

Abstract
A novel saccharide host containing four boronic acid recognition units on a single DNA duplex terminus was constructed. This construct allowed boronic acid sugar recognition in the context of double stranded DNA to be established while highlighting the benefits of multivalency. Following the solid-phase synthesis of a bis-boronic acid tag, two end-functionalized oligonucleotides with complementary sequences were functionalized through amide ligation. By annealing the boronic acid-DNA conjugates, a tetra-boronic acid DNA duplex was assembled. The saccharide binding ability of this tetra-boronic acid host was revealed through cellulose paper chromatography in the absence and presence of various saccharides. While no appreciable saccharide binding was seen in the case of a bis-boronic DNA conjugate, the increased migration of the tetra-boronic acid host relative to the control sequences in the presence of selected monosaccharides underscored the importance of multivalent effects. We thus identified a requirement for multiple recognition sites in these conjugate systems and expect the results to facilitate future efforts toward applying synthetic recognition systems to the realm of macromolecules.
INTRODUCTION
The selective binding of saccharides, particularly oligosaccharides, in a physiological environment remains an unsolved problem, despite the biological importance and medicinal relevance of these compounds (1). In small molecule systems, the binding of saccharides has been most commonly achieved through the use of boronic acids, which form stable cyclic boronate esters with saccharide diols (2, 3). However, the efficacy of boronic acid recognition units has rarely been investigated in the context of the macromolecules that would be present in a biological system (4, 5). One specific concern is whether multivalent interactions (i.e. the use of multiple boronic acid subunits) will be required to achieve efficient binding in such an environment. In order to investigate the viability of boronic acid-based saccharide recognition in the context of DNA, we designed a system that allows for the evaluation of the effect of zero, two, or four boronic acid units located at one terminus of a short (20 base pair) DNA duplex. The oligonucleotide derivatization scheme was based on a standard peptide coupling reaction between a bis-boronic acid-functionalized dipeptide and two amine-functionalized single strand (ss) oligonucleotides with complementary base sequences (Scheme 1). Three different constructs were prepared by mixing the boronic acid-ssDNA conjugates with the complementary unfunctionalized and functionalized sequences. Evaluation of the saccharide binding ability of these constructs was performed through cellulose paper chromatography, a technique that requires minute amounts of material (< 1 pmol) and boasts a long history in DNA characterization (6–10). The mobility of these constructs in the presence and absence of various small saccharides was investigated. In these experiments, the tetra-boronic acid DNA duplex displayed significant interactions with saccharides while a bis-boronic acid DNA duplex does not appear to bind saccharides appreciably. We thus conclude that boronic acid-sugar recognition is viable in the context of DNA provided a critical number of binding subunits is used, in this case four boronic acid moieties. The observed importance of multivalency may prove critical in future embodiments of hybrid small molecule-biomacromolecule recognition systems.
Scheme 1.

Construction of host.
EXPERIMENTAL PROCEDURES
Materials and instrumentation
All chemicals were purchased from Aldrich Chemical Co., Acros Organics or EM and used without further purification unless otherwise noted. All amino acid derivatives and coupling reagents were purchased from Novabiochem. All solvents were of reagent grade quality and purchased commercially. Dry CH2Cl2 (DCM) refers to solvent freshly distilled over CaH2. Dry DMF and methanol were obtained by passing high grade commercial material through two columns of molecular sieves as part of an SG Water USA solvent delivery system. All water used was filtered through a Millipure filtration system. TBE buffer refers to a solution of 89 mM Tris, 89 mM boric acid, and 2 mM EDTA at pH 8.3. Oligonucleotides were purchased from Integrated DNA Technologies (IDT) and gel-purified prior to use. Other biological reagents were purchased or prepared as indicated. NMR spectra were recorded on Varian INOVA 600 or Varian Mercury 400 instruments. Small molecule UV-Vis spectra were recorded on a Beckman DU 640 spectrophotometer. Oligonucleotide UV-Vis spectra were recorded on a ThermoScientific NanoDrop 1000 spectrophotometer. HR electrospray ionization (ESI) mass spectra were recorded on a Varian 9.4T QFT-ESI ICR system. Gel and paper imaging was performed using a GE Storm 840 Imager and quantified using the ImageQuant software program. HPLC purification was performed on Beckman Gold system equipped with a Waters Xterra MS C18 column (2.5 μm) kept at 60°C. Compounds were eluted using a 0.1 M triethylammonium acetate (TEAA) - acetonitrile solvent gradient.
N6,N-Bis((2-dihydroxyborono)benzyl)-lysinoglycine
Fluorenylmethyloxycarbonyl (Fmoc)-Gly-Wang resin (4.00 g, 2.44 mmol) was swollen in dry DCM (40 mL) by shaking over the course of one hour. Fmoc Deprotection: After filtration to remove the solvent, a solution of 20% piperidine in DMF was added to the resin, and the mixture was shaken for 20 minutes. The solvent was removed, and the piperidine treatment repeated one more time. Wash: The resin was washed three times each with 40 mL of the following solvents in succession: DMF, DCM, methanol, DMF, and DCM. Coupling: To the washed resin was added a solution of Fmoc-Lys(Fmoc)-OH (8.65 g, 14.64 mmol), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 5.55 g, 14.64 mmol), 1-hydroxybenzotriazole (HOBt, 2.24 g, 14.64 mmol) and Hünig's base (4.25 mL, 24.4 mmol) in 1:1 DMF:DCM (40 mL), and the mixture was allowed to shake at room temperature overnight. The resin was then taken through the steps of washing, Fmoc deprotection, and a second washing as described above. The resin was then dried in vacuo for three hours. Reductive Amination: A solution of formylphenyl boronic acid (3.66 g, 24.4 mmol) and anhydrous methanol (4 mL) in freshly distilled DCM (40 mL) was added to the dry resin. After shaking overnight at room temperature, 10.3 g of sodium triacetoxyborohydride was added slowly. After addition of acetic acid (0.8 mL), the mixture was shaken for 10 hours. The resin was washed as above, and the reductive amination procedure was repeated one additional time. The resin was washed and dried in vacuo for four hours. A solution of 2.5% water and 2.5% triethylsilane in TFA was added to the resin, and the mixture was shaken for three hours. The supernatant was drained and collected, and the resin rinsed two times with TFA (40 mL). The TFA fractions were combined and the solvent removed in vacuo. The residue was further purified via chromatography on a 10 g C18 SepPak (eluting 0–95% methanol in water) to give a white solid (186.4 mg, 40% yield over four steps). M.p. > 230°C (decomposition). 1H NMR (400 MHz, DCl (2.3 mol %) in D2O) δ (ppm) = 0.087–1.01 (m, 2H, C-CH2-C), 1.12–1.27 (m, 2H, C-CH2-C), 1.32–1.53 (m, 2H, C-CH2-C), 2.57 (t, 2H, N-CH2-C), 3.30–3.53 (m, 3H, N-CH2-C), 3.78–3.98 (m, 4H, N-CH2-C), 6.90–7.06 (m, C(Ph)-H), 7.27 (d, 2H, C(Ph)-H). LRMS (ESI+): 471 (M+). 13C NMR (150 MHz, DCl (2.3 mol %) in D2O) δ (ppm) = 20.9, 24.6, 29.4, 40.8, 46.2, 51.1, 51.6, 59.2, 128.8, 129.0, 130.9, 131.1, 131.6, 134.0, 135.0, 135.2,1 168.6, 171.9. HRMS (ESI+): calcd. m/z for C22H28B2N3O5+ [M+H-2(H2O)]+ 436.22096; found m/z 436.22122.
General precipitation procedure
To a solution of oligodeoxynucleotide in water was added 0.1× volume 3 M aqueous sodium acetate (pH 5.2) and 2.3× volume absolute ethanol. After addition of glycogen (2 μL, 40 μg), the sample was cooled at −80°C for 30 minutes and then centrifuged for 45 minutes at 4°C. The supernatant was removed and the pellet washed carefully with 70% ethanol in water. The pellet was dried under vacuum and then suspended in 20 μL of water.
General denaturing polyacrylamide gel electrophoresis (PAGE) procedure
Polymerization was accomplished via the addition of 120 μL 10% v/v ammonium persulfate (APS) and 30 μL of N,N,N',N'-tetramethylethylenediamine (TEMED) to a solution of 10–20% acrylamide and urea (7 M) in TBE buffer (25 mL). The solution was immediately poured into a gel frame constructed with 0.75 mm spacers and allowed to polymerize for 1 hour. Samples were denatured via the addition of 1× volume of 2× stop dye (98% formamide, 20 mM EDTA, 0.05% (w/v) bromophenol blue), followed by heating at 65°C for three minutes. Electrophoresis was then performed in TBE buffer until the dye reached the bottom of the gel. The gel was then stained by shaking in a solution of SybrGold (Molecular Probes) in TBE buffer and imaged. Radiolabeled products were visualized by exposing the gel to a phosphorscreen overnight.
General gel purification procedure
Purchased oligonucleotides were purified through denaturing PAGE using 20% acrylamide. The product band was visualized by irradiation of the gel on a TLC plate. The band was then removed using a razor blade, and the gel chunks incubated with water overnight at 37°C. The supernatant was removed and subjected to standard precipitation conditions.
Oligonucleotide coupling procedure
The carboxylic acid tag (BBA, 2.61 mg, 5.54 μmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC, 1.59 mg, 8.31 μmol) were dissolved in DMF (0.55 mL). Hünig's base (1.37 μL, 8.31 μmol) was added and the solution allowed to react at room temperature for 2 hours with rotational mixing. To an aliquot of the activated solution (100 μL) was added 35 μL DMF followed by 10 nmol of the amine-functionalized oligonucleotide in water (NH2-DNA1 or NH2-DNA1c) and enough additional water to produce a 9:1 DMF:water solvent ratio (150 μL final volume). The final reaction mixture was allowed to react at room temperature with rotational mixing. After 24 hours, the mixture was diluted with 60 μL of 0.1 M TEAA and heated to 85°C for 3 minutes. The solution was immediately filtered using an Ultrafree-MC microcentrifuge filter tube (0.45 μm, Millipore). The resulting solution was divided into two equal aliquots, and each aliquot was submitted to HPLC purification. Purification of BBA-DNA1 and BBA-DNA1c was performed using a 5% – 25% 0.1 M TEAA - acetonitrile gradient. Fractions corresponding to product or starting material peaks were collected in 15 mL conical centrifuge tubes (VWR). After freezing at −80°C, the samples were submitted to lyophilization. The dry products were then resuspended in 300 μL of water and precipitated according to standard procedures. The isolated yield of each reaction was estimated to be ca. 10% (1 nmol) as inferred from absorbance spectroscopy. The purity of the samples was analyzed through PAGE (10% acrylamide, 7 M urea).
General procedure for the 5'-32P-end labeling of oligonucleotides
Oligonucleotide substrate (20 pmol) was brought to 1 μM in Forward Reaction Buffer (New England Biolabs). To this solution was added γ-32P-ATP (1 μL, 23.8 μmol, 0.143 mCi, Perkin Elmer) and T4 polynucleotide kinase (20 units, New England Biotechnologies). The reaction mixture was incubated for 1 hour at 37°C and immediately subjected to denaturing gel purification. Band visualization was achieved by first obtaining a phosphorimage of the gel. The printed image was then aligned with the gel beneath a glass plate and the band excised. The remainder of the gel purification proceeded as described above.
Duplex formation procedure
An equimolar mixture of functionalized or unfunctionalized DNA-1 and DNA-1c was heated to 80°C in a standard PCR buffer (1.5 mM MgCl2, 50 mM KCl, 10 mM Tris, pH 8.3). The solution was then slowly cooled at a rate of 0.1°C per second.
Native PAGE procedure
Polymerization was accomplished by addition of 300 μL 10% v/v APS and 30 μL TEMED to a solution of 20% acrylamide and 5% v/v glycerol in 0.25× TBE buffer (50 mL). The solution was immediately poured into a gel frame constructed with 1.5 mm spacers and allowed to polymerize for 1 hour. The gel was preheated by running at 120 V for 90 minutes in 0.25× TBE buffer. Samples were combined with 6× Orange G (disodium (8Z)-7-oxo-8-(phenylhydrazinylidene) naphthalene-1,3-disulfonate) dye (Molecular Probes). Electrophoresis was then performed in 0.25× TBE buffer at 200 V with fan cooling until the dye had progressed 2/3 of the distance to the bottom of the gel. The gel was then stained by shaking in a solution of SybrGold (Molecular Probes) in TBE buffer and visualized through fluorescence imaging.
Cellulose paper chromatography procedure
Duplexes were first synthesized using radiolabeled samples of BBA-DNA1c or DNA1c. Approximately 0.5 pmoles of the duplexes or radiolabeled CELAPT 14.11 (a cellulose aptamer described in ref. (11)) were spotted separately using a pipetter ca. 2 cm above the bottom of 10 × 15 cm cellulose blotting paper (VWR, grade 703). The paper was then suspended in a large glass chromatography chamber containing PBS (150 mM NaCl, 10 mM Na2HPO4, pH 7.4) or other buffers as indicated. Displacement assays were performed in the PBS buffer with added 5 mM MgCl2 and 0.1 M saccharide, with the final solution adjusted, if necessary, to pH 7.4. After the buffer migrated approximately 8 cm, the paper was removed, dried, and exposed to a phosphorscreen. The retention factor (r.f.) of the duplexes was determined by plotting the intensity of pixels along a line from the origin to the solvent front in the ImageQuant software. The pixel positions were then normalized to the total distance from the origin to solvent front. The data was plotted in the Origin software program and the peaks fit to Gaussian curves. The peak maxima were taken to be the r.f. values.
RESULTS
Design of boronic acid-DNA conjugates
In order to explore the efficacy and valency of boronic acid recognition units in the context of DNA, three constructs were designed based on the general procedure outlined in Scheme 1. The construct shown, BBA-DNA1:BBA-DNA1c, contains four boronic acids while a construct consisting of DNA1 and BBA-DNA1c (DNA1:BBA-DNA1c) only contains two such binding units. These functionalized duplexes could then be compared to the native duplex (DNA1:DNA1c). To synthesize the BBA-DNA1 and BBA-DNA1c conjugates, we focused our functionalization efforts on amide bond formation reactions due to the robustness of such techniques in bioconjugation reactions and the ready availability of primary amine-functionalized oligonucleotides (12). The oligonucleotide sequences (Scheme 2) were selected to achieve duplex stability at room temperature and minimize the probability of sequence mismatch. The solid-phase synthesis of a bis-boronic acid tag containing a free carboxylic acid moiety was designed based on previous examples of solid phase-functionalization published by the Anslyn group (13).
Scheme 2.

Oligonucleotide substrates.
Synthesis of bis-boronic acid recognition unit
The synthesis of BBA was performed as shown in Scheme 3. General solid-phase synthesis procedures were performed according to manufacturer recommendations (Novabiochem). First, Wang resin functionalized with an Fmoc-protected glycine residue (1) was submitted to standard Fmoc cleavage conditions using a piperidine/DMF solution. The coupling of a bis-Fmoc-protected lysine residue was performed using HBTU as the coupling agent with HOBt and Hünig's base as additives. The Fmoc groups were then removed to expose two primary amine reaction sites (4). Boronic acid functionalization was achieved through the reductive amination of two equivalents of 2-formylphenylboronic acid using triacetoxyborohydride. The resin was then submitted to TFA cleavage conditions, which yielded the desired bis-boronic acid dipeptide (BBA) after purification.
Scheme 3.

Synthesis of BBA.
Conjugation of bis-boronic acid tag (BBA) with oligonucleotides
The carboxylic acid moiety of BBA was first activated with EDC and Hünig's base in anhydrous DMF. This solution was then added to an aqueous solution of DNA1 or DNA1c containing 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer at pH 7.4, and the reaction was monitored by PAGE (Figure 1). Staining with SybrGold revealed a new band with reduced mobility relative to the starting oligonucleotide. Quantification of the band intensity was used to estimate the degree of reaction completion. The maximum observed conversion was estimated to be 35% in these reactions. The new material could also be distinguished as a unique peak via reverse-phase HPLC analysis (Figure 2). The product was isolated through preparative reverse-phase HPLC and precipitated with ethanol to remove any remaining organic material. PAGE gel shift analysis was used to confirm the purity of the isolated material. The concentration of the product was determined from the absorbance at 260 nm.2 These reactions routinely resulted in an isolated yield of 10%.
Figure 1.
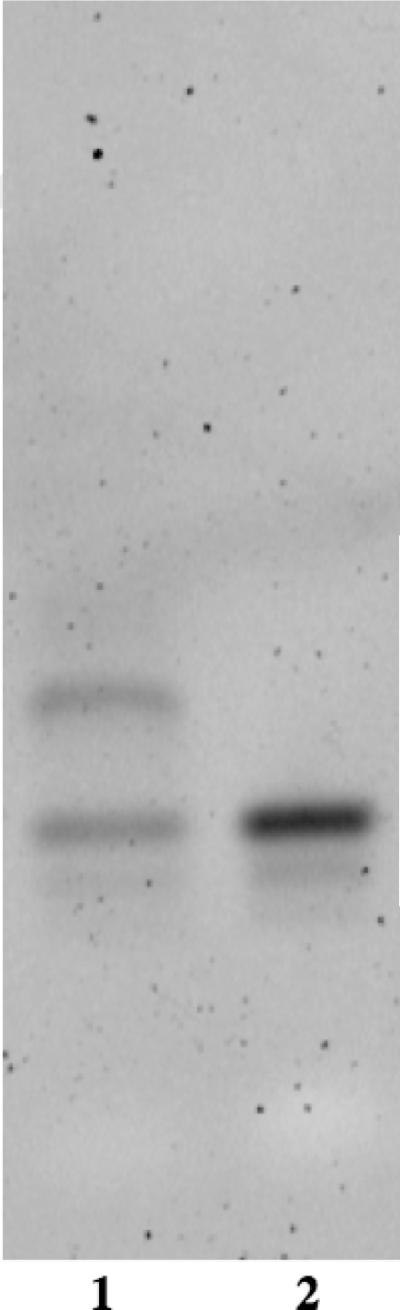
Gel mobility assays for the product of the BBA-DNA1 conjugation reaction.
Lanes (5 ng oligonucleotide): 1 - BBA-DNA1 / NH2-DNA1 reaction mixture; 2 - NH2-DNA1. Conditions: 10% acrylamide, 7 M urea PAGE gel at 0.75 mm thickness visualized with SybrGold stain.
Figure 2.
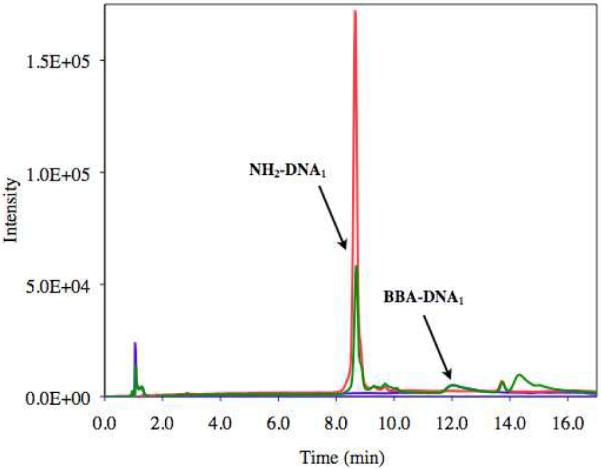
HPLC assay for BBA-DNA1 conjugation reaction.
a) Red: NH2-DNA1; Green: Reaction mixture; Conditions: reverse phase (C18) column; 0.1 M triethylammonium acetate (TEAA):acetonitrile eluant.
Formation of functionalized duplexes
The ability of the synthesized DNA conjugates to form double stranded duplex structures was analyzed through gel shift analysis on a non-denaturing (native) polyacrylamide gel (Figure 3). In order to anneal the appropriate conjugates, a 1:1 mixture of BBA-DNA1 and BBA-DNA1c was heated to 80°C for 10 minutes in a standard PCR buffer solution. The solution was then slowly cooled to 20°C. After the addition of Orange G dye, the reaction solution was loaded onto a native polyacrylamide gel. Control solutions of DNA1 alone and BBA-DNA1 alone were submitted to identical conditions. A significant reduction in electrophoretic mobility was observed for the solution of BBA-DNA1:BBA-DNA1c over the single BBA-DNA1, as expected for the formation of a DNA duplex. This procedure was repeated with a 1:1 mixture of DNA1 and BBA-DNA1c to form the bis-boronic acid duplex.
Figure 3.
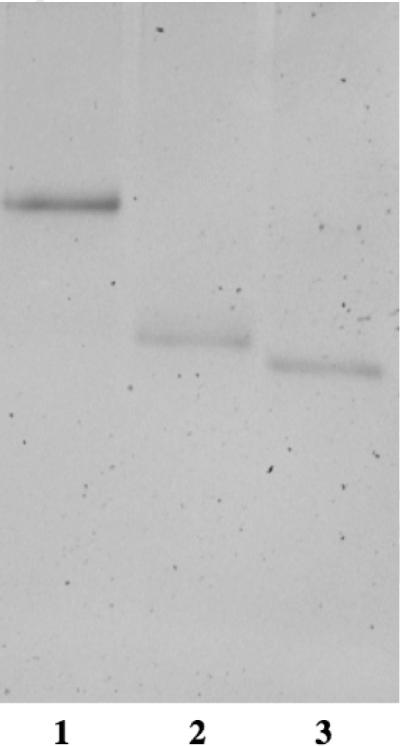
Gel mobility analysis of the product of the BBA-DNA1:BBA-DNA1c duplex forming reaction. Lanes (5 ng): 1 - BBA-DNA1:BBA-DNA1c; 2 - BBA-DNA1; 3 - NH2-DNA1. All samples were submitted to the duplex formation procedure. Conditions: 20% acrylamide PAGE gel containing 5% glycerol (v/v) at 1.5 mm thickness visualized with SybrGold stain.
Cellulose paper chromatography of functionalized duplexes
The retention of the native duplex (DNA1:DNA1c), the bis-boronic acid duplex (DNA1:BBA-DNA1c), and the tetra-boronic acid duplex (BBA-DNA1:BBA-DNA1c) on cellulose paper chromatography was explored as an indicator of boronic acid - diol interactions. It was expected that the boronic acid functionalized duplexes would migrate a shorter distance than the native duplex due to increased binding to the hydroxyl groups available on the cellulose paper. The migration of the synthesized duplexes was compared to that of a cellulose aptamer (CELAPT14.11) developed by the Breaker laboratory and shown to display greatly reduced mobility on cellulose paper as compared to random DNA sequences (11). Both BBA-DNA1c and DNA1c sequences were labeled with phosphorous-32 at the 5'-terminus to allow for the use of phosphorimaging methods.
In the initial assay, the duplex and aptamer solutions, respectively, were spotted on a sheet of cellulose paper using a single channel pipetter, and the paper was then developed in a PBS buffer. As seen in Figure 4, CELAPT14.11 (the positive control) was largely retained at the origin (A) while the native duplex DNA1:DNA1c (the negative control) traveled with the solvent front (C). The bis-boronic acid duplex DNA1:BBA-DNA1c also traveled with the solvent front while the tetra-boronic acid duplex BBA-DNA1:BBA-DNA1c was slightly retained (B).
Figure 4.

Paper chromatography in PBS buffer. Lanes (0.25 pmol): 1 - BBA-DNA1:BBA-DNA1c; 2 - DNA1:BBA-DNA1c; 3 - DNA1:DNA1c; 4 - CELAPT14.11.
The above result provided initial support for the notion that the tetra-boronic acid species interacts with cellulose (a saccharide-rich substrate). This prompted the study of a number of buffer conditions in the hopes of optimizing the response. The conditions included 2× PBS, 0.5× PBS, PBS in the absence of NaCl, and PBS in the presence of 5 mM MgCl2. The most significant differentiation between BBA-DNA1:BBA-DNA1c and DNA1:DNA1c was observed in the PBS + MgCl2 buffer system (Figure 5, images for other conditions shown in the S.I.). The migration of these duplexes was also tested in a Tris buffer (20 mM Tris, 100 mM NaCl, 5 mM MgCl2) at pH values of 7.4, 8.2, and 9.0. No significant dependence on the pH was seen under these conditions (see S.I.). The PBS + MgCl2 buffer system was thus chosen for further analysis.
Figure 5.

Paper chromatography in PBS buffer with 5 mM MgCl2. Lanes (0.25 pmol): 1 - BBA-DNA1:BBA-DNA1c; 2 - DNA1:BBA-DNA1c; 3 - DNA1:DNA1c; 4 - CELAPT14.11.
Saccharide displacement studies
We next investigated the ability of various saccharides to displace the tetra-boronic acid duplex BBA-DNA1:BBA-DNA1c from the cellulose paper. Solutions of DNA1:DNA1c, DNA1:BBA-DNA1c, and CELAPT14.11 were again included as controls. The BBA-DNA1:BBA-DNA1c signal was observed to migrate differently when developed in various 0.1 M saccharide solutions (Figure 6 and S.I.). For each saccharide, the position of the host signal relative to the origin and solvent front (i.e. the retention factor (r.f.)) was then determined. First, the ImageQuant program was used to draw a single-pixel line from the sample origin to the solvent front. The intensity of each pixel on the line and its relative position were then extracted and re-plotted as a chromatogram in the Origin software program (14). Using this program, the major peak of each chromatogram was fit to a Gaussian curve. The intensity maximum of the calculated Gaussian curve was taken as the r.f. value (Table 1). Due to the difficulty associated with resolving peaks close to the origin, only peaks with r.f. values greater than 0.15 were subject to analysis. Measurements for maltose, galactose, and trehalose were conducted in triplicate to allow the generation of error bars (Table 1). These errors were found to be approximately 15%.
Figure 6.

Paper chromatography in PBS buffer with 5 mM MgCl2 and 0.1 M of the indicated saccharide. Lanes (0.25 pmol): 1 - BBA-DNA1:BBA-DNA1c; 2 - DNA1:BBA-DNA1c; 3 - DNA1:DNA1c; 4 - CELAPT14.11.
Table 1.
Cellulose paper chromatography retention factors for BBA-DNA1:BBA-DNA1c in the presence of various saccharides in PBS buffer at pH 7.4.
| r.f. | Description | r.f. | Description | ||
|---|---|---|---|---|---|
| None | < 0.15 | - | Cellobiose | < 0.15 | β-Glc-(1→4)-Glc |
| Ribose | < 0.15 | Rib | Sucrose | < 0.15 | αGlc-(1→ 2)-β-Fru |
| Fructose | < 0.15 | Fru | αLactose | 0.19 | β-Gal-(1→4)-α-Glc |
| Glucose | < 0.15 | Glc | βLactose | 0.22 | β-Gal-(1→4)-β-Glc |
| Galactosea | 0.21 (0.03) | Gal | Lactulose | < 0.15 | β-Gal-(1→4)-Fru |
| Maltosea | 0.33 (0.01) | α-Glc-(1→4)-Glc | Melibiose | < 0.15 | α-Gal-(1 → 6)-Glc |
| Trehalosea | 0.77 (0.12) | α-Glc-(1→ 1)-α-Glc | Raffinose | < 0.15 | α-Gal-(1→6)-α-Glc-(1→ 2)-β-Fru |
Average of three trials (standard deviation).
DISCUSSION
DNA duplexes containing zero, two, or four boronic acid units, respectively, were designed in order to investigate the sugar recognition ability of boronic acid groups in the presence of nucleic acid structures. The synthesis of these hosts relied on the endfunctionalization of complementary single stranded oligodeoxynucleotides through amide bond ligation. Hybridization of these strands was expected to allow for the determination of whether the proximity of boronic acid moieties influences the valency of binding and the concomitant recognition of saccharides under these conditions. After radiolabeling and hybridization, the sugar recognition ability of these hosts was tested through cellulose paper chromatography in the absence and presence of various saccharides.
Construction of boronic acid-DNA conjugate
The synthesis of the boronic acid tag focused on the solid phase derivatization of a dipeptide containing a terminal lysine residue. With the goal of optimizing previously reported conditions for the solid phase reductive amination of 2-formylphenylboronic acid (13), we developed a procedure based on reports by Hindsgaul and co-workers for the solid phase synthesis of carbohydrates (15). The successful final reaction used dichloromethane to swell the resin and also featured sodium triacetoxyborohydride as the reducing agent, a safer alternative to the sodium cyanoborohydride agent previously reported. These conditions allowed the final bis-boronic acid dipeptide tag (BBA) to be isolated as a white powder.
Conditions for the DNA conjugation reaction were optimized for this system from reports by Cravatt and co-workers that used N-hydroxysuccinimide to facilitate activation of the carboxylic acid group (16); however, this additive did not facilitate the coupling of the boronic acid tag in our hands. In the present system, successful conjugation of the tag was achieved through activation with EDC and a non-nucleophilic base in dry DMF. A large excess of this solution was then added to a concentrated aliquot of the oligonucleotide in aqueous solution, thus minimizing the water content while retaining solubility of the nucleotide. HPLC purification followed by ethanol precipitation allowed for the isolation of the desired conjugate in ~ 1 nmole quantities. Nearly complete duplex formation was observed through native PAGE experiments, a finding that provided support for the conclusion that the final receptor construct had been formed. However, extensive efforts to further characterize these system through mass spectrometry were unsuccessful. This is ascribed to the interference caused by the boronic acid functionality.
Boronic acid valency requirement
Simple phenylboronic acids generally display dissociation constants with saccharides in the millimolar range in aqueous media, with higher binding observed at lower salt concentrations (17). However, few reports have analyzed boronic acid-saccharide interactions in the context of biopolymers. These factors raise concerns about the utility of such receptors in a physiological environment and led us to investigate the possible impact of avidity in DNA-structured hosts. While the functionalized duplexes were not able to be retained on traditional saccharide resins at low concentrations, paper chromatography experiments revealed significant retention of the tetra-boronic acid duplex BBA-DNA1:BBA-DNA1c on cellulose paper even at high salt concentrations. In contrast, the native (unfunctionalized) duplex DNA1:DNA1c traveled with the solvent front. Perhaps even more intriguing is the fact that the bis-boronic acid duplex was not retained by the cellulose paper.
From the above results, we infer that BBA-DNA1:BBA-DNA1c interacts with cellulose more strongly than the DNA1:DNA1c, presumably due to the paired boronic acid recognition units. The different behavior of the differently functionalized duplexes may be attributable to an avidity effect in which four boronic acid recognition units promote significant saccharide interactions under the conditions of our experiment while two boronic acid units do not. The need for multiple convergent boronic acid units to achieve saccharide binding will be critical to the design of physiologically relevant boronic acid receptors.
BBA-DNA1:BBA-DNA1c displacement from cellulose by saccharides
In order to probe further the nature of the tetra-boronic acid duplex BBA-DNA1:BBA-DNA1c with saccharides, cellulose chromatography was performed in concentrated sugar solutions. It was expected that competition of the small molecules for the BBA sites would cause this duplex to adhere less well to the cellulose support. In accord with such expectations, clear displacement was observed in the presence of galactose, maltose, trehalose, and both α and β-lactose. However, little to no displacement was observed with the other saccharides tested (Table 1). To the extent that the degree of displacement can be taken as an indicator of relative binding affinity, the strength of the saccharide-BBA-DNA1:BBA-DNA1c interactions does not follow the order generally observed for simple mono-boronic acid receptors (17). We ascribe these differences to the effects of multivalency and the interactions between two or more boronic acid units of the functionalized duplex and certain saccharides (18). For instance, a lack of migration is seen in the presence of fructose, the monosaccharide that is generally bound the most effectively by mono-boronic acid receptors. The monosaccharide galactose, on the other hand, boasts two 1,2-cis diol units when present as either the α-pyranoside or α-furanoside conformer. The increased migration observed in the presence of galactose supports binding with two boronic acid units, and subsequent studies will further explore this possibility (19, 20). A large r.f. was also observed for BBA-DNA1:BBA-DNA1c in the presence of trehalose, an effect that mimics what was seen in the case of CELAPT14.11, the anti-cellulose aptamer used as a positive control. Similarly, both our host and the aptamer were unresponsive to cellobiose, despite the similarity of this disaccharide and the cellulose matrix. The similar binding patterns for CELAPT14.11 and our system provides support for our interpretation that the supramolecular host is interacting with sugar moieties, although non-specific effects or some other fortuitous basis for this concordance cannot be fully ruled out at the present time.
CONCLUSION
While boronic acids show great promise as recognition units for biologically important saccharide targets, the efficacy of these groups in the context of biological macromolecules has not been extensively studied. To address this issue, we attached sugar binding boronic acids at the terminus of a DNA duplex. It was found that boronic acid recognition units are compatible with traditional amide-based bioconjugation reactions and that boronic acid-DNA conjugates readily undergo duplex formation. The ability of the tetra-boronic acid duplexes to interact with sugar-rich substrates was confirmed through cellulose paper chromatography. The lack of binding displayed by the bis-boronic acid duplex and the native duplex provides support for the intuitively appealing notion that multivalent interactions are required to achieve an appreciable degree of conjugate-cellulose interaction. Further evidence for saccharide binding came from displacement studies, which revealed that some mono and disaccharides could compete effectively with cellulose. The bioconjugate host was able to select specific saccharides among the various analytes tested and did so with a pattern that deviated from the previously reported preferences of monoboronic acid receptors. Trehalose was found to effect the migration of the tetra-boronic acid duplex, just as it did with CELAPT14.11, an aptamer selected to bind cellulose. However, in the case of other smaller saccharides notable differences were seen for the present synthetic conjugates and this latter aptamer. This speaks to the selectivity of our conjugates.
In conclusion, we have demonstrated that the recognition of saccharides can be achieved via the use of boronic acid-functionalized DNA duplexes, although multiple recognition sites are required for success. The present results thus provide a blue print for the effective translation of synthetic recognition systems into the context of macromolecules.
Supplementary Material
ACKNOWLEDGEMENT
The authors gratefully acknowledge the contributions of Matt Levy and Gwen Stovall. Funding was provided by the National Institutes of Health (CA-68682 to J.L.S.), the Robert A. Welch (F-1018 to J.L.S., F-1654 to A.D.E., F-1151 to E.V.A.), the National Science Foundation (CHE-0716049 to E.V.A.), and the Office of Naval Research (N0014-09-1-1087 to A.D.E.).
Footnotes
Supporting Information Available: Additional cellulose paper chromatography images and example of curve fitting line. This material is available free of charge via the Internet at http://pubs.acs.org.
The similarity of the two phenyl rings leads to significant overlap in the aromatic region of the spectrum. As a result, not all peaks could be cleary identified.
The molar absorptivity of BBA at 260 nm was found to be 719 M−1 cm−1 in 10 mM phosphate buffer at pH 7.4. This value was less than 0.5% of the oligonucleotide molar absorptivities reported by the manufacturer at this wavelength. Concentration estimates of the BBA-DNA conjugate solutions were thus based on the molar absorptivities of the oligonucleotide sequences.
REFERENCES
- (1).Varki A, Sharon N. Historical Background and Overview. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 2009. Chapter 1. [PubMed] [Google Scholar]
- (2).Mader HS, Wolfbeis OS. Boronic acid based probes for microdetermination of saccharides and glycosylated biomolecules. Microchim. Acta. 2008;162:1–34. [Google Scholar]
- (3).Jin S, Cheng Y, Reid S, Li M, Wang B. Carbohydrate recognition by boronolectins, small molecules, and lectins. Med. Res. Rev. 2010;30:171–257. doi: 10.1002/med.20155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Li M, Lin N, Huang Z, Du L, Altier C, Fang H, Wang B. Selecting Aptamers for a Glycoprotein through the Incorporation of the Boronic Acid Moiety. J. Am. Chem. Soc. 2008;130:12636–12638. doi: 10.1021/ja801510d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Yang X, Dai C, Molina AD, Wang B. Boronic acid-modified DNA that changes fluorescent properties upon carbohydrate binding. Chem. Commun. 2010;46:1073–1075. doi: 10.1039/b921163b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vischer E, Chargaff E. The separation and characterization of purines in minute amounts of nucleic acid hydrolysates. J. Biol. Chem. 1947;168:781–782. [PubMed] [Google Scholar]
- (7).Markham R, Smith JD. Chromatography of nucleic acid derivatives. Nature. 1949;163:250–251. doi: 10.1038/163250b0. [DOI] [PubMed] [Google Scholar]
- (8).Lawley PD. The relative reactivities of deoxyribonucleotides and of the bases of DNA towards alkylating agents. Biochim. Biophys. Acta. 1957;26:450–451. doi: 10.1016/0006-3002(57)90042-2. [DOI] [PubMed] [Google Scholar]
- (9).Schnitzler BE, Thebo PL, Tomley FM, Uggla A, Shirley MW. PCR identification of chicken Eimeria: a simplified read-out. Avian Pathol. 1999;28:89–93. doi: 10.1080/03079459995091. [DOI] [PubMed] [Google Scholar]
- (10).Zhang S, Li BW, Weil GJ. Paper chromatography hybridization: a rapid method for detection of Onchocerca volvulus DNA amplified by PCR. Am. J. Trop. Med. Hyg. 2000;63:85–9. doi: 10.4269/ajtmh.2000.63.85. [DOI] [PubMed] [Google Scholar]
- (11).Boese BJ, Breaker RR. In vitro selection and characterization of cellulose-binding DNA aptamers. Nucleic Acids Res. 2007;35:6378–6388. doi: 10.1093/nar/gkm708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12). http://www.idtdna.com/catalog/Modifications/.
- (13).Edwards NY, Sager TW, McDevitt JT, Anslyn EV. Boronic acid based peptidic receptors for pattern-based saccharide sensing in neutral aqueous media, an application in real-life samples. J. Am. Chem. Soc. 2007;129:13575–13583. doi: 10.1021/ja073939u. [DOI] [PubMed] [Google Scholar]
- (14). http://www.originlab.com/.
- (15).Hummel G, Jobron L, Hindsgaul O. Solid-phase synthesis of a 1-thio-β-D-GlcNAc carbohydrate mimetic library. J. Carbohydr. Chem. 2003;22:781–800. [Google Scholar]
- (16).Adam GC, Sorensen EJ, Cravatt BF. Trifunctional chemical probes for the consolidated detection and identification of enzyme activities from complex proteomes. Mol. Cell Proteomics. 2002;1:828–835. doi: 10.1074/mcp.t200007-mcp200. [DOI] [PubMed] [Google Scholar]
- (17).Springsteen G, Wang B. A detailed examination of boronic acid-diol complexation. Tetrahedron. 2002;58:5291–5300. [Google Scholar]
- (18).Sandanayake KRAS, Nakashima K, Shinkai S. Specific Recognition of Disaccharides by Trans-3,3'-Stilbenediboronic Acid - Rigidification and Fluorescence Enhancement of the Stilbene Skeleton Upon Formation of a Sugar-Stilbene Macrocycle. Chem. Commun. 1994:1621–1622. [Google Scholar]
- (19).Morin GT, Paugam MF, Hughes MP, Smith BD. Boronic Acids Mediate Glycoside Transport through a Liquid Organic Membrane Via Reversible Formation of Trigonal Boronate Esters. J. Org. Chem. 1994;59:2724–2728. [Google Scholar]
- (20).Gray CW, Johnson LL, Walker BT, Sleevi MC, Campbell AS, Plourde R, Houston TA. Specific sensing between inositol epimers by a bis(boronate) Bioorg. Med. Chem. Lett. 2005;15:5416–5418. doi: 10.1016/j.bmcl.2005.08.112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.