl, pH 8.0) was mixed equally with 25 mg mL−1 bovine RNase A (Sigma-Aldrich). The complex was run through a 10/30GL Superdex-75 size exclusion column (GE Healthcare), and the peak corresponding to the VHH/RNase A complex was collected. Crystallization was performed by the hanging drop method by mixing 2 μL of the 20 mg mL−1 VHH-RNase A solution with 2 μL of 0.2M lithium sulfate monohydrate, 0.1M bis-tris pH 5.5, 25% w/v polyethylene glycol 3,350 (Hampton Research, Aliso Viejo, CA).
X-Ray data were collected at Southeast Regional Collaborative Access Team (SER-CAT), 22-BM beamline at the Advanced Photon Source, Argonne National Laboratories. Glycerol (20%) was used as a cryoprotectant. Data were indexed, merged, and scaled using HKL-2000.42 The CCP4 suite was used to generate the initial electron density, as well as perform molecular replacement (phaser).43 The initial search model for molecular replacement was the anti-RNase A VHH/RNase A complex, PDB ID: 2P49. A single VHH#24/RNase A complex was found within the asymmetric unit. Data were refined using CCP4's Refmac. Model building was performed using the program coot.44 The three complementary determining regions were initially removed from the structure and subsequently rebuild. Residues 16-22 of RNase A are not included in the model due to lack of electron density. These residues are also missing in other anti-RNase A VHH/RNase A crystal structures.9,10 Crystallographic statistics are presented in Table 1. The structure is assigned PDB ID code 3QSK. Structure models are generated with Pymol.45
Table I.
Crystallography statistics for the VHH#24/RNase A complex
| Data collection | |
| Space group | P1211 |
| a, b, c | 40.79, 54.68, 48.79 |
| α, β, γ | 90, 109.24, 90 |
| Resolution range | 20–1.75 Å |
| Number of reflections | 176379 |
| I/σ | 21.46 |
| Rmerge | 0.047 |
| Completeness (%) | 99.9 |
| Redundancy | 3.7 |
| Refinement | |
| Rwork/Rfree | 19.08/24.47 |
| Average residues/chain | |
| VHH | 123 |
| RNase A | 129 |
| Water | 91 |
| RMSD | |
| Bond lengths (Å) | 0.0147 |
| Bond angles (°) | 1.5242 |
| Chiral volume | 0.1083 |
| Ramachandran plot statistics | |
| Preferred (%) | 229 (97.86) |
| Additional allowed (%) | 3 (1.28) |
| Outliers (%) | 2 (0.85) |
Acknowledgments
The authors thank Dr. R. Walter for assistance in X-ray data collection and Dr. S. Koide for providing the initial anti-RNase A VHH phagemid vector. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Glossary
Abbreviations:
- ITC
isothermal titration calorimetry
- VHH-wt
wild-type anti-RNase A VHH
- VHH-10
pH clone #10
- VHH-24
pH clone #24
- ASA
solvent accessible surface area
- ΔASA
Change in solvent accessible surface area
Supplementary material
References
- 1.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol. 2007;373:924–940. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Stratton MM, Loh SN. Converting a protein into a switch for biosensing and functional regulation. Protein Sci. 2011;20:19–29. doi: 10.1002/pro.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith MT, Mackenzie DW, Meiering EM. Dissecting the molecular determinants of ligand-binding-induced macromolecular switching using thermodynamic cycles. Protein Eng. 2011;24:213–217. doi: 10.1093/protein/gzq099. [DOI] [PubMed] [Google Scholar]
- 4.Perutz MF, Kilmartin JV, Nishikura K, Fogg JH, Butler PJG, Rollema HS. Identification of residues contributing to the Bohr effect of human-hemoglobin. J Mol Biol. 1980;138:649–670. doi: 10.1016/s0022-2836(80)80022-2. [DOI] [PubMed] [Google Scholar]
- 5.Ascenzi P, Aducci P, Amiconi G, Ballio A, Guaragna A, Menegatti E, Schnebli HP, Bolognesi M. Binding of the recombinant proteinase inhibitor eglin c from leech Hirudo medicinalis to serine (pro)enzymes: a comparative thermodynamic study. J Mol Recognition. 1991;4:113–119. doi: 10.1002/jmr.300040402. [DOI] [PubMed] [Google Scholar]
- 6.Vincent JP, Lazdunski M. The interaction between alpha-chymotrypsin and pancreatic trypsin inhibitor (Kunitz inhibitor). Kinetic and thermodynamic properties. Eur J Biochem. 1973;38:365–372. doi: 10.1111/j.1432-1033.1973.tb03069.x. [DOI] [PubMed] [Google Scholar]
- 7.Keeler C, Jablonski EM, Albert YB, Taylor BD, Myszka DG, Clevenger CV, Hodsdon ME. The kinetics of binding human prolactin, but not growth hormone, to the prolactin receptor vary over a physiologic pH range. Biochemistry. 2007;46:2398–2410. doi: 10.1021/bi061958v. [DOI] [PubMed] [Google Scholar]
- 8.Kulkarni MV, Tettamanzi MC, Murphy JW, Keeler C, Myszka DG, Chayen NE, Lolis EJ, Hodsdon ME. Two independent histidines, one in human prolactin and one in its receptor, are critical for pH-dependent receptor recognition and activation. J Biol Chem. 2010;285:38524–38533. doi: 10.1074/jbc.M110.172072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decanniere K, Desmyter A, Lauwereys M, Ghahroudi MA, Muyldermans S, Wyns L. A single-domain antibody fragment in complex with RNase A: non-canonical loop structures and nanomolar affinity using two CDR loops. Structure. 1999;7:361–370. doi: 10.1016/s0969-2126(99)80049-5. [DOI] [PubMed] [Google Scholar]
- 10.Koide A, Tereshko V, Uysal S, Margalef K, Kossiakoff AA, Koide S. Exploring the capacity of minimalist protein interfaces: Interface energetics and affinity maturation to picomolar K(D) of a single-domain antibody with a flat paratope. J Mol Biol. 2007;373:941–953. doi: 10.1016/j.jmb.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ambroggio XI, Kuhlman B. Design of protein conformational switches. Curr Opin Struct Biol. 2006;16:525–530. doi: 10.1016/j.sbi.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 12.Buskirk AR, Liu DR. Creating small-molecule-dependent switches to modulate biological functions. Chem Biol. 2005;12:151–161. doi: 10.1016/j.chembiol.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Tawfik DS, Chap R, Eshhar Z, Green BS. Ph on-off switching of antibody hapten binding by site-specific chemical modification of tyrosine. Protein Eng. 1994;7:431–434. doi: 10.1093/protein/7.3.431. [DOI] [PubMed] [Google Scholar]
- 14.Morag E, Bayer EA, Wilchek M. Reversibility of biotin-binding by selective modification of tyrosine in avidin. Biochem J. 1996;316:193–199. doi: 10.1042/bj3160193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marttila AT, Hytonen VP, Laitinen OH, Bayer EA, Wilchek M, Kulomaa MS. Mutation of the important Tyr–33 residue of chicken avidfunctional and structural consequences. Biochem J. 2003;369:249–254. doi: 10.1042/BJ20020886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarkar CA, Lowenhaupt K, Horan T, Boone TC, Tidor B, Lauffenburger DA. Rational cytokine design for increased lifetime and enhanced potency using pH-activated “histidine switching.”. Nat Biotechnol. 2002;20:908–913. doi: 10.1038/nbt725. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe H, Matsumaru H, Ooishi A, Feng YW, Odahara T, Suto K, Honda S. Optimizing pH response of affinity between protein G and IgG Fc how electrostatic modulations affect protein-protein interactions. J Biol Chem. 2009;284:12373–12383. doi: 10.1074/jbc.M809236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linder M, Nevanen T, Teeri TT. Design of a pH-dependent cellulose-binding domain. Febs Lett. 1999;447:13–16. doi: 10.1016/s0014-5793(99)00253-7. [DOI] [PubMed] [Google Scholar]
- 19.Nordlund HR, Hytonen VP, Laitinen OH, Uotila STH, Niskanen EA, Savolainen J, Porkka E, Kulomaa MS. Introduction of histidine residues into avidin subunit interfaces allows pH-dependent regulation of quaternary structure and biotin binding. Febs Lett. 2003;555:449–454. doi: 10.1016/s0014-5793(03)01302-4. [DOI] [PubMed] [Google Scholar]
- 20.Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, Wakabayashi T, Hayasaka A, Kadono S, Miyazaki T, Haraya K, Sekimori Y, Kojima T, Nabuchi Y, Aso Y, Kawabe Y, Hattori K. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol. 2010;28:1203–1207. doi: 10.1038/nbt.1691. [DOI] [PubMed] [Google Scholar]
- 21.Isom DG, Cannon BR, Castaneda CA, Robinson A, Bertrand GME. High tolerance for ionizable residues in the hydrophobic interior of proteins. Proc Natl Acad Sci USA. 2008;105:17784–17788. doi: 10.1073/pnas.0805113105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno BE. Large shifts in pK(a) values of lysine residues buried inside a protein. Proc Natl Acad Sci USA. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isom DG, Castaneda CA, Velu PD, Garcia-Moreno B. Charges in the hydrophobic interior of proteins. Proc Natl Acad Sci USA. 2010;107:16096–16100. doi: 10.1073/pnas.1004213107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edgcomb SP, Murphy KP. Variability in the pKa of histidine side-chains correlates with burial within proteins. Proteins. 2002;49:1–6. doi: 10.1002/prot.10177. [DOI] [PubMed] [Google Scholar]
- 25.Cunningham BC, Wells JA. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- 26.Weiss GA, Watanabe CK, Zhong A, Goddard A, Sidhu SS. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc Natl Acad Sci USA. 2000;97:8950–8954. doi: 10.1073/pnas.160252097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerstner RB, Carter P, Lowman HB. Sequence plasticity in the antigen-binding site of a therapeutic anti-HER2 antibody. J Mol Biol. 2002;321:851–862. doi: 10.1016/s0022-2836(02)00677-0. [DOI] [PubMed] [Google Scholar]
- 28.Kouadio JL, Horn JR, Pal G, Kossiakoff AA. Shotgun alanine scanning shows that growth hormone can bind productively to its receptor through a drastically minimized interface. J Biol Chem. 2005;280:25524–25532. doi: 10.1074/jbc.M502167200. [DOI] [PubMed] [Google Scholar]
- 29.Sidhu SS, Weiss GA. Constructing phage display libraries by oligonucleotide-directed mutagenesis. In: Clackson T, Lowman HB, editors. Phage display: a practical approach. Oxford: Oxford University Press; 2004. pp. 27–41. [Google Scholar]
- 30.Baker BM, Murphy KP. Evaluation of linked protonation effects in protein binding reactions using isothermal titration calorimetry. Biophys J. 1996;71:2049–2055. doi: 10.1016/S0006-3495(96)79403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atwell S, Ultsch M, De Vos AM, Wells JA. Structural plasticity in a remodeled protein-protein interface. Science. 1997;278:1125–1128. doi: 10.1126/science.278.5340.1125. [DOI] [PubMed] [Google Scholar]
- 32.Lipovsek D, Pluckthun A. In-vitro protein evolution by ribosome display and mRNA display. J Immunol Methods. 2004;290:51–67. doi: 10.1016/j.jim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 33.Mian IS, Bradwell AR, Olson AJ. Structure, function and properties of antibody binding sites. J Mol Biol. 1991;217:133–151. doi: 10.1016/0022-2836(91)90617-f. [DOI] [PubMed] [Google Scholar]
- 34.Pal G, Kossiakoff AA, Sidhu SS. The functional binding epitope of a high affinity variant of human growth hormone mapped by shotgun alanine-scanning mutagenesis: insights into the mechanisms responsible for improved affinity. J Mol Biol. 2003;332:195–204. doi: 10.1016/s0022-2836(03)00898-2. [DOI] [PubMed] [Google Scholar]
- 35.Tereshko V, Uysal S, Koide A, Margalef K, Koide S, Kossiakoff AA. Toward chaperone-assisted crystallography: protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold. Protein Sci. 2008;17:1175–1187. doi: 10.1110/ps.034892.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, Fuh G. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science. 2009;323:1610–1614. doi: 10.1126/science.1165480. [DOI] [PubMed] [Google Scholar]
- 37.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 38.Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 39.Sonneson GJ, Horn JR. Hapten-induced dimerization of a single-domain VHH camelid antibody. Biochemistry. 2009;48:6693–6695. doi: 10.1021/bi900862r. [DOI] [PubMed] [Google Scholar]
- 40.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turnbull WB, Daranas AH. On the value of c: can low affinity systems be studied by isothermal titration calorimetry? J Am Chem Soc. 2003;125:14859–14866. doi: 10.1021/ja036166s. [DOI] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Collaborative Computational Project N. The CCP4 suite: Programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.