

Abstract
Neuropathological and brain imaging studies suggest that schizophrenia may result from neurodevelopmental defects. Cytoarchitectural studies indicate cellular abnormalities suggestive of a disruption in neuronal connectivity in schizophrenia, particularly in the dorsolateral prefrontal cortex. Yet, the molecular mechanisms underlying these findings remain unclear. To identify molecular substrates associated with schizophrenia, DNA microarray analysis was used to assay gene expression levels in postmortem dorsolateral prefrontal cortex of schizophrenic and control patients. Genes determined to have altered expression levels in schizophrenics relative to controls are involved in a number of biological processes, including synaptic plasticity, neuronal development, neurotransmission, and signal transduction. Most notable was the differential expression of myelination-related genes suggesting a disruption in oligodendrocyte function in schizophrenia.
Schizophrenia is a severe psychiatric disorder characterized by hallucinations, delusions, disorganized thought, and various cognitive impairments. Neuropathological and neuroimaging studies have reported a number of anatomical alterations associated with the disease (1, 2). Cellular aberrations, such as decreased neuronal size, increased cellular packing density, and distortions in neuronal orientation, have been observed in immunocytochemical and ultrastructural studies (1, 2). Biochemical and RNA analyses have shown alterations in various neurotransmitter pathways and presynaptic components (2, 3). Polygenic models of inheritance and linkage analysis studies have postulated that several genes confer susceptibility to schizophrenia (4). Although some insights into the etiology of schizophrenia have been developed from these studies, an understanding of the disease on the molecular level remains elusive. Efforts to identify molecular aberrations associated with the disease may be confounded by the subtle structural and cellular changes that occur and the polygenic nature of schizophrenia.
Given these difficulties, methods designed to survey global alterations in mRNA expression have the advantage of sampling a large portion of the genome in search of genes consistently dysregulated in schizophrenia. DNA microarray analysis is one such technique that allows for the quantitative measurement of the transcriptional expression of several thousand genes simultaneously (5). This approach was used here to profile the gene expression patterns in schizophrenia and control samples. We report that the expression levels of genes involved in neuronal myelination, development, synaptic plasticity, neurotransmission, and signal transduction were altered in the dorsolateral prefrontal cortex of schizophrenia brain tissue. Implications drawn from these changes in gene expression provide insights into the etiology of schizophrenia.
Methods
Sample Information and Preparation of Total RNA.
Postmortem dorsolateral prefrontal cortex (left hemisphere, Broadman area 46; ref. 6) from control and medicated (typical neuroleptics) schizophrenic patients were dissected while frozen. All dissections were performed blind to diagnosis. A region corresponding to Bm46 (ref above) and measuring ≈1.5 cm along the cortical surface was dissected from 0.5- to 0.8-cm-thick coronal tissue blocks. Every effort was made to leave no more than a 1-mm white matter ribbon along the medial aspects of the dissected block. The entire tissue block was dry-homogenized and aliquotted to avoid possible differences among samples because of dissection variation. None of the patients died with a coma longer than 12 h, and all died of natural causes. Patients classified as schizophrenics had all been residents of Pilgrim Psychiatric Center (Long Island, NY) and were diagnosed with chronic intractable schizophrenia. Controls were derived from nursing home residents who, on extensive medical chart review and care-giver interview, evidenced no neurological or neuropsychiatric diseases and who died of natural causes (myocardial infarction, various non-brain non-hepatic cancers, and congestive heart failure). Patients were diagnosed antemortem and assessed by a team of research clinicians according to Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria. Specimens from all schizophrenic and control cases did not show evidence of any significant neuropathology (7). Samples selected for analysis were restricted to chronic intractable schizophrenia patients, each with at least 35 years of hospitalization, in an effort to reduce the heterogeneity associated with the disease. Samples from the 12 schizophrenia patients on neuroleptic (anti-psychotic) medications [9 males and 3 females, average age 72.1 ± 11.7 yr, average postmortem interval (PMI) 14.0 ± 8.5 h] and 12 control patients (4 males and 8 females, average age 78.7 ± 13.6 yr, average PMI 9.3 ± 6.5 h), which met these inclusion and exclusion criteria and possessed high quality RNA in tissue samples (described below), were chosen for the study. Samples from four additional schizophrenic patients (3 males and 1 female, average age 79.3 ± 7.6 yr, average PMI 13.9 ± 7.3 h) that had been off neuroleptic medications for 6, 9, 11, and 124 weeks were also examined. Differences in the age and PMI of the schizophrenic and control groups were statistically insignificant (two-tailed Student's t test). No correlation was observed between PMI and RNA quality. Total RNA was purified from ≈100 mg of each sample with TRIzol Reagent (GIBCO/BRL) following the manufacturer's protocol. RNA was then purified by using the RNeasy mini kit (Qiagen, Chatsworth, CA). The quality of total RNA was assessed by agarose gel electrophoresis (visual absence of significant 28S and 18S band degradation) and by spectrophotometry.
Microarray Procedure.
Microarray analysis was performed essentially as previously described (8). Briefly, 8 μg total RNA was used to synthesize cDNA that was then used as a template to generate biotinylated cRNA. cRNA was fragmented and added to Affymetrix (Santa Clara, CA) HuGeneFL chips (contains probes for over 6,000 human genes) as described in the standard protocol outlined in the Gene Chip Expression Analysis Technical Manual (Affymetrix). After sample hybridization, microarrays were washed and scanned with a laser scanner (Agilent, Palo Alto, CA). Each sample was profiled in duplicate, with cRNA prepared separately from total RNA. RNA quality was also assessed by examination of the 3′ to 5′ ratios for human actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) oligonucleotides on Affymetrix Test 2 chips. Relative quantitative reverse transcription (RT)-PCR by using QuantumRNA 18S Internal Standards (Ambion, Austin, TX) was performed as described in the manufacturer's protocol to validate the changes in gene expression detected by microarray analysis. Seven of the 89 genes listed in Table 1 were tested and confirmed to have similar changes in expression levels, as determined by microarray analysis (data not shown).
Table 1.
Genes differentially expressed in chronic schizophrenia Fold-ΔGeneAcc. no.P valueFold-ΔGeneAcc. no.P value
 |
Genes with altered expression levels in schizophrenia samples in comparison with control samples. Genes are clustered into groups by biological function. Genes not classified are also listed. The mean fold-change of each gene in schizophrenic samples relative to control samples as well as the P value and GenBank accession number are indicated. Genes with decreases in expression levels are highlighted in red.
Statistical Analysis.
Replicate data for the same sample were weighted gene-wise by using inverse squared standard error as weights (9). An unpaired two-group comparison for each probe set was performed, considering both measurement error and variation among individuals. Genes were determined to have altered expression levels in the schizophrenic versus the control group based on the following criteria: (i) P < 0.05, (ii) 1.4-fold or greater change in the expression levels between the means of the two groups, (iii) an absolute difference between the means of the expression levels of the two groups greater than 50, and (iv) a sum of the fractions of the presence in each group greater than 1.0. The fold-change criterion is based on subtle differences in expression levels observed in other schizophrenic brain studies (2). ANOVA performed on the filtered gene set revealed no significant interaction (P > 0.05) between sex and patient identify, suggesting that male and female patients had similar changes in gene expression between the control and patient groups. Classification of myelination-related genes (see Table 2, which is published as supplemental data on the PNAS web site, www.pnas.org) for the permutation-based analysis, as well as functional grouping, was based on literature searches for genes shown to play a role in glia-mediated myelination.
Hierarchical Clustering.
After filtering, genes were clustered and ordered by a hierarchical clustering algorithm (10) by using an average linkage method (11). Briefly, the expression values for a gene across the 24 samples were standardized to have mean 0 and standard deviation 1 by linear transformation, and the distance between two genes was defined as 1 − r where r is the standard correlation coefficient between the 24 standardized values of two genes. Two genes with the closest distance were first merged into a supergene and connected by branches with length representing their distance, and were then deleted for future merging. The expression level of the newly formed supergene is the average of standardized expression levels of the two genes (average-linkage) for each sample. Then the next pair of genes (supergenes) with the smallest distance were merged, and the process was repeated 88 times to merge all 89 genes. The software (dchip) used to implement model-based expression calculations, two-group comparison, and clustering is available on request (W.H.W.)
Results
The dorsolateral prefrontal cortex (DLPFC) has been implicated by a number of studies in the pathology of schizophrenia (12, 13). DNA microarray analysis was performed on postmortem tissue from the DLPFC of 12 chronic schizophrenic and 12 control patients to identify changes in gene transcription associated with the disease. Selection of tissue from those patients who had chronic intractable schizophrenia was intended to reduce the heterogeneity associated with schizophrenia and to maximize disease-associated gene expression differences. A model-based metaanalysis approach was used to compute the gene expression values and confidence interval for the fold-change of each gene in the profiled tissue (9). Classification of genes having altered expression levels in schizophrenics relative to controls was based on a number of criteria, including fold-change and statistical analyses. Genes found to have altered expression levels in schizophrenia were clustered into groups by biological function. Identification of functionally grouped genes may imply that abnormal regulation of particular biological processes, biochemical pathways, or cell types is prevalent in the disease state. Table 1 lists 89 genes found to have altered expression levels in schizophrenic samples in comparison with control samples.
A striking finding was the identification of five genes whose expression is enriched in myelin-forming oligodendrocytes, all of which were transcriptionally down-regulated in schizophrenia. In contrast, genes in all of the other groups were predominantly up-regulated in expression. These five genes have been implicated in the formation and maintenance of myelin sheaths, which are critical for efficient axonal signal propagation and provide extrinsic trophic signals that affect the development and long-term survival of axons (14). MAL, myelin and lymphocyte protein, and the actin-capping protein gelsolin are expressed in oligodendrocytes and localized in compact myelin (15, 16). Altered expression of 2′,3′-cyclic nucleotide 3′-phosphodiesterase, myelin-associated glycoprotein (MAG), and transferrin has been shown to result in aberrant oligodendrocyte-mediated myelination and development (17–19). MAG is also believed to regulate the interaction between myelinating cells and axons (20). Dysregulation of these genes suggests a disruption in normal oligodendrocyte function. The neuregulin receptor Her3 (ErbB3), which is involved in Schwann cell development and myelination (21), was also found to be down-regulated in schizophrenia. Down-regulation of Her3 may likewise affect oligodendrocytes as neuregulin promotes oligodendrocyte survival and the proliferation of their precursor cells in culture (22).
Thirty-five transcripts that can be detected by the microarrays were classified as being involved in myelination, six of which had decreased expression levels in schizophrenia. Given that there are 235 − 1 subsets of myelination-related genes, the possibility exists that the set of six shown to be differentially expressed are false-positives (i.e., type-1 errors). The probability of the six myelination-related genes passing the filtering criteria by chance was therefore assessed by using a permutation-based analysis. The analysis was performed by randomly partitioning the 24 patient and control samples into two groups of 12 samples. The permutation of the grouping was performed 200,000 times. This sample size is large enough to yield a standard error of 0.00021 for the estimated P value. For each permutation, the 35 myelination-related genes were analyzed based on the filtering criteria. The sum of t-statistics of the genes in the filtered set (−17.60 for the original six genes) is used as the statistic to assess the significance of the filtered set. A P value of 0.00852 was calculated based on these results, which is highly suggestive of the involvement of myelination-related genes in schizophrenia. This analysis took into account the possibility that myelination-related genes may be coregulated in a given pathway instead of being regulated independently of one another. Such an approach provides a much more conservative estimation of the probability than would be obtained by simply considering the genes to be only regulated independently.
Evidence from a number of studies has led to the hypothesis that schizophrenia is a neurodevelopmental disorder with synaptic pathology that could become most apparent in early adulthood (23). Several genes involved in neuronal development and plasticity were also up-regulated in schizophrenics compared with controls. Myristoylated alanine-rich C kinase substrate (MARCKS), growth-associated protein-43 (GAP-43), superior cervical ganglia-10 (SCG-10), and neuroserpin are involved in various aspects of neuronal development. In addition, these genes may also play a functional role beyond early development by modulating synaptic plasticity (24, 25). The results are consistent with previous reports of increased GAP-43 protein levels in the frontal cortices of schizophrenics (26). The light chain of kinesin could likewise be classified in this group for its role in the transport of various axonal components that might modulate neuronal plasticity (27).
Other groups of functionally clustered genes are involved in neurotransmission. Four genes regulating the γ-aminobutyric acid (GABA) neurotransmission pathway were found up-regulated in schizophrenia. The localization of GABA in diverse neuronal populations whose numbers may be differentially altered in schizophrenia (28) suggests that the transcriptional changes detected here reflect the disruption of GABAergic neurotransmission in different DLPFC cell groups. Several neuropeptides were also found to have altered expression levels. Each of these neuropeptides has been reported to have a broad range of functions, which makes it difficult to understand the effects of their dysregulation. In addition, genes that may be involved in neuropeptide processing and release had changed in expression levels. These results support previous reports of disruptions in various neurotransmission systems in schizophrenia (3, 29).
Dopamine is known to bind to G protein-coupled receptors, thereby activating various downstream protein kinase and phosphatase signaling pathways (30). A number of genes involved in these pathways were dysregulated in schizophrenics. These include genes in the protein kinase A pathway (protein kinase A RII subunit, adenylyl cyclase-associated protein 2) and protein kinase C (beta isozyme). Various calcium-regulated components of these pathways such as calmodulin, calcineurin A, and the calcineurin regulator ZAKI-4 were also up-regulated in schizophrenics, as was the calcineurin substrate inositol 1,4,5-triphosphosphate receptor. Both cAMP-dependent protein kinase A (PKA) and calcineurin-related pathways converge on the regulation of dopamine- and cAMP-regulated phosphoprotein of Mr 32,000 (DARPP-32) phosphorylation, a key regulator of protein phosphatase 1 (30). Although not present on the microarray for detection at the mRNA level in the present study, DARPP-32 protein has been shown to be decreased in the prefrontal cortex of schizophrenics (P. Greengard, personal communication). Dysregulation of genes involved in dopamine receptor-mediated signaling is therefore consistent with the hypothesis that schizophrenia may be a functional disorder of hyperdopaminergic activity (31).
Interestingly, several genes differentially expressed in the brain tissue of the schizophrenics are involved in the regulation of the cytoskeleton. NF-L and NF-M are components of neurofilaments, whereas profilin II regulates actin filament formation. Gelsolin, MARCKS, GAP-43, and SCG10 may also regulate the cytoskeleton by directing and stabilizing axons and dendrites as well as cell shape, size, and polarity. Alterations in cellular architecture by modulatory elements of the cytoskeleton could explain some of the morphometric changes reported in different schizophrenic brain regions (2).
The relative expression levels of the 89 genes in each sample are displayed in Fig. 1. Genes are clustered by their relative expression pattern over the 24 patient samples, with their correlations depicted in the dendrogram. Overall, there appears to be a distinction in the gene expression pattern between control and schizophrenic samples. As expected, genes found to be down-regulated in schizophrenia tissue relative to control tissue cluster within a separate branch from the genes found to be up-regulated in the disease state. Five of the six myelination-related genes cluster near one another, suggesting a similarity in their transcriptional regulation. Other gene expression relationships are also evident from this cluster analysis. For example, glutamate decarboxylase-65 (GAD65) and GAD67, as well as somatostatin I and neuropeptide Y, cluster closely to one another, as would be expected based on known metabolic or anatomical relationships. None of the 89 genes are differentially expressed consistently in all schizophrenic samples in comparison to all control samples. Linear discriminant analysis (LDA; ref. 32) was applied to assess the distinction between the schizophrenic and control samples based on their expression profiles. This method seeks the linear combination of variables that maximizes the ratio of between-group variance and within-group variance by using the grouping information. Implicitly, the linear weights used by LDA depend on how a gene separates in the two groups and how this gene correlates with the other genes. LDA was applied to the data matrix of 24 (samples) by 89 (probe sets). The results show that schizophrenic and control samples can be reasonably segregated into two distinct groups based on the pattern of their expression profiles (Fig. 2A). LDA was also performed for the six myelination-related genes alone (Fig. 2B). A comparable degree of separation of control and schizophrenic samples was found relative to the analysis performed by using all 89 genes. LDA was repeated by using all 35 of the myelination-related genes that can be detected by the microarray. This analysis takes an unbiased approach because the genes used to fit the LDA are not derived by the selection criteria, but by the virtue of their functional relation to the six myelination-related genes identified in the screen. Using all 35 myelination-related genes markedly improved the capacity of the analysis to discriminate the two patient groups (Fig. 2C). Improvement in the LDA by the addition of other myelination-related genes suggests that their expression levels also correlate with the disease state. LDA using other functional groups identified in the screen resulted in poorer group segregation (data not shown). These results, together with the permutation-based analysis, clearly provide evidence of the involvement of the myelination pathway in schizophrenia.
Figure 1.

Relative expression levels of the 89 genes differentially expressed in schizophrenic samples relative to control samples. Each column displays the gene expression levels in individual samples and each row corresponds to the individual genes. The expression values for each gene are normalized to have a mean of 0 and a standard deviation of 1. Expression levels are color coded relative to the mean: blue for values less than the mean, red for values greater than the mean. The dendrogram on the left illustrates the final clustering tree resulting from hierarchical clustering of gene expression values. The branch lengths of the tree reflect the degree of similarity of gene expression values across the 24 samples.
Figure 2.
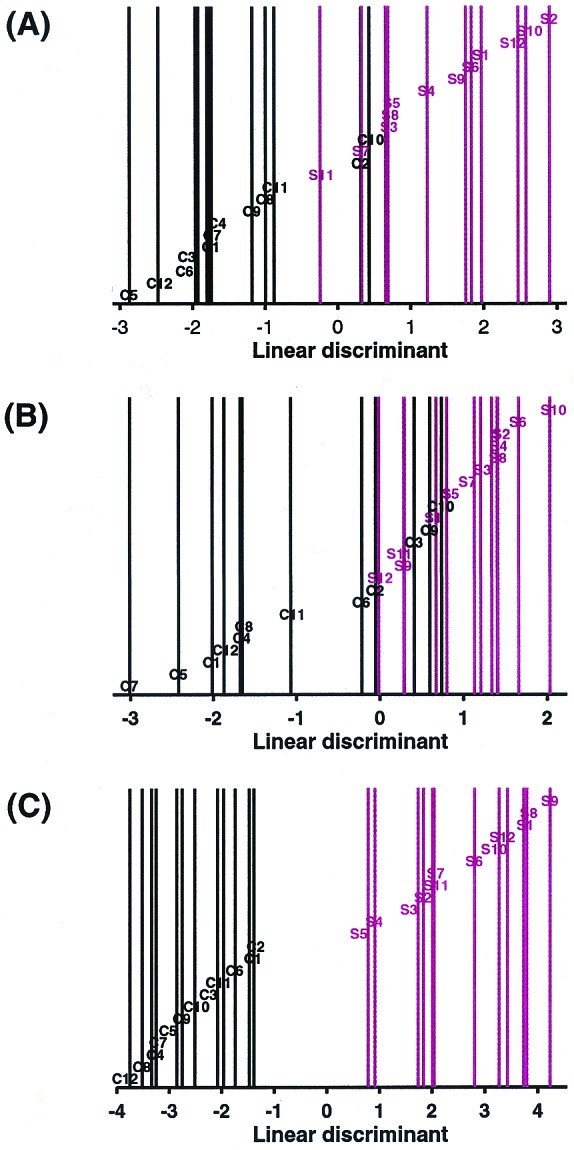
Linear discriminant analysis of schizophrenic and control samples. LDA was applied to the data matrix of the 24 samples by (A) the 89 gene set, (B) the six myelination-related gene set, and (C) the 35 myelination-related gene set. Linear discriminants were determined for each sample and plotted. C = control, S = schizophrenic. Control samples are coded in black and schizophrenic samples in red. Sample identities correspond to those in Fig. 1.
The schizophrenic patients in this study were being treated with neuroleptic (anti-psychotic) medications until the time of death. Neuroleptics are known to inhibit activation of dopamine receptor-mediated signal transduction. It is possible that treatment with neuroleptics may result in the observed changes in gene expression, such as those involved in dopamine signaling. To address this possibility, DLPFC samples from four additional schizophrenic patients that were medication free for 6 weeks or more were profiled. Statistical analysis (P < 0.05, Student's t test) was performed on the genes listed in Table 1 to compare the expression levels in schizophrenics off medication with those on medication. None of the 89 genes were found to have statistically significant differences in expression levels in comparisons between schizophrenics on and off medications. Thus, medication may not account for the observed gene expression changes in schizophrenia. The low sample number of schizophrenics off medication, however, may have limited the analysis of these genes. Therefore, we cannot rule out the possibility that the expression levels of some genes may be modulated by neuroleptics.
Discussion
We have identified a number of functionally clustered groups of genes with altered expression levels in schizophrenics. The most notable of these groups of genes is involved in central nervous system myelination and points to the possibility of oligodendrocytes as a specific cell type that is functionally deficient in schizophrenia. Dysregulation of the myelination-related genes is unlikely to be due to neuroleptic medications. Animal studies of the effects of neuroleptic treatments on glial cells have reported discrete increases in proliferation, presumably of astrocytes and microglia (33). Yet, the myelination-related genes identified here are predominantly expressed in oligodendrocytes. Furthermore, dysregulation of several of these genes has been shown to inhibit oligodendrocyte development and function, rather than promote cellular proliferation (17–19). In addition, none of the 89 genes had statistically significant differences in expression levels in comparisons between schizophrenics on and off medications.
While this manuscript was in preparation, Mirnics et al. (34) reported a microarray analysis of the prefrontal cortex of six schizophrenic patients. This report indicated an alteration in the expression levels of genes involved in the regulation of presynaptic function. Although we did observe changes in the expression levels of some genes involved in presynaptic function, a few reasons may account for the discrepancies in the results between the two reports. First, the patient cohort studied here represents a population of elderly schizophrenics with such profound impairment as to necessitate lifelong hospitalization, whereas the Mirnics et al. cohort were community-dwelling schizophrenics dying at young or middle age. Furthermore, although the Mirnics et al. cohort was representative of the population who on death were referred to the Medical examiner office, the cohort described here represent individuals who had been residents of long-term hospital units and who died in old age of natural causes. In addition, the microarray types used by Mirnics et al. contain probes for only one of the six myelination genes identified in this study and therefore would not have detected the changes in gene expression observed here. Because schizophrenia is a heterogenous disorder with a varying clinical presentation over the lifetime of the patient, studies performed at both middle and old age have equal relevance in determining the underlying etiology.
Oligodendrocytes increase neuronal conduction velocity through their insulating properties and provide extrinsic trophic factors that promote neuronal maturation and axonal survival (35). Given that myelinating glia have been reported to regulate the axonal cytoskeleton (35), we hypothesize that alterations in oligodendrocyte–axon interactions may underlie the subtle cytoarchitectural changes found in schizophrenia (2). This hypothesis is consistent with previous reports that suggest a possible role for myelination in schizophrenia. Myelination of the prefrontal cortex has been observed to occur in late adolescence and early adulthood, which is typically the age of onset of schizophrenia (36, 37). Brain imaging studies have found subtle white matter abnormalities in schizophrenics (38, 39), whereas metachromatic leukodystrophy, a demyelination disorder, is associated with a schizophrenic-like psychoses (40). Immunohistochemical staining of the schizophrenic brain tissues may help to verify a disruption in neuronal myelination. It will also be of interest to perform studies on demyelination mouse models with mild phenotypes to look for a schizophrenic-like behavior (e.g., prepulse inhibition). In addition, microarray analyses of other brain regions from schizophrenic patients, as well as tissues from patients with different psychiatric conditions (e.g., depression and anxiety), are warranted to further validate the results. The finding of a group of genes involved in oligodendrocyte function having differential expression in the DLPFC of schizophrenics provides a new area for future studies and demonstrates the utility of genome-scale expression profiling for the discovery of the etiological basis of neuropsychiatric disorders.
Supplementary Material
Acknowledgments
We thank D. Lockhart, J. Hogenesch, and M. Cooke for helpful discussions, and S. Kay and H. C. Hemmings, Jr. for comments on the manuscript. The Schizophrenia Brain Bank and some of the studies reported were supported by Merit Review and Mental Illness Research, Education and Clinical Centers (MIRECC; Department of Veterans Affairs) awards (to V.H.) and by U.S. Public Health Service Grant MH45212 (to K.L.D.).
Abbreviations
- PMI
postmortem interval
- DLPFC
dorsolateral prefrontal cortex
- LDA
linear discriminant analysis
References
- 1.Arnold S E, Trojanowski J Q. Acta Neuropathol (Berl) 1996;92:217–231. doi: 10.1007/s004010050512. [DOI] [PubMed] [Google Scholar]
- 2.Harrison P J. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- 3.Benes F M. Brain Res Brain Res Rev. 2000;31:251–269. doi: 10.1016/s0165-0173(99)00041-7. [DOI] [PubMed] [Google Scholar]
- 4.Owen M J. Brain Res Brain Res Rev. 2000;31:179–186. doi: 10.1016/s0165-0173(99)00035-1. [DOI] [PubMed] [Google Scholar]
- 5.Lipshutz R J, Fodor S P, Gingeras T R, Lockhart D J. Nat Genet. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- 6.Rajkowska G, Goldman-Rakic P S. Cereb Cortex. 1995;5:307–322. doi: 10.1093/cercor/5.4.307. [DOI] [PubMed] [Google Scholar]
- 7.Purohit D P, Perl D P, Haroutunian V, Powchik P, Davidson M, Davis K L. Arch Gen Psychiatry. 1998;55:205–211. doi: 10.1001/archpsyc.55.3.205. [DOI] [PubMed] [Google Scholar]
- 8.Lockhart D J, Dong H, Byrne M C, Follettie M T, Gallo M V, Chee M S, Mittmann M, Wang C, Kobayashi M, Horton H, Brown E L. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Wong W H. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. . (First Published December 26, 2000; 10.1073/pnas.011404098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisen M B, Spellman P T, Brown P O, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sokal R R, Michener C D. Univ Kans Sci Bull. 1958;38:1409–1438. [Google Scholar]
- 12.Goldman-Rakic P S, Selemon L D. Schizophr Bull. 1997;23:437–458. doi: 10.1093/schbul/23.3.437. [DOI] [PubMed] [Google Scholar]
- 13.Bunney W E, Bunney B G. Brain Res Brain Res Rev. 2000;31:138–146. doi: 10.1016/s0165-0173(99)00031-4. [DOI] [PubMed] [Google Scholar]
- 14.Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab M H, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave K A. Science. 1998;280:1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- 15.Frank M. Prog Neurobiol. 2000;60:531–544. doi: 10.1016/s0301-0082(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka J, Sobue K. J Neurosci. 1994;14:1038–1052. doi: 10.1523/JNEUROSCI.14-03-01038.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gravel M, Peterson J, Yong V W, Kottis V, Trapp B, Braun P E. Mol Cell Neurosci. 1996;7:453–466. doi: 10.1006/mcne.1996.0033. [DOI] [PubMed] [Google Scholar]
- 18.Schachner M, Bartsch U. Glia. 2000;29:154–165. doi: 10.1002/(sici)1098-1136(20000115)29:2<154::aid-glia9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Espinosa de los Monteros A, Kumar S, Zhao P, Huang C J, Nazarian R, Pan T, Scully S, Chang R, de Vellis J. Neurochem Res. 1999;24:235–248. doi: 10.1007/s11064-004-1826-2. [DOI] [PubMed] [Google Scholar]
- 20.Trapp B D. Ann N Y Acad Sci. 1990;605:29–43. doi: 10.1111/j.1749-6632.1990.tb42378.x. [DOI] [PubMed] [Google Scholar]
- 21.Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin G R, Birchmeier C. Nature (London) 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- 22.Barres B A, Raff M C. J Cell Biol. 1999;147:1123–1128. doi: 10.1083/jcb.147.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raedler T J, Knable M B, Weinberger D R. Curr Opin Neurobiol. 1998;8:157–161. doi: 10.1016/s0959-4388(98)80019-6. [DOI] [PubMed] [Google Scholar]
- 24.McNamara R K, Lenox R H. J Comp Neurol. 1997;379:48–71. [PubMed] [Google Scholar]
- 25.Aigner L, Arber S, Kapfhammer J P, Laux T, Schneider C, Botteri F, Brenner H R, Caroni P. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 26.Perrone-Bizzozero N I, Sower A C, Bird E D, Benowitz L I, Ivins K J, Neve R L. Proc Natl Acad Sci USA. 1996;93:14182–14187. doi: 10.1073/pnas.93.24.14182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gindhart J G, Jr, Desai C J, Beushausen S, Zinn K, Goldstein L S. J Cell Biol. 1998;141:443–454. doi: 10.1083/jcb.141.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daviss S R, Lewis D A. Psychiatry Res. 1995;59:81–96. doi: 10.1016/0165-1781(95)02720-3. [DOI] [PubMed] [Google Scholar]
- 29.Benes F M, Vincent S L, Alsterberg G, Bird E D, SanGiovanni J P. J Neurosci. 1992;12:924–929. doi: 10.1523/JNEUROSCI.12-03-00924.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greengard P, Allen P B, Nairn A C. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 31.Davis K L, Kahn R S, Ko G, Davidson M. Am J Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 32.Venables W N, Ripley B D. Modern Applied Statistics with. 1997. s-plus (Springer, New York). [Google Scholar]
- 33.Selemon L D, Lidow M S, Goldman-Rakic P S. Biol Psychiatry. 1999;46:161–172. doi: 10.1016/s0006-3223(99)00113-4. [DOI] [PubMed] [Google Scholar]
- 34.Mirnics K, Middleton F A, Marquez A, Lewis D A, Levitt P. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 35.Bjartmar C, Yin X, Trapp B D. J Neurocytol. 1999;28:383–395. doi: 10.1023/a:1007010205037. [DOI] [PubMed] [Google Scholar]
- 36.Yakovlev P I, Lecours A R. In: Regional Development of the Brain in Early Life. Minkowski A, editor. Oxford: Blackwell; 1967. pp. 3–70. [Google Scholar]
- 37.Benes F M. Schizophr Bull. 1989;15:585–593. doi: 10.1093/schbul/15.4.585. [DOI] [PubMed] [Google Scholar]
- 38.Lim K O, Adalsteinsson E, Spielman D, Sullivan E V, Rosenbloom M J, Pfefferbaum A. Arch Gen Psychiatry. 1998;55:346–352. doi: 10.1001/archpsyc.55.4.346. [DOI] [PubMed] [Google Scholar]
- 39.Andreasen N C, Arndt S, Swayze V, 2nd, Cizadlo T, Flaum M, O'Leary D, Ehrhardt J C, Yuh W T. Science. 1994;266:294–298. doi: 10.1126/science.7939669. [DOI] [PubMed] [Google Scholar]
- 40.Hyde T M, Ziegler J C, Weinberger D R. Arch Neurol. 1992;49:401–406. doi: 10.1001/archneur.1992.00530280095028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.