

Summary
A bootstrap based methodology is introduced for analyzing repeated measures/longitudinal microarray gene expression data over ordered categories. The proposed non-parametric procedure uses order-restricted inference to compare gene expressions among ordered experimental conditions. The null distribution for determining significance is derived by suitably bootstrapping the residuals. The procedure addresses two potential sources of correlation in the data, namely, (a) correlations among genes within a chip (“intra-chip” correlation), and (b) correlation within subject due to repeated/longitudinal measurements (“temporal” correlation). To make the procedure computationally efficient, the adaptive bootstrap methodology of Guo and Peddada (2008) is implemented such that the resulting procedure controls the false discovery rate (FDR) at the desired nominal level.
Key Words and Phases: Bootstrap residuals, dose-response, gene expression, heteroscedastic gene expression data, longitudinal data, ordered categories, ORIOGEN, time course
1. INTRODUCTION
Microarray gene expression studies are routinely conducted by researchers to understand changes in expression of thousands of genes under various experimental conditions. In fields such as agriculture, toxicology, cancer research, etc. a common problem of interest is to investigate changes in gene expression over ordered categories. Some examples of ordered categories include dose groups, time points, cancer stages etc. For example, Tamoto et al. (2004) studied the changes in gene expression at various stages of esophageal cancer, while Hamadeh et al. (2004) investigated the effect of Furon on the gene expression of rat liver at various doses of the compound. In a time-course study, Blanding et al. (2007) investigated the changes in gene expression in corn when the plants were exposed to different levels of ultraviolet radiation. Frequently, a goal of such ordered categorical experimental designs is to select statistically significant genes and to cluster them according to their patterns of expression over the ordered categories. As described by several authors (e.g. Blanding et al., 2007) such clusters provide insight into possible co-regulation or functional relationships among genes. For convenience, throughout this article we shall use the terms “ordered categories”, “dose groups” and “time points” interchangeably.
A typical experimental design consists of a sample of n independent microarrays obtained in the ith ordered category and each array consists of M probes representing several thousands of genes. Thus the data derived from microarray experiments are typically high dimensional (in several thousands) with relatively small sample size. This makes data analysis a challenge as the usual notion of Type I error needs modification and classical methods of analyses need to be modified suitably. The focus of this paper is the case when C, the number of ordered categories, is “small” unlike the usual “long series” time course experiments conducted in the context of cell-cycle experiments. There is no clear definition for what is considered to be “large” or “small” although many authors have used C > 10 to denote “large” (Conesa et al., 2006).
For convenience, throughout this paper we shall use the term “probe” and “gene” interchangeably. Also, for convenience we assume that all categories have the same number of subjects or experimental units n (i.e. same sample size). As usually done in other traditional experiments, two types of microarray experiments are commonly conducted: Design I: the samples are independent across ordered categories, and Design II: the samples are correlated across ordered categories as in the case of longitudinal or repeated measurement studies. It is important to recognize that there are two potential sources of correlations. The first source is due to “within chip/intra-chip” correlation among genes. This may be due to some artifact of how each chip was handled or may be due to the natural correlation among genes at a given experimental condition (or category). This intra-chip correlation structure can potentially exist in both Design I and in Design II. The second source of correlation is the temporal correlation in the expression of a gene which arises in Design II.
Due to the underlying correlation structures, statistical methods used for analyzing data from the two designs are different. Numerous statistical and computational methods have been proposed in the literature for analyzing time course gene expression data obtained under Design I. Although some of these methods account for the intra-chip correlation, many do not. Some popular methods of analyses for Design I include, the classical regression based methodology (Liu et al., 2005, Conesa et al., 2006), splines based methods (Luan and Li 2003, 2004, Storey et al., 2005), order-restricted inference based methods (Peddada et al., 2003, 2005, Hu et al., 2005, Simmons and Peddada, 2007, Liu et al. 2009), and Bayesian methods (Ishwaran and Rao, 2003, 2005a, 2005b, Jensen et al., 2009).
As with any longitudinal study, the longitudinal microarray experiments provide an opportunity for researchers to understand the changes in expression over time while accounting for any subject effect. Also, such experiments allow the investigator to estimate inter and intra individual variability in the gene expression. Recently Karlovich et al. (2009) conducted a longitudinal microarray study of normal healthy men and women of different ages to evaluate the changes in the gene expression in venous blood over a period of six months. Their study confirmed that the expression of most of the important genes remained stable during the six month period. In Ferre et al. (2007), the authors conducted a longitudinal study to investigate the changes in gene expression over time in piglets that were given intramuscular injections. Using the resulting gene expression data they identified several pathways involved in post-injection muscle injury. Other interesting longitudinal gene expression data sets may be found at the NCBI’s website: http:/www.ncbi.nlm.nih.gov/projects/geo/.
Several methods have been proposed in the literature to analyze longitudinal gene expression data utilizing ideas from linear mixed models or hierarchical Bayesian models to describe the covariance structure. For example, based on Brumback and Rice (1998), Storey et al. (2005) modeled the covariance structure using a linear mixed effects model based on B-Splines. Karlovich et al. (2009) used the standard linear mixed effects model approach, while Marot et al. (2009) extended the structural mixed model approach of Jaffrézic et al. (2007) to longitudinal data.
It is important to note that the intra-chip correlations and temporal correlations may arise in a complex unknown manner and hence are often difficult to model. As a result, often times the correlation models are misspecified. For instance, it may not even be appropriate to assume that all genes have the same temporal correlation structure due to potential heterogeneity among genes. Consequently, it is important to have a methodology which is robust to any hidden/correlation structure among the genes. Motivated by this requirement, in this paper we use a simple bootstrap methodology that accounts for the above correlations non-parametrically without making any modeling assumptions. The proposed methodology, described in Section 2, uses order-restricted inference based test statistics introduced in Peddada et al. (2003, 2005). It is designed to control the false discovery rate (FDR) at the pre-specified nominal level. Results of a small simulation study are provided in Section 3. The proposed method is illustrated in Section 4 using a published data set. Concluding remarks and future research problems in this area are provided in Section 5.
2. ANALYSIS OF GENE EXPRESSION DATA USING ORDER RESTRICTED INFERENCE
2.1 Notations and motivation
Let Yg,c,i denote the observed expression of the gth gene g = l,2, …, G, in the cth category, c = 1,2, …, C, for the ith experimental unit, i = 1,2, …, n, with E(Yg,c,i) = μg,c. Throughout this paper the terms “profile” and “pattern” will be used interchangeably. A gene g is said to have a “flat” or constant profile across the categories if μg,1=μg,2= … =μg,C. This Profile will be denoted by P0. Typically, researchers are interested in (a) identifying genes that do not have a flat pattern profile across categories, and (b) clustering genes with similar profiles/patterns. For example, μg,1 ≤ μg,2 ≤ … ≤ μg,c represents a non-decreasing pattern in gene expression over the C ordered categories, whereas μg,1 ≤ μg,2 ≤ … ≤ μg,C ≥ μg,c+1 ≥ … ≥ μg,C represents an umbrella profile with peak at the cth category. In practice a researcher may be interested in clustering genes that belong to a subset of patterns/profiles which can be enumerated before performing the analysis. These patterns may typically consist of (i) a non-decreasing pattern, (ii) umbrella patterns with peaks at c = 2,3, …, C−1, (iii) a non-increasing pattern, and (iv) inverted umbrella patterns with minimum at c = 2,3, …, C −1. Patterns such as cyclical patterns are of interest some times, but other irregular/arbitrary patterns are not very common. Thus in this paper, and in the companion software that soon will be released (ORIOGEN 3.0), we limit to monotonic patterns, umbrella patterns and single cycle patterns and do not include other arbitrary patterns. Let the collection of all non-null patterns be denoted by 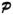 . Then, for each individual gene g one may consider testing the following hypotheses using the classical likelihood ratio test (LRT) provided that the structure of the true temporal covariance matrix is known:
. Then, for each individual gene g one may consider testing the following hypotheses using the classical likelihood ratio test (LRT) provided that the structure of the true temporal covariance matrix is known:
Against
| (1) |
However, even if one were to know the structure of the temporal covariance matrix, the sample size is typically not large enough to use the asymptotic critical values. Furthermore, it is important to note that such a test does not account for the within intra-chip correlation. Hence one needs to use some version of a resampling procedure to test (1) for all genes.
A possible alternative approach to this problem is to use a parametric model to describe the relationship between the mean expression μg,c and c. For example, in a time course experiment or a dose-response study one may consider a regression model to describe the relationship (cf. Liu et al., 2005) and impose a mixed model structure to account for dependence in the data. However, such a model requires the experimenter to know a priori the relationship between the two variables for all genes. Another alternative is to use variations to splines with mixed models as in Storey et al. (2005). Such procedures are generally attractive because of their simplicity but they induce a very specific dependence structure that may not be correct and/or difficult to verify for all genes. Furthermore, such model-based approaches may not be appropriate when c represents an ordered category, such as “tumor stage”. For these reasons, Peddada et al. (2003, 2005) used order restricted inference where profiles/patterns among the means are described in terms of mathematical inequalities instead of a parametric model. Hence in this paper we continue with the nonparametric methodology developed in Peddada et al. (2003, 2005) and in ORIOGEN 2.2.1, making no distributional or modeling assumptions and thus providing a large amount of flexibility. As one would expect, a nonparametric procedure such as the procedure proposed here will have smaller power than a parametric procedure when the underlying model is correctly specified. However, since it is hard to justify a particular parametric model in gene expression studies, a nonparametric procedure, such as the one proposed here, may be preferable.
Corresponding to a given inequality pattern, two parameters are said to be linked if the inequality between them is specified by the inequality pattern. For example, in an umbrella pattern μg,1 ≤ μg,2 ≤ … ≤ μg,c ≥ μg,c+1≥ … ≥ μg,C the parameters μg,1,μg,2, …, μg,c are linked to one another and similarly μg,c,μg,c+1, …, μg,C are linked to one another. However, none of the parameters in the collection {μg,1, μg,2 μg,c−1}are linked to any of the parameters in the collection {μg,c+1, … μg,C}. A parameter in a given inequality patterns is said to be nodal if it is linked with all parameters { μg,1, μg,2, …, μg,C}. Thus in the above umbrella pattern μg,c is the only nodal parameter, whereas in a non-decreasing pattern every parameter is a nodal. For a given pattern P ∈ , using the estimators derived in Hwang and Peddada (1994), we compute TP the studentized distance between the farthest linked parameters in P. Thus for example, in the case of umbrella pattern P, μg,1 ≤ μg,2 ≤ … ≤ μg,c ≥ μg,c+1 ≥ … ≥ μg,C the statistic Tg, P is given by
| (2) |
where μ̂g,i,i = 1,2, … C, is the constrained estimator of μg,c under the constraint P derived according to the Hwang and Peddada (1994) methodology, ignoring correlations in the data. It is important to note that one could use restricted maximum likelihood estimators (RMLE) of μg,c instead of the point estimators proposed in Hwang and Peddada (1994) (ignoring the correlation structure). Perhaps iterative algorithms along the lines of Shi (1994), Shi and Jiang (1998) and Hoferkamp and Peddada (2002) may be developed to the present context which not only estimate the mean parameters but also estimate the underlying covariance matrix iteratively. Although in theory such an iterative algorithm may be appealing, it can be computationally expensive. Furthermore, as noted in Hwang and Peddada (1994), the RMLE need not perform well as an estimator for individual components of the mean vector. For these reasons, we use the estimator proposed in Hwang and Peddada (1994), ignoring correlations in the data. As usual, denotes the pooled sample variance for the gth gene g = 1,2, … G. Again, one may replace by the true standard error of the numerator in (2). However, the derivation of such a quantity is not straightforward and since our test relies on bootstrap the choice of the correct denominator is very important. As in Peddada et al. (2003, 2005), for a gene g, the test statistic for testing (1) is defined as:
| (3) |
Thus neither in the computation of the order-restricted estimators, nor in the construction of the above test statistic do we incorporate the underlying correlation structure. However, in the spirit of the popular generalized estimating equations (GEE) methodology, the correlation in the data will be accounted in the derivation of the bootstrap null distribution (3), which is described in Section 2.2.
Once the null hypothesis is rejected for a gene g according to the methodology described in Section 2.2, we assign it to the pattern P in which corresponds to
.
2.2 The bootstrap methodology
In Peddada et al. (2003, 2005) and in ORIOGEN 2.2.1, for a gene g, the null distribution of was derived by assigning simple random samples (with replacement) of size n, from the combined sample of size n × C, to each ordered category and by computing the statistic . The process was repeated a large number of times B which yielded the null distribution for gene g. The above bootstrap methodology assumes that for a gene g, under the null hypothesis, its expressions across the C categories are independently and identically distributed. In the context of longitudinal data such an assumption is not valid. Under the null hypothesis we can only assume that the means across categories are equal, but cannot assume independence or that the variances are equal. A simple method to relax these assumptions is by bootstrapping the residuals as shown below. Bootstrapping residuals is widely used in literature because of the flexibility it provides to deal with heterogeneity in the data (Efron and Tibshirani, 1993).
Let denote the grand sample mean of the gth gene g = 1,2, …, G, averaged over all experimental units and categories and let Ȳg,c denote its sample mean in the cth category, c = 1,2, …, C. Let ε̂g,c,i = Yg,c,i −Ȳg,c,. denote the residual corresponding to the ith experimental unit in the cth category. Let the jth simple random sample of n experimental units (with replacement) from{l,2, …, n} be denoted by {j1, j2, …, jn}. Thus we are resampling the experimental units. Then for gene g, the bootstrap sample that honors the null hypothesis while maintaining any dependence structure in the data is given by , i = 1,2, …, n, c = 1,2, …, C. Using this bootstrap sample we construct the statistic which yields the bootstrap null distribution. Rather than fixing the number of bootstrap samples B a priori, we use the adaptive bootstrap methodology of Guo and Peddada (2008) which reduces the total number of bootstraps while controlling the false discovery rate at the desired nominal level α.
Although this manuscript is written for analyzing longitudinal gene expression data, the residual bootstrap methodology described above is readily applicable for analyzing cross-sectional ordered gene expression data when the variance of expression for a gene is not constant across ordered categories (known as heteroscedasticity). Since the sample experimental unit is not observed in all categories in a cross-sectional data, the above bootstrap sampling scheme can be modified as follows. As above, let denote the grand sample mean of the gth gene g = 1,2, …, G, averaged over all experimental units and categories and let Ȳg,c denote its sample mean in the cth category, c = 1,2, …, C. Let ε̂g,c,i = Yg,c,i − Ȳg,c,. denote the residual corresponding to the ith experimental unit in the cth category. For the cth category, let jth simple random sample of n experimental units (with replacement) from {1,2, …, n} be denoted by {jc,1 jc,2, …, Jc,n}. Thus we are resampling the experimental units within each category separately rather than using the same set of sampling units across all categories as was done above for the longitudinal data. Then for gene g, the bootstrap sample that honors the null hypothesis while maintaining any dependence structure in the data is given by , i = 1,2, …, n, c = 1,2, …, C. Using this bootstrap sample we construct the bootstrap statistic which yields the bootstrap null distribution. Thus the proposed methodology is applicable for heteroscedastic gene expression data.
3. SIMULATION STUDY
A simulation study was conducted to evaluate the performance of the proposed methodology in terms of the control of false discovery rate (FDR) and the power in identifying the true non-nulls. We compared our procedure with the EDGE procedure developed by Storey et al. (2005). We generated data according to the following linear mixed effects model which is somewhat consistent with the underlying assumptions of Storey’s methodology (Storey et al., 2005).
Yg,c,i =θg,c + αi + εg,c,i, g = 1,2, …, G, c = 1,2, …, C, i = 1,2, …, n, where
| (4) |
| (5) |
Note that (4) induces intra-chip dependence as well as temporal dependence and (5) induces heteroscedasticity. All random variables described above are assumed to be independently distributed. We considered an experiment where microarray data were obtained on a random sample of n = 6 subjects over C = 6 ordered categories. Each microarray chip consisted of G = 16000 probes of which 12000 had null pattern (i.e., θg,c = 0 g = 1,2, …, 12000, c = 1,2, …, 6) and 4000 had non-null patterns described in Table 1. Each non-null pattern had 400 probes. Patterns of parameters in equations (4) and (5) that describe the variability and correlation structure are provided in Table 2. Although a broad collection of patterns of various parameters were investigated, we provide a small subset of our simulation study.
Table 1.
Patterns of mean expressions of non-null probes
| Pattern name | Mean vector (θ1, θ2, θ3, θ4, θ5, θ6) |
|---|---|
| Increasing | (0,0.5,1,1.5,2,2.5) |
| Deceasing | (0,−0.5,−1,−1.5,−2,−2.5) |
| Umbrella Peak at 2 | (0,0.5,0,−0.5,−1,−1.5) |
| Umbrella Peak at 3 | (0,0.5,1,0.5,0,−0.5) |
| Umbrella Peak at 4 | (0,0.5,1,1.5,1,0.5) |
| Umbrella Peak at 5 | (0,0.5,1,1.5,2,1.5) |
| Inverted Umbrella Min at 2 | (0,−0.5,0,0.5,1,1.5) |
| Inverted Umbrella Min at 3 | (0,−0.5,−1,0−.5,0,0.5) |
| Inverted Umbrella Min at 4 | (0,−0.5,−1,−1.5,−1,−0.5) |
| Inverted Umbrella Min at 5 | (0,−0.5,−1,−1.5,−2,−1.5) |
Table 2.
Patterns of parameters describing different variance structures. Within each case, for each value of σ, 6000 null probes and 2000 non-null probes were selected. Thus each case consists of a total of 16000 probes per microarray.
| Case | (d1,g,c, d2,g,c ) | σ | Case | (d1,g,c, d2,g,c) | σ | ||
|---|---|---|---|---|---|---|---|
| (a1,a2) = (5,0.2) | (a1, a2) = (5,1) | ||||||
| Null | Non-null | Null | Non-null | ||||
| 1 | (c, 0.2) | ( ) | 0.2 |
5 | (2, .1c) | (2, .1| .1 + θg,c |) | 0.2 |
| 1 | 1 | ||||||
| 2 | (c, 0.2) | (c, 1/c) | 0.2 |
6 | (2, .1c) | (2, .1| .1 + θg,c |2) | 0.2 |
| 1 | 1 | ||||||
| 3 | (c2, 0.2) | ( ) | 0.2 |
7 | (2, .1c2) | (2, 1| .1 + θg,c |) | 0.2 |
| 1 | 1 | ||||||
| 4 | (c2, 0.2) | ( ) | 0.2 |
8 | (2, .1c2) | (2, .1| .1 + θg,c |2) | 0.2 |
| 1 | 1 | ||||||
A software package called ORIOGEN 3.0 was developed by SDP and programmed by SH for the methodology described in this paper and is currently in use. We used this software for simulations reported in this paper. The results are based on 10,000 bootstrap samples per gene. The nominal FDR level was set at 0.05. Similarly, EDGE 1.1.291 was used to evaluate the procedure of Storey et al. (2005). Throughout this paper EDGE 1.1.291 will be referred to EDGE and ORIOGEN 3.0 will be referred to as ORIOGEN. Since EDGE uses q-values, we evaluated EDGE at a nominal q-value of 0.05, which in our experience approximates an FDR nominal level of 0.05. We implemented EDGE using 500 bootstrap samples.
Based on the results of the simulation study for the 8 cases summarized in Table 3 and others (not reported in the paper) we find that the proposed methodology controls the FDR at the nominal level of 0.05, although it can be conservative. In our experience its FDR never exceeded the nominal level. Although the EDGE procedure of Storey et al. (2005) enjoys larger power than the proposed methodology, it tends to have an inflated FDR especially when there is a significant amount of heteroscedasticity among the null genes.
Table 3.
FDR and Power comparisons between ORIOGEN and EDGE
| Case | ORIOGEN | EDGE | ||
|---|---|---|---|---|
| FDR | Power | FDR | Power | |
| 1 | 0.022 | 0.795 | 0.054 | 0.926 |
| 2 | 0.020 | 0.616 | 0.048 | 0.765 |
| 3 | 0.038*(.051, .035, .028, .041, .036) | 0.79*( .82, .81, .74, .80, .78) | 0.091*(.096, .082, .097, .091, .090) | 0.918*( .919, .935, .912, .911, .912) |
| 4 | 0.043 | 0.640 | 0.098 | 0.712 |
| 5 | 0.015 | 0.937 | 0.052 | 0.989 |
| 6 | 0.020 | 0.774 | 0.051 | 0.918 |
| 7 | 0.027 | 0.940 | 0.080 | 0.988 |
| 8 | 0.033 | 0.774 | 0.085 | 0.916 |
average of the numbers that are in the parenthesis
Note that in an ideal simulation study we should repeat this simulation for several thousand random realizations for each case considered in Table 2. However, neither ORIOGEN nor EDGE is designed to run a simulation study and it is practically impossible to run ORIOGEN and EDGE one random realization at a time for a large number of times. However, we arbitrarily selected case 3 in Table 2 and generated 5 sets of microarrays with 16000 genes each and ran ORIOGEN and EDGE 5 times each. The estimated FDRs and powers for each of the 5 sets of random realizations are provided in Table 3 in the row corresponding to case 3. From these numbers it is clear that the FDR estimates for ORIOGEN and EDGE are fairly stable with standard deviations of 0.0085 and 0.0061, respectively.
4. ILLUSTRATION
We also illustrate the proposed methodology by re-analyzing the data of Ferre et al. (2007). They observed that patients receiving treatments/drugs by intramuscular (IM) injections may experience some adverse reactions or outcomes. Among infants IM injections could lead to serious complications. Since not much was known about the molecular pathways involved in local skeletal muscle injury due to IM injections, Ferre et al. conducted a longitudinal gene expression study using 10 male piglets weighing between 23 and 32 kg. Each piglet was injected with 4ml of Propylene Glycol at 4 time points (6 hours, 2 days, 7 days, and 21 days before sacrifice) in the lumbar region and cDNA microarrays were obtained using injected muscles and the non-injected sites. Thus on each piglet 5 microarray chips were obtained, one at the baseline and one each at 6 hours, 2 days, 7 days and 21 days before sacrifice, respectively. In the analysis reported in their paper, Ferre et al. analyzed 1651 clones that correspond to genes with known accession numbers. They discovered 324 genes to have “variable expression” using a p-value of 0.0125. Of these 324, they found 153 to have a differential expression with a fold change more than 1.5 (in either direction) in at least one time interval. The standard t-test was used for making each pair-wise comparison between neighboring time intervals. Using these results, a “global” pattern of expression for each gene over the 21 day time interval was provided. Thus, among genes that were declared to be significant, Ferre et al. were interested in identifying their pattern of expression over time. Furthermore from the summaries provided in their paper, it seems that the authors were interested in clustering genes with similar pattern of expression. Professors Lefebvre and Liaubet, authors of the above publication, provided us the log-transformed normalized gene expression data, which is also available at http:/www.ncbi.nlm.nih.gov/geo/. with GEO accession number GSE3217, so that we may re-analyze the data using the methodology described in this paper.
Intrinsically Ferre et al. (2007) were interested in identifying the time course pattern for each gene. For this reason, we considered the following patterns for each gene:
-
Increasing pattern:
P1: μg,base ≤ μg,6hrs ≤ μg,2days ≤ μg,7 days ≤ μg,21days
-
Umbrella pattern with peak at 6 hours:
P2:μg,base ≤ μg,6hrs ≥ μg,2days ≥ μg,7days ≥ μ g,21 day
-
Umbrella pattern with peak at day 2:
P3: μg,base ≤ μg,6hrs ≤ μg,2days ≥ μg,7days ≥ μg,21days
-
Umbrella pattern with peak at day 7:
P4: μg,base ≤ μg,6hrs ≤ μg,2days ≤ μg,7days ≥ μg,21days
-
Decreasing pattern:
P5: μg,base ≥ μg,6hrs ≥ μg,2days ≥ μg,7days ≥ μg,21days
-
Inverted umbrella pattern with minimum at 6 hours:
P6: μg,base ≥ μg,6hrs ≤ μg,2days ≤ μg,7days ≤ μg,21days
-
Inverted umbrella pattern with minimum at day 2:
P7: μg,base ≥ μg,6hrs ≥ μg,2days ≤ μg,7days ≤ μg,21days
-
Inverted umbrella pattern with minimum at day 7:
P8: μg,base ≥ μg,6hrs ≥ μg,2days ≥ μg,7days ≤ μg,21days
Thus for each gene g we tested H0: μg,1 = μg,6hrs = μg,2days = μg,7days = μg,21days) against the alternative hypothesis Hg,a: (μg,base,μg,6hrs,μg,2days,μg,7days,μg,21days)′∈ where
. If the null hypothesis was rejected then the gene was assigned to the pattern Pj with the largest goodness of fit statistic defined in (1).
Since multiple hypotheses were being tested, we selected significant genes at an FDR of 0.05 using 10,000 bootstrap samples. Our procedure discovered a total of 429 probes. In Table 4 we provide the list of all genes selected by ORIOGEN according to the 8 different time course patterns. In many cases multiple probes of the same gene were selected. In many cases the probes of the same gene displayed different time course patterns of expression. In Table 4 corresponding to each gene we provide the number of probes that were identified in a given pattern. There were several probes for which the gene names were not known. The plot of mean expression of each probe was plotted according to the pattern of expression. These plots are provided in Figure 1.
Table 4.
List of selected genes clustered according to the pattern of expression.
| Clones with decreasing pattern | Clones with increasing pattern over time | ||
|---|---|---|---|
| Gene | Gene Id | Gene | Gene Id |
| 26S proteasome non-ATPase regulatory subunit 2 | Psmd2 | 6-phosphofructokinase type C | PFKP |
| Actin alpha skeletal muscle | Acta1 (2) * | 6S ribosomal protein L27a | Rpl27a |
| Band 4.1-like protein 2 | EPB41L2 * | Calcyclin | S1A6 |
| Creatine kinase M chain | CKM * | Cell division protein kinase 4 | CDK4 |
| Cytochrome c oxidase polypeptide VIII heart mitochondrial precursor | COX8H (2) * | Collagen alpha 1 | COL6A1 * |
| Collagen alpha 2 | COL1A2 * | ||
| Fructose-bisphosphate aldolase A | ALDOA (5) * | DNA replication licensing factor MCM2 | MCM2 |
| Glyceraldehyde-3-phosphate dehydrogenase | GAPD (2) * | DNA-binding protein inhibitor ID-3 | ID3 |
| Glycerol-3-phosphate dehydrogenase [NAD+] cytoplasmic | Gpd1 * | Fibronectin | FN1 * |
| Galectin-1 | LGALS1 (2) | ||
| Glycogen phosphorylase muscle form | PYGM * | Histone H3.3 | |
| Importin 13 | Ipo13 | Homo sapiens annexin A2 | ANXA2 |
| Myosin heavy chain skeletal muscle juvenile | MYH4 * | Hydroxyacylglutathione hydrolase | Hagh |
| Myosin light chain 1 skeletal muscle isoform | MYL1 | Ligand of Numb-protein X 2 | Lnx2 |
| Myosin light polypeptide 3 | MYL3 | Lipid phosphate phosphohydrolase 1 lipoprotein lipase | PPAP2A LPL |
| Myosin regulatory light chain 2, skeletal muscle isoform | Mylpf (2) * | Protein phosphatase 1 regulatory subunit 12A | PPP1R12A |
| NADH-ubiquinone oxidoreductase MLRQ subunit | NDUFA4 | Septin 4 | 4 S E P |
| Phosphoglycerate mutase 2 | PGAM2 (2) * | Splicing factor 3B subunit 1 | SF3B1 |
| Protein C2orf142 homolog precursor | C2orf142 | Tumor suppressor p53-binding protein 1 | |
| Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | ATP2A1 * | Tyrosine-protein kinase JAK1 Vimentine |
JAK1 (2) |
| Selenoprotein W | SEPW1 | Zinc finger protein 14 | ZNF14 |
| Triosephosphate isomerase | TPI1 | ||
| Tropomyosin 1 alpha chain | TPM1 (3) * | Clones with minimum at 6 hours | |
| Tropomyosin alpha 3 chain | TPM3 | ||
| Troponin C skeletal muscle | TNNC2 * | 72 kDa type IV collagenase precursor | MMP2 * |
| Troponin C slow skeletal and cardiac muscles | Collagen alpha 1 | COL1A1 * | |
| Troponin T fast skeletal muscle isoforms | Collagen alpha 2 | COL1A2 * | |
| Troponin T slow skeletal muscle isoforms | ? (3) | Jerky protein | Jrk |
| Pyruvate kinase isozymes M1/M2 secreted protein, acidic, cysteine-rich | PKM2 * | ||
| Clones with peak at 6 hours | Sus scrofa H19 gene | SPARC * | |
| 4S ribosomal protein S16 | Rps16 * | ||
| Ankyrin 1 | Ank1 | Clones with minimum at day 2 | |
| Ankyrin repeat domain protein 2 | Ankrd2 (2) * | ||
| Ankyrin repeat domain protein 6 | Ankrd6 * | Actin alpha skeletal muscle | Acta1 (4) * |
| BAG-family molecular chaperone regulator-3 | BAG3 * | Alpha-actinin 3 | Acta3 (2) * |
| Cytochrome c oxidase polypeptide III | MT-CO3 * | Conserved oligomeric Golgi complex component 4 | COG4 |
| GTP-binding protein RAD | RRAD * | Creatine kinase M chain | CKM * |
| Heat shock 7 kDa protein 1B | HSPA1B * | Enigma homolog | Enh |
| Multisynthetase complex auxiliary component p38 | JTV1 * | Fructose-bisphosphate aldolase A | ALDOA (2) * |
| Oligodendrocyte transcription factor 2 | Olig2 | Glycerol-3-phosphate dehydrogenase [NAD+] cytoplasmic | Gpd1 * |
| Pantophysin | SYPL * | ||
| Tyrosine-protein kinase JAK1 | JAK1 | Glycogen phosphorylase muscle form | PYGM * |
| Ubiquitin-conjugating enzyme | CDC34 | Muscle type phosphofructokinase | M-PFK * |
| E2-32 kDa complementing | Myoglobin | MB | |
| Myosin heavy chain cardiac muscle beta isoform | MYH7 (2) * | ||
| Clones with peak at day 2 | Myosin heavy chain skeletal muscle adult 2 | MYH2 * | |
| Myosin heavy chain skeletal muscle juvenile | MYH4 * | ||
| Adenylyl cyclase-associated protein 1 | CAP1 | Myosin regulatory light chain 2 ventricular/cardiac muscle isoform | MYL2 |
| Alpha crystallin B chain | |||
| Beta-2-microglobulin precursor | B2M (2) * | Myosin regulatory light chain 2, skeletal muscle isoform | Mylpf * |
| Calgizzarin | S1A11 | Nebulin | NEB * |
| Calnexin precursor | CANX | Phosphoglycerate mutase 2 | PGAM2 (5) * |
| Calpain small subunit 1 | CAPNS1 * | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | ATP2A1 * |
| Cofilin non-muscle isoform | CFL1 (3) * | Selenoprotein W | SEPW1 |
| Desmin | DES * | Triosephosphate isomerase | TPI1 * |
| DNA repair protein RAD51 homolog 1 | RAD51 | Troponin C skeletal muscle | TNNC2 (2) * |
| Dynein light chain 2A cytoplasmic | Dncl2a | Troponin T fast skeletal muscle isoforms beta/alpha | ? (3) |
| Ferritin heavy chain | FTH * | Ubiquitin-like protein FUBI | FAU |
| Ferritin light chain | FTL (2) * | ||
| ferritin, heavy polypeptide 1 | FTH1 | Clones with minimum at day 7 | |
| Gamma-aminobutyric acid | Gabarap | ||
| receptor associated protein | 4S ribosomal protein S2 | Rps2 | |
| Glia maturation factor gamma | GMFG (2) * | 5′ | Nt5m |
| Heat shock protein HSP 9-alpha | HSPCA * | 6-phosphofructokinase muscle type | PFKM |
| High mobility group protein 2 | HMGB2 | Actin alpha skeletal muscle | Acta1 (32) * |
| Legumain precursor | LGMN * | Actin aortic smooth muscle | Acta2 |
| Peptidyl-prolyl cis-trans isomerase A | PPIA * | Adenylate kinase isoenzyme 1 | AK1 |
| Protein K4 | Alpha-actinin 3 | Acta3 (6) * | |
| Tubulin alpha-ubiquitous chain | ATP synthase delta chain mitochondrial precursor | ATP5D | |
| U4/U6 small nuclear ribonucleoprotein Prp3 | Prpf3 (2) | Band 4.1-like protein 2 | EPB41L2 * |
| Beta enolase | ENO3 (11) * | ||
| Clones with peak at day 7 | Calsequestrin skeletal muscle isoform precursor | CASQ1 | |
| 26S protease regulatory subunit 6B | Carboxy-terminal domain RNA polymerase II polypeptide A small phosphatase 2 | CTDSP2 * | |
| 6S ribosomal protein L18 | RPL18 * | Creatine kinase M chain | CKM (16) * |
| ADP-ribosylation factor-like protein 7 | Arl7 | Cytochrome c oxidase polypeptide VIa-heart mitochondrial precursor | COX6A2 |
| Calmodulin | CALM2 | ||
| calponin 2 | CNN2 | Cytochrome c1 heme protein mitochondrial precursor | CYC1 |
| Co-chaperone protein HscB mitochondrial precursor | HSCB | FKSG26 protein | FKSG26 * |
| Fructose-bisphosphate aldolase A | ALDOA (6) * | ||
| Collagen alpha 1 | COL6A1 * | Glyceraldehyde-3-phosphate dehydrogenase | GAPD (12) * |
| Elongation factor 1-alpha 1 | EEF1A1 * | Glycogen phosphorylase muscle form | PYGM (4) * |
| Enabled protein homolog | ENAH | GPD; | |
| Eukaryotic translation initiation factor 2 subunit 1 | Eif2s1 | Hypothetical protein B495.5 in chromosome II | |
| Formin-binding protein 3 | FNBP3 | 11Importin 13 | IPO13 |
| Guanine nucleotide-binding protein G | GNB2 | L-lactate dehydrogenase B chain | LDHB |
| Heterogeneous nuclear ribonucleoprotein K | Myomesin 2 | MYOM2 | |
| IGF1; | Myosin heavy chain cardiac muscle beta isoform | MYH7 (2) * | |
| Leukocyte elastase inhibitor | SERPINB1 | Myosin heavy chain skeletal muscle adult 1 | MYH1 (7) * |
| Metastasis-associated protein MTA1 | Mta1 | Myosin heavy chain skeletal muscle juvenile | MYH4 (9) * |
| Palmitoyl-protein thioesterase 2 precursor | PPT2 | Myosin light polypeptide 3 | MYL3 (2) |
| Pig complement cytolysis inhibitor | Myosin-binding protein C fast-type | MYBPC2 (2) * | |
| Pleckstrin 2 | PLEK2 | NADH-ubiquinone oxidoreductase 19 kDa subunit | NDUFA8* |
| Potential carboxypeptidase-like | CPXM2 | Oxytocin receptor | OXTR |
| protein X2 precursor | Peroxiredoxin 2 | ||
| Retinol dehydrogenase type III | Phosphoglycerate mutase 2 | PGAM2 (6) * | |
| Retrovirus-related Pol polyprotein LINE-1 | Pol | Proteasome activator complex subunit 3 | PSME3 |
| S1 calcium-binding protein A16 | S1A16 | Protein C1orf8 precursor | C1orf8 |
| Syntaxin-1 | STX1 | Protein kinase C and casein kinase | PACSIN3 |
| TBC1 domain family, member 15 | TBC1D15 | substrate in neurons protein 3 | |
| Thiosulfate sulfurtransferase | TST | Protein phosphatase inhibitor 1 | Ppp1r1a |
| Thymosine beta 4 | TMSB4 * | Sarcoplasmic/endoplasmic reticulum calcium ATPase 1 | ATP2A1 * |
| Tryptophanyl-tRNA synthetase | WARS | Sterol regulatory element binding protein-1 | SREBF1 |
| Tubulin alpha-3 chain | TUBA3 | Telethonin | TCAP |
| Tropomyosin 1 alpha chain | TPM1 (4) * | ||
| Troponin C skeletal muscle | TNNC2 (22) * | ||
| Troponin I fast skeletal muscle | Tnni2 | ||
| Troponin T fast skeletal muscle isoforms | ? (2) | ||
| Troponin T fast skeletal muscle isoforms beta/alpha | |||
| Ubiquitin-conjugating enzyme E2-32 kDa complementing | CDC34 |
genes also identified by Ferre et al. (2007)
If the number of clones are more than 1 then the number is within parenthesis ( ) next to the Gene Id
Figure 1.

Time course plots of mean expression of all significant probes clustered according to their pattern.
Genes that were also identified by Ferre et al. (2007) are denoted by the asterisk (*) in Table 4. Interestingly, more than 85% of the genes identified by Ferre et al. were also identified by our procedure. The gene expression patterns were also consistent between the two methods, although the patterns shown by Ferre et al are less specific than the ORIOGEN patterns. For example, the Ferre et al pattern indicated by a single down arrow followed by three horizontal arrows could fall into any of the ORIOGEN patterns with a decreasing first segment: overall decreasing pattern or any of the inverted umbrella patterns. Several genes identified by our methodology were not identified by Ferre et al. (2007). These are also provided in Table 4 without the asterisk. There were a few probes whose gene names were ambiguous and hence were not listed in tables provided in this paper.
A large proportion of genes selected by ORIOGEN displayed either an umbrella pattern in expression with maximum at day 7 or an inverted umbrella with minimum at day 7. This is interesting in view of the pathology findings reported in Ferre et al. (2007). On day 21, the authors observed dense fibrous and collagenous tissues in the injected areas with re-generating myocites, consistent with a repairing process following muscle injury. The top 10 biological functions (At FDR = 0.05) these genes involved in are provided in Figure 3 (using Ingenuity Pathway Analysis). The lengths of horizontal bars represent the log-p value associated with the category. As expected, these genes are largely involved in functions such as skeletal muscle development, tissue morphology, tissue development etc. These results are obtained using Ingenuity Pathway Analysis software (Ingenuity Inc.)
Figure 3.

Top ten biological functions of genes with umbrella or inverted umbrella pattern at day 7.
Ferre et al. (2007), who did not control for multiple testing, found only two genes to be differentially expressed between day 7 and 21. Consequently, it is not surprising that in the Principal Component Analysis (PCA) plot in Figure 2, based on genes with fold change exceeding 1.5, day 7 and day 21 samples are clustered together. Samples from the remaining time points seem to be well clustered according within their groups. Although we see two potential outliers at the baseline which cluster with 6 hours time point. Here fold change for a given pattern is defined to be the exponential of the mean distance between the farthest linked points. For example, in the case of umbrella pattern with peak at 6 hours, for a gene g, we define fold change as exp(max(μ̂g,6hrs − μ̂g,base, μ̂g,6hrs − μ̂g,21days)).
Figure 2.

PCA plots based on probes selected by ORIOGEN with fold change larger than 1.5
Our procedure did not identify 21 genes that were identified by the original authors (see Table 5). However, it is interesting to see that according our test these genes had p-values ranging from 0.012 to 0.392.
Table 5.
Genes that were identified by Ferre et al. (2007) and not by ORIOGEN
| Gene Name | Gene ID | P-value according to ORIOGEN |
|---|---|---|
| 4S ribosomal protein S11 | Rps11 | 0.146 |
| 4S ribosomal protein S15 | Rps15 | 0.117 |
| 4S ribosomal protein S19 | Rps19 | 0.392 |
| 4S ribosomal protein S26 | Rps26 | 0.043 |
| 4S ribosomal protein S3 | RPS3 | 0.045 |
| 4S ribosomal protein S5 | RPS5 | 0.058 |
| 6S ribosomal protein L11 | Rpl11 | 0.062 |
| 6S ribosomal protein L19 | Rpl19 | 0.012 |
| 6S ribosomal protein L23a | Rpl23a | 0.046 |
| 6S ribosomal protein L8 | Rpl8 | 0.069 |
| Actinin alpha 3 | ACTN3 | 0.068 |
| ATP synthase alpha chain heart isoform mitochondrial precursor | ATP5A1 | 0.062 |
| Carboxypeptidase D precursor | CPD | 0.036 |
| Cytochrome c oxidase polypeptide VIIa-liver/heart mitochondrial precursor | COX7A2 | 0.015 |
| Inhibitor of carbonic anhydrase precursor | ICA | 0.034 |
| Major seminal plasma glycoprotein PSP-I precursor | 0.027 | |
| NF-kappaB inhibitor-like protein 1 | NFKBIL1 | 0.036 |
| Nucleoprotein TPR | TPR | 0.060 |
| Peptidyl-prolyl cis-trans isomerase like 2 | PPIL2 | 0.017 |
| Trifunctional purine biosynthetic protein adenosine-3 [Includes: Phosphoribosylamine--glycine ligase | GART | 0.021 |
| Tropomyosin beta chain | Tpm2 | 0.018 |
Note that there are several differences between the two methods. Firstly, the two methods are testing different types of hypotheses and hence it is not surprising to find some differences. Our method tests for trend in expression over time whereas the original authors performed pairwise comparisons. Secondly, our method controls the overall FDR whereas the original paper did not control for multiple testing. Lastly, our method accounts for the heteroscedasticity, any correlations within chip and also accounts for within animal correlation over time.
5. DISCUSSION
In this article we described a nonparametric bootstrap methodology based on the residuals for analyzing gene expression data which accounts for potential correlations among genes within a chip as well as temporal correlations due to repeated measurements on the same subject. The proposed methodology uses order-restricted inference based techniques developed in Peddada et al. (2003 in Peddada et al. (2006). A methodology is also proposed which is suitable for cross-sectional data when the variance within gene is not constant across ordered categories (heteroscedasticity). One could argue that it would be better to use order restricted estimates that account for the underlying covariance matrices. However there are three problems with that approach, firstly, as demonstrated in Hwang and Peddada (1994), the classical restricted maximum likelihood estimators (RMLE) may fail even if the covariance matrix is known. Secondly, typically the covariance matrix is unknown, and in gene expression studies it needs to be estimated using substantially smaller sample size relative to the number of genes. Thirdly, the correlation structure among the genes is typically unknown to the researcher which makes it difficult to model. Our hope (which is confirmed in our modest simulation study) is that bootstrapping the residuals results in a methodology which is robust to underlying correlation structures. However, we do believe that there is room for improvement in the proposed methodology and is worth exploring new methods. For instance, one could explore modifications to the proposed nonparametric bootstrap by using a suitable parametric bootstrap. Secondly, the test statistic may be modified by using a better denominator than used in this paper. Note that the standard deviation used in the denominator is not a consistent estimator of the true variance estimator of the numerator. Unfortunately, it is practically a challenging problem to derive the moments of order restricted estimates. Consequently, we are using a pooled sample variance estimator. In a bootstrap setting it may be reasonable to use such a scaling factor. However, we believe there is an opportunity to improve the methodology by using a better scaling factor than one used here. The resulting methodologies would have applications in the analysis of other high dimensional data. It would also be interesting to extend the proposed methodology to deal with designs with more than one explanatory variable, possibly some having continuous covariates. The present framework would allow such generalizations, although they may be non-trivial.
Acknowledgments
The research of SDP was supported [in part] by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01 ES101744-04). The research of OD was partially supported by the Israeli Science Foundation Grant 875/09 and this research was conducted while the author was visiting SDP at NIEHS. The research of SH was supported by the NIEHS contract N01ES55547. The authors thank Drs. Keith Shockley and Pierre Bushel (both at NIEHS, RTP, North Carolina, USA) for their comments which improved the presentation of this paper. The authors are also thankful to Professors Hervé Lefebvre (National Veterinary School of Toulouse, Toulouse, France) and Dr Laurence Liaubet (INRA, Toulouse, France) for providing the data which were analyzed in this paper.
Contributor Information
Shyamal D. Peddada, Biostatistics Branch, NIEHS, NIH, T. W. Alexander Dr. NC, 27709.
Shawn F. Harris, SRA International, Inc. Durham, NC
Ori Davidov, Department of Statistics, University of Haifa, Haifa, Israel.
References
- 1.Blanding CR, Simmons SJ, Casati P, Walbot V, Stapleton AE. Coordinated regulation of maize genes during increasing exposure to ultraviolet radiation: Identification of ultraviolet-responsive genes, functional processes and associated potential promoter motifs. Plant Biotechnology Journal. 2007;5:677–695. doi: 10.1111/j.1467-7652.2007.00282.x. [DOI] [PubMed] [Google Scholar]
- 2.Brumback BA, Rice JA. Smoothing spline models for the analysis of nested and crossed samples of curves. J Am Statist Assoc. 1998;93:961–976. [Google Scholar]
- 3.Conesa A, Nueda M, Ferrer A, Talon M. maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics. 2006;22(9):1096–1102. doi: 10.1093/bioinformatics/btl056. [DOI] [PubMed] [Google Scholar]
- 4.Efron B, Tisbshirani R. An Introduction to the Bootstrap. Chapman & Hall; New York, NY: 1993. [Google Scholar]
- 5.Ferre P, Liaubet L, Concordet D, SanCristobal M, Uro-Coste E, Tosser-Klopp G, Bonnet A, Toutain P, Hatey F, Lefebvre H. Longitudinal Analysis of Gene Expression in Porcine Skeletal Muscle After Post-Injection Local Injury. Pharmaceutical Research. 2007;24:1480–1489. doi: 10.1007/s11095-007-9266-8. [DOI] [PubMed] [Google Scholar]
- 6.Guo Wenge, Peddada Shyamal. Adaptive Choice of the Number of Bootstrap Samples in Large Scale Multiple Testing. Statistical Applications in Genetics and Molecular Biology. 2008;7(1):Article 13. doi: 10.2202/1544-6115.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamadeh HK, Jayadev S, Gaillard ET, Huang Q, Stoll R, Blanchard K, Chou J, Tucker CJ, Collins J, Maronpot R, Bushel P, Afshari C. Integration of Clinical and Gene Expression Endpoints to Explore Furan-Mediated Hepatotoxicity. Mutation Research. 2004;549:169–183. doi: 10.1016/j.mrfmmm.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 8.Hoferkamp C, Peddada SD. Estimation of parameters in linear models with heteroscedastic variances subject to order restrictions. J Multivariate Analysis. 2002;82:65–87. [Google Scholar]
- 9.Hu J, Kapoor M, Zhang W, Hamilton S, Coombes K. Analysis of dose-response effects on gene expression data with comparison of two microarray platforms. Bioinformatics. 2005;21:3524–3529. doi: 10.1093/bioinformatics/bti592. [DOI] [PubMed] [Google Scholar]
- 10.Hwang JTG, Peddada SD. Confidence Interval Estimation Subject to Order Restrictions. Annals of Statistics. 1994;22:67–93. [Google Scholar]
- 11.Ishwaran H, Rao J. Detecting differentially expressed genes in microarrays using Bayesian model selection. J Amer Stat Assoc. 2003;98:438–455. [Google Scholar]
- 12.Ishwaran H, Rao J. Spike and slab variable selection for multigroup microarray data. J Amer Stat Assoc. 2005a;100:764–780. [Google Scholar]
- 13.Ishwaran H, Rao J. Spike and slab variable selection: frequentist and Bayesian strategies. Ann Stat. 2005b;33:730–773. [Google Scholar]
- 14.Jaffrézic F, Marot G, Degrelle S, Hue I, Foulley J. A structural mixed model for variances in differential gene expression studies. Genetical Research. 2007;89:19–25. doi: 10.1017/S0016672307008646. [DOI] [PubMed] [Google Scholar]
- 15.Jensen S, Soi S, Wang L. A Bayesian approach to efficient differential allocation for resampling-based significance testing. BMC Bioinformatics. 2009;2009:10, 198. doi: 10.1186/1471-2105-10-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karlovich C, Duchateau-Nguyen G, Johnson A, McLoughlin P, Navarro M, Fleurbaey C, Steiner L, Tessier M, Nguyen T, Wilhelm-Seiler M, Caulfield J. A longitudinal study of gene expression in healthy individuals. BMC Medical Genomics. 2009;2:33. doi: 10.1186/1755-8794-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H, Tarima S, Borders A, Getchell T, Getchell M, Stromberg A. Quadratic regression analysis for gene discovery and pattern recognition for non-cyclic short time-course microarray experiments. BMC Bioinformatics. 2005;6:106. doi: 10.1186/1471-2105-6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu T, Lin N, Shi N, Zhang B. Information criterion-based clustering with order-restricted candidate profiles in short time-course microarray experiments. BMC Bioinformatics. 2009;10:146. doi: 10.1186/1471-2105-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luan Y, Li H. Clustering of time-course gene expression data using a mixed-effects model with B-splines. Bioinformatics. 2003;19:474–482. doi: 10.1093/bioinformatics/btg014. [DOI] [PubMed] [Google Scholar]
- 20.Luan Y, Li H. Model-based methods for identifying periodically expressed genes based on time course microarray gene expression data. Bioinformatics. 2004;20:332–339. doi: 10.1093/bioinformatics/btg413. [DOI] [PubMed] [Google Scholar]
- 21.Marot G, Foulley J, Jaffrézic A structural mixed model to shrink covariance matrices for time-course differential gene expression studies. Computational Statistics and Data Analysis. 2009;53:1630–1638. [Google Scholar]
- 22.Peddada SD, Lobenhofer L, Li L, Afshari C, Weinberg C, Umbach D. Gene selection and clustering for time-course and dose-response microarray experiments using order-restricted inference. Bioinformatics. 2003;19:834–841. doi: 10.1093/bioinformatics/btg093. [DOI] [PubMed] [Google Scholar]
- 23.Peddada SD, Harris S, Zajd J, Harvey E. ORIOGEN: Order Restricted Inference for Ordered Gene Expression data. Bioinformatics. 2005;21:3933–3934. doi: 10.1093/bioinformatics/bti637. [DOI] [PubMed] [Google Scholar]
- 24.Shi NZ. Maximum likelihood estimation of means and variances from normal populations under simultaneous order restrictions. J Multivariate Analysis. 1994;50:282–293. doi: 10.1006/jmva.1994.104311. [DOI] [Google Scholar]
- 25.Shi NZ, Jiang H. Maximum likelihood estimation of isotonic normal means with unknown variances. J Multivariate Analysis. 1998;64:183–195. doi: 10.1006/jmva.1997.1717. [DOI] [Google Scholar]
- 26.Simmons S, Peddada SD. Order-restricted inference for ordered gene expression (ORIOGEN) data under heteroscedastic variances. Bioinformation. 2007;1(10):414–419. doi: 10.6026/97320630001414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storey J, Xiao W, Leek J, Tompkins R, Davis R. Significance analysis of time course microarray experiments. Proc Nat Acad Sci. 2005;102:12837–12842. doi: 10.1073/pnas.0504609102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamoto E, Tada M, Murakawa K, Takada M, Shindo G, Teramoto K, Matsunaga A, Komuro K, Kanai M, Kawakami A, Fujiwara Y, Kobayashi N, Shirata K, Nishimura N, Okushiba S, Kondo S, Hamada J, Yoshiki T, Moriuchi T, Katoh H. Gene-expression profile changes correlated with tumor progression and lymph node metastasis in esophageal cancer. Clinical Cancer Research. 2004;10:3629–3638. doi: 10.1158/1078-0432.CCR-04-0048. [DOI] [PubMed] [Google Scholar]