

Abstract
Chemotherapy strategies thus far reported can result in both side effects and drug resistance. To address both of these issues at the cellular level, we report a molecular engineering strategy which employs polymeric aptamers to induce selective cytotoxicity inside target cells. The polymeric aptamers, composed of both multiple cell-based aptamers and a high ratio of dye-labeled short DNA, exploit the target recognition capability of the aptamer, enhanced cell internalization via multivalent effects, and cellular disruption by the polymeric conjugate. Importantly, the polymer backbone built into the conjugate is cytotoxic only inside cells. As a result, selective cytotoxicity is achieved equally in both normal cancer cells and drug-resistant cells. Control assays have confirmed the nontoxicity of the aptamer itself, but they have also shown that the physical properties of the polymer backbone contribute to target cell cytotoxicity. Therefore, our approach may shed new light on drug design and drug delivery.
Keywords: aptamers, polymerization, cytotoxicity
Introduction
A major concern in cancer therapeutics is the nonspecific effect of cancer drugs, which kill healthy as well as diseased cells. Thus, both in vitro and in vivo methods to achieve selective drug targeting are actively sought. Research in our laboratory has focused on the use of anti-cell aptamers to reach this goal. Similar to anti-small molecule or anti-protein aptamers, an anti-cell aptamer is a short length of single-stranded DNA (ssDNA) which binds specifically to a certain type of cancer cells.1,2 Using the method known as Cell-based Systematic Evolution of Ligands by Exponential enrichment (Cell-SELEX), a panel of aptamer probes can be selected without prior knowledge of the cell's molecular signature.3,4 When cell-based selection is coupled with their natural binding affinity, specificity, and easy modification, aptamers have shown the capacity to both efficiently recognize target cells and deliver therapeutic agents, including chemical drugs, toxins, small interfering RNAs (siRNAs) and nanomaterial-encapsulated drugs.5–9
The requirements of specific targeting and drug delivery have been met through many novel drug-conjugate formulations. However, issues of drug toxicity and resistance still present obstacles to the full realization of aptamer-directed cancer therapy.10–12 During the past few decades, polymer therapeutics, with such potential benefits as biocompatibility, have addressed these limitations by efficiently delivering conventional drugs or by integrating chemotherapy with hyperthermia methods.13–19 Furthermore, new strategies and molecular entities are continuously being introduced to counteract or diminish the side effects of drugs.20,21 However, when multiple functionalities are involved, the fabrication of the conjugates becomes correspondingly complicated and can compromise the efficacy of these drug candidates.
The next generation of cancer molecular therapy is expected to bring entirely new treatment modalities, including triggered release of cytotoxic molecules, cellular disruption, the delivery of genetic materials, and the use of heat.22–24 Among these methods, cellular disruption offers exceptional potential in treating drug-resistant cancer cells if specific uptake can be guaranteed. Therefore, we envisioned an anticancer system that obviates the drug component by utilizing the toxicity of the polymer itself after it has been selectively internalized, as facilitated by multiple cell-based aptamers. The cytotoxicity of the polymer backbone most likely arises from the cellular disruption caused by its physical size and flexibility. This paper reports the construction of a model polymeric aptamer system and the evaluation of its potential for selective anticancer therapy at the cellular level.
Acrydite™ is an attachment chemistry based on an acrylic phosphoramidite that can be added to oligonucleotides as a 5'- modification at the time of synthesis. Acrydite-modified oligonucleotides can be further incorporated into polyacrylamide during polymerization.25–27 As shown schematically in Figure 1, the conjugate was assembled by polymerization of three components using a one-step procedure. 1) A reporting element, 5'-acrydite-T10-dye-3', is introduced to maintain the appropriate configuration of the individual aptamers and provide a tracking signal for both targeting and internalization. 2) Multiple targeting elements, 5'-acrydite-aptamers, on the polymer chain facilitate cellular delivery by multivalent binding. 3) Polyacrylamide was selected as the polymer backbone based on its stability and biocompatibility.28–30 Overall, the polymeric aptamer conjugate is superior to conventional drug treatments because, as described below, the conjugate can kill both normal and drug-resistant cancer cells, yet has little effect on nontarget cells.
Figure 1. Schematic of polymeric aptamer synthesis.
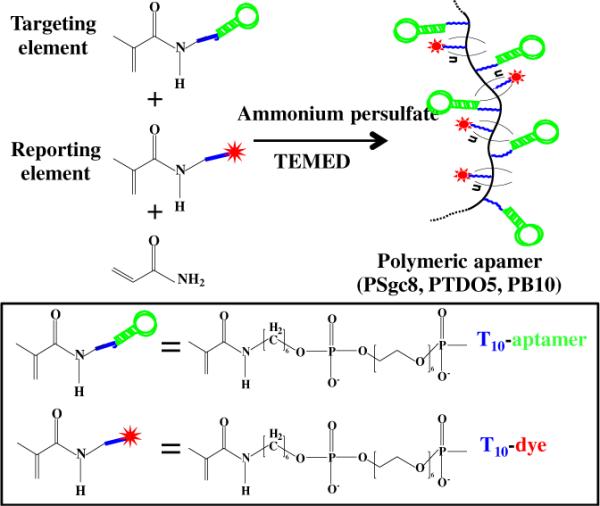
Polymerization is utilized to engineer the flexible molecular probe with multiple dye-labeled reporting elements and targeting elements.
Results and Discussion
Synthesis of the polymeric aptamer conjugate
The free aptamers used for this work were previously selected for different cancer cell lines, and they have all demonstrated high specificity and affinity.3,31 The aptamers were first modified with acrydite at the 5'-end (Table 1). After polymerization, the polymeric aptamers were purified by reversed phase HPLC to remove unbound monomer. As displayed in Figure 2a, there were three peaks, named 1, 2, and 3, corresponding to three different components from the synthesis.
Table 1.
Aptamer sequences used for the polymeric aptamer
| T10-Sgc8c | 5'-TTT TTT TTT TAT CTA ACT GCT GCG CCG CCG GGA AAA TAC TGT ACG GTT AGA-3' |
| Reporting element | 5'-TTTTTTTTTT-FAM/TMR-3' |
| T10-T2-KK1B10 | 5'-TTT TTT TTT TAC AGC AGA TCA GTC TAT CTT CTC CTG ATG GGT TCC TAT TTA TAG GTG AAG CTG T-3' |
| T10-TDO5 | 5'-TTT TTT TTT TCA TCC TAT ATA GTT CGG TGG CTG TTC ATA TTC TCC TCT CAA-3' |
Figure 2. Identification of purified polymeric aptamer where.

a) shows an original HPLC chromatogram with three elution bands, and b) displays the flow cytometry binding test. These flow results prove the functionality of different components in binding to the same target cells. Only fraction 1 in a) gives a positive binding, and the distribution of purified polymeric aptamer is displayed by Dynamic Light Scattering measurements in c).
The binding abilities of the three chromatographic fractions plus the unpurified product (0) were tested by cell cytometry. As shown in Figure 2b, compared to signals from negative Ramos cells, only component 1 gave a positive shift when incubated with CEM cells. We used a competition experiment to rule out the binding from polymer (Figure S1). Therefore, considering the spectral properties, component 1 was determined to be the purified polymeric aptamer, while 2 and 3 corresponded to free aptamer (Sgc8c) and dye-labeled T10, respectively. Light scattering experiments indicated that the size distribution was 368±94 nm with polydispersity of 0.223±0.083 in binding buffer (Figure 2c). The average MW of the polymeric aptamer was determined by fluorescence correlation spectroscopy (FCS)32 to be about 8.3×106 g/mol (shown in Supporting Information, Figure S2a, b). Furthermore, the calculated molar ratio of aptamer to reporting element was 1:20 (Experimental section, quantitation). According to the above-noted measurements, an estimated average of ~90 aptamers were present on one polymer chain. Using this strategy, aptamers T2-KK1B10, Sgc8c and TDO5, which specifically bind to K562/K562/D, CEM and Ramos cells, respectively,3,31 were incorporated into polymeric conjugates designated PB10, PSgc8 and PTDO5.
Improved binding affinity by multivalency
As shown in Figure 3, after subtracting the control signal from Ramos cells at each concentration, PSgc8 demonstrated a higher binding signal plateau (b: 75 a.u.) compared to that of free sgc8c (a: 10 a.u.) by 7.5-fold when added to target cells, CCRF-CEM. Because of its significant signal amplification, PSgc8 was also able to detect cancer cells at 1 nM aptamer concentration (inset of B, calculated at aptamer monomer concentration), while the free aptamer at an equally low concentration failed to yield a detectable signal. The improved binding ability of PB10 to K562/D cells was also demonstrated (Figure S3), indicating that the polymeric aptamer gains its specific targeting function by the presence of multiple aptamers. This is consistent with the multivalent binding shown by other molecular probe systems.33–35
Figure 3. Binding affinity of fluorescein-labeled sgc8c a) and Psgc8 probe b) to CEM cells.

The mean fluorescence intensity of target cells was obtained by subtracting the mean fluorescence intensity by nonspecific binding of each probe with Ramos cells. Inset shows corresponding binding images at 1 nM aptamer, respectively.
Specific internalization
Although little is known about cellular internalization and trafficking of polymers, some reports recently presented several approaches, including conjugation of artificial translocation domain RGD peptide and antibody to the polymer, as a means of localizing the polymer carrier.36,37 In the present study, the dye-labeled reporting element was directly used for tracking the entire conjugate. To determine the specific cellular uptake following binding, trypsin treatment and lysosensor co-location of different cancer cells were observed by confocal microscopy. In contrast to results for the negative cell line (Ramos; Figure 4, d, e and f), PB10 appeared in the lysosomes of K562/D cells with high efficiency after 180 min of incubation (Figure 4c). In another experiment, the fluorescence signal from PSgc8 inside CEM cells could still be observed after trypsin was added. Since the target protein on the cell membrane was removed by trypsin, the signal must have come from PSgc8 inside the cell (Figure S4). As expected, the uptake efficiency was found to be dose-dependent up to 250 nM of aptamers. Based on having previously shown the internalization capability of Sgc8,38 the specific cellular uptake of PSgc8 and PB10 demonstrated here proves that aptamers can guide the internalization of the macromolecule conjugates after the aptamer binds to the cell surface and that the multi-binding benefits from the polymeric design facilitate the entire process. It should be noted that the weak binding ability of TDO5 at 37 °C results in the equally weak uptake of PTD05 by Ramos, indicating that initial specific binding to the surface is necessary for polymeric aptamer internalization.
Figure 4. Internalization of PB10 by K562/D cells (upper panel) and Ramos cells (lower panel).
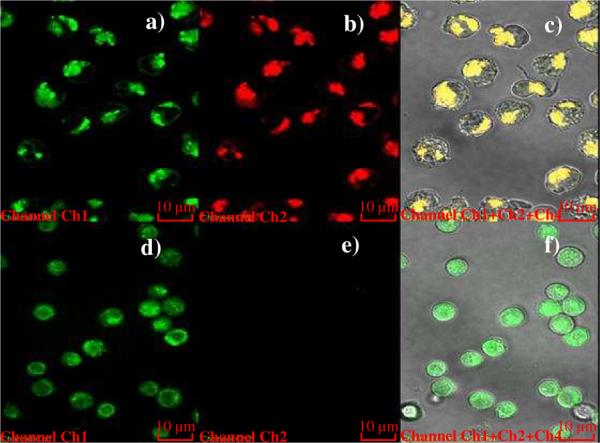
a) and d) display the fluorescence from the lysosensor, indicating that both kinds of cells can uptake lysosensor. Frames b) and e) capture the red signal of PB10, which can only be observed inside K562/D cells. c) and f) merge the signal of both lysosensor and PB10 to show that only K562/D cells uptake PB10.
Selective cytotoxicity of polymeric aptamers
The in vitro cytotoxicity of the polymeric aptamer conjugates was measured as a function of aptamer concentration using the (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay. As shown in Figure 5a, treatment of CEM (target) and Ramos (control) cells with increasing concentrations of PSgc8 leads to >50% inactivity of CEM cells after 48 h of incubation, while Ramos cells maintain relatively high viability. Also, as shown in Figure 5b, the cytotoxicity of PB10 toward both K562 and drug-resistant K562/D cell lines increases with increasing polymer concentration. Based on these results, the metabolically active fraction of K562/K562D cell population is 0.62 ± 0.10 at 135 nM aptamer. In contrast, Ramos cells in the same experiment retained 0.81± 0.09 viability. The results are consistent among all the cell lines tested (Figure S5), indicating that the polymeric conjugate can bypass the P-glycoprotein (P-gp) on the cell membrane of the drug-resistant cell line K562/D and interrupt cell metabolism. P-gp is a drug efflux transporter that reduces intracellular levels of a number of structurally related drugs.39,40
Figure 5. Cell viability test using MTS assay.

The in vitro cytotoxicity was measured after 48 h exposure to PSgc8 a) and PB10 b) with variable aptamer concentrations.
Because the MTS assay measures only cell metabolic activity, flow cytometry was utilized to ascertain the selective cytotoxicity of polymeric aptamers. Figure 6 shows two different cell populations after the aptamer-treated cells were double-stained with annexin V-FITC (Fluorescein Isothiocyanate) and PI (Propidium Iodide). This procedure differentiated live cells (not stained with either annexin V-488 or PI) from apoptotic cells (stained with both reagents). Apoptotic cells accounted for a noticeable fraction of K562/D (Figure 6b: 40.55%) and CEM (Figure 6d: 23.51%) cells after incubation with PB10 and PSgc8, respectively. In contrast, for the negative conjugate, necrosis is observed for only 12.24% (Figure 6a) and 6.65% (Figure 6c) of the cells with a 3.3- and 3.5-fold reduction in cytotoxicity, respectively. Therefore, the data obtained using the MTS assay directly correlate to the data obtained by double-stain analysis, proving selective cytotoxicity.
Figure 6. Cell viability test by using flow cytometry.

K562/D (upper) and CEM (lower) cells were treated with either PSgc8 (a), d)) or PB10 (b), c)). The Annexin V-positive and PI-positive populations indicate the proportion of apoptotic cells.
Possible mechanisms of cytotoxicity
In order to investigate possible reasons for the selective cytotoxicity of polymeric aptamers, two experiments were designed: cell proliferation screening of free aptamers to different cell lines and transfection studies using lipofectamine vector. The results of the first experiment (Figure S3) confirmed that the free aptamers do not induce obvious cytotoxicity in any of the cell lines, even at a concentration of 5 μM, suggesting that membrane protein bound by aptamer has little effect on cell viability. In the second experiment, transfection using lipofectamine vector can transfer the entire conjugate, including nonspecific PTDO5 and PSgc8, to K562/D, CEM and Ramos. Therefore, it should be easy to determine whether the polymeric backbone has an effect in inducing cytotoxicity once it enters cells by this passive delivery route. The deduced cytotoxic selectivity in every group can be observed in Figure 7. In detail, cell viability of CEM, K562/D and Ramos treated with PTDO5 decreases from 0.91± 0.09 to 0.72±0.11 after adding the lipofectamine vectors. Therefore, this result strongly supports the important cytotoxicity role played by the polyacrylamide backbone by virtue of its physical size, primary amine functionality and flexibility.41,42 It is worth pointing out that this strategy can even induce cellular disruption in drug-resistant cells by a universal mechanism, regardless of the binding receptor of the aptamer. In other words, the combination of aptamer and polymer fully utilizes the advantages of each part, i.e., selectivity and tolerable toxicity, to overcome drug-resistant cancer cells.
Figure 7. Cytotoxicity induced by transfection of polymeric aptamer.
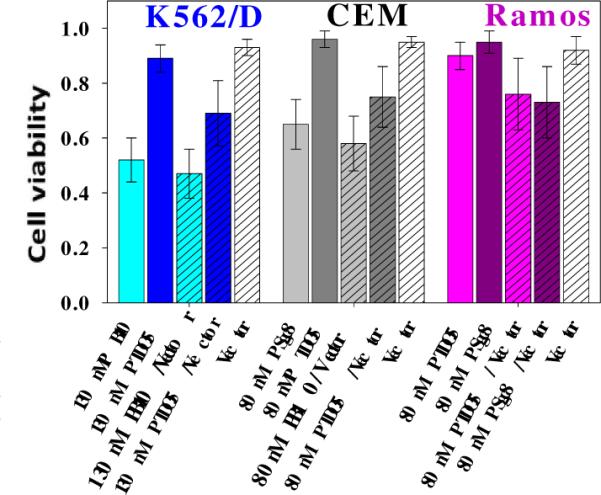
Cytotoxicity was compared before (solid) and after (hatched) using Lipofectamine to conduct passive delivery of polymeric aptamer into different cells.
Conclusion
In summary, by synthesis of polymeric aptamers that can specifically bind and be internalized by target cells, selective cytotoxicity was achieved. Because of the selectivity of the aptamer, the toxic effect of the polymeric backbone is observed only upon internalization by the target cells, including drug-resistant cells. Moreover, the effect of the conjugate on drug-resistant cells further demonstrates that cellular disruption is involved in the cytotoxicity. The polymeric backbone design facilitates multiple binding and uptake, and it is also proved to be cytotoxic after selective internalization. Another advantage of the polymer backbone arises from the potential for synthesis and processing of materials with tailored structures and enhanced properties. These features provide tremendous opportunities for refining and improving the design of the conjugate for application in vivo. Thus, our approach might find potential applications in new drug development, existing drug improvement, and drug delivery for therapy.
Experimental Section
Cell lines
CCRF-CEM (human acute lymphoblastic leukemia), Ramos, (human Burkitt's lymphoma), and K562 (chronic myelogenous leukemia) were purchased from ATCC; the doxorubicin-resistant K562 cell line (K562/D) was kindly provided by Dr. Ruoping Tang and Prof. Troy A. A. Harkness of the Department of Anatomy and Cell Biology, College of Medicine, University of Saskatchewan. All cells were cultured in RPMI 1640 medium (ATCC) supplemented with 10% FBS and 100 IU/mL penicillin-streptomycin (Cellgro).
Sample preparation
DNA sequences with acrydite coupled at the 5'-end, as shown in Table 1, were synthesized using the ABI3400 DNA/RNA synthesizer (Applied Biosystems, Foster City, CA). A ProStar HPLC (Varian, Walnut Creek, CA) with a C18 column (Econosil, 5, 250 mm) from Alltech (Deerfield, IL) was used to purify all fabricated DNA. The molecular weight obtained by MS (ThermoFinnigan (San Jose, CA) LCQ with electrospray ionization) for Sgc8c and acrydite-sgc8c was 16246 and 16469.4 respectively. The shift of 223.4 indicates a successful coupling of acrydite (MW: 247.2). For each synthesis, the first HPLC peak was quantified using a Cary Bio-300 UV spectrometer (Varian, Walnut Creek, CA).
Polymerization
1) Stock solutions of targeting element (5'-acrydite-aptamer) and reporting element (5'-acrydite-T10-dye-3') were prepared separately at 19 mM and 54 mM DNA concentration in Millipore water. Initiator and catalyst solutions were freshly prepared by adding 0.05 g ammonium persulfate (Fisher) and 25 mL tetramethylethylenediamine (TEMED) (Fisher) into 0.5 mL H2O, respectively. Air bubbles were removed using a vacuum canister for 10 min. Then 4% aqueous acrylamide was mixed with the aptamer and reporting elements with a ratio of acrylamide: aptamer: reporting element of 105: 1: 20, followed by addition of 3 % initiator and catalyst. The full mixture was kept in a vacuum system for 80 min at room temperature in the dark.
2) Polyacrylamide used for the control experiment was polymerized according to the protocol below. Acrylamide (2.5 g) was dissolved in water, and the total volume was adjusted to 50 ml (5% W/W). The monomer solution was degassed in vacuum for 10 min. Then TEMED (0.25 ml) and APS (0.5 ml) water solutions, all with the concentration of 10% (w/w), were added into the monomer solution in vacuum. Polymerization proceeded overnight at room temperature.
Purification
1) To obtain the purified conjugate, the mixture was centrifuged at 14000 rpm for 5 min. Then, HPLC was performed using a gradient from 13% acetonitrile (ACN) and 87% 0.10 M Triethylammonium acetate buffer (TEAA) to 39% ACN in 32 min.
2) Purification of polyacrylamide. The polymerization mixture (usually with conversion greater than 99.9%) from each of the polymerization methods was diluted with water from 5% (w/w) to 2 – 3% (w/w) concentration and added dropwise into a large excess of methanol (1.5 L). The precipitated polymer was collected and washed with methanol (10 times).
Quantitation
Approximate MW of PSgc8: Fluorescence Correlation Spectroscopy (FCS) was used to test the diffusion time of Rhodamine-123, free aptamer, Alexa488_antiPTK7, and Psgc8. Since the characteristic diffusion time of each species is related to its size and molecular weight, a calculation can be performed to determine the probe MW (Supporting Information).
Approximate ratio of aptamer to reporting element in polymeric aptamer: The absorbances of 5'-acrydite-T10-FAM-3' at 260 nm and 490 nm were used to calculate the molar absorptivity of FAM at 490 nm in this system. Then, the absorbances of the polymeric aptamers at 260 nm and 490 nm were recorded. Using equations (a) and (b), a 1:20 ratio of aptamer to reporting element was determined.
Flow cytometry analysis and confocal imaging
Cells were grown at a concentration of 2 × 106 mL−1 before the experiments were conducted. For the free aptamer and polymeric aptamer binding affinity measurement, cells (1 × 106 mL−1) were first washed with washing buffer (500 μL) at 4 °C, followed by staining on ice with different probes at a series of concentrations in binding buffer (200 μL) containing 10 % FBS for 20 min. After that, cells were washed again with washing buffer (500 μL) three times and suspended in 200 μL binding buffer for fluorescence detection on a FACScan cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA). The fluorescence was determined by counting 10,000 events, and data were analyzed with WinMDI software. All of the experiments for the binding assay were repeated three times. For confocal imaging, the treatment process for cell incubation was the same as described above. Considering the low stability of fluorescence dye FAM, the label was changed to carboxytetramethylrhodamine (TAMRA) in the initial synthesis. An Olympus IX-81 inverted microscope was used to image the binding effect, at 5-mW, 543-nm, with a He-Ne laser as the excitation source for TAMRA.
Competition binding test
To monitor the binding ability of pure polyacrylmide (without any aptamer or dye), a competition experiment was carried out. Briefly, 0.03 × 1 % (w/w) pure polymer was incubated with cells for 20 min on ice. Then, 2 μM aptamer (FAM-labeled) was added for 15 min further incubation. Before flow cytometric analysis, cells were washed twice with washing buffer and suspended in washing buffer (0.2 mL).
Specific internalization
Co-localization with lysosensor: To trace the cellular uptake of polymeric aptamer, K562 and Ramos cells were incubated with 100 nM of PB10 for different times: 30 min, 90 min, 180 min and overnight. Lysosensor (1 μM, Invitrogen) was added to each sample and incubated for one hour before imaging. The lysosensor traces the endocentric vesicles and eventually accumulates within the lysosomes.
Trypsin treatment: First, two batches of Psgc8 were incubated with CEM and Ramos cells (control), respectively, for 20 min on ice. After washing twice with washing buffer (500 μL) to remove the FBS (Fetal Bovine Serum), which may quench the function of Trypsin, one batch of cells was incubated with Trypsin (500 μL, 0.05 %)/EDTA (0.53 mM) in HBSS at 37 °C for 20 min. After the incubation, 50 μL FBS was added, and the cells were washed with washing buffer (500 μL) once again and suspended in binding buffer. This experiment was designed to verify the effect of Trypsin treatment on the surface-binding probe. As displayed in Figure S8, bound probe on the cell surface is removed after treatment. Meanwhile, control cells show minimal nonspecific binding.
Cell viability
MTS assays: Cytotoxicity of polymeric aptamer in four kinds of cells was determined by MTS assays using a commercially available CellTiter 96 aqueous cell proliferation assay (Promega). Before the experiment, 80000 K562, K562/D cells and 800000 CEM and Ramos cells were seeded in wells of a 96-well plate and were incubated with increasing concentrations of the polymeric aptamer in 200 μL of 1640/FBS. The medium was removed after 24 h or 48 h and replaced with a mixture containing 100 μL of fresh 1640 and 20 μL of MTS reagent solution. The absorbance of each sample was then measured at 505 nm to determine cell viability. The results are expressed as the mean percentage of cell viability relative to untreated cells. Each concentration was tested at least 3 times, and differences were considered significant at P< 0.1.
Staining cells with PI and Annexin488 for flow cytometry: Cells were treated with 80 nM polymeric aptamer and control probe for 24 h. Cells were then washed with PBS and resuspended in 100 μL of 1× annexin-binding buffer. 5 μL of Alexa Fluor® 488, annexin V, and 2 μg/ml of PI were added (Invitrogen), and the mixture was left at RT for 15 min. After incubation, PI fluorescence was detected in the FL3 channel of the cytometer, and annexin V was monitored in FL1.
Transfection
To determine if the polymer backbone affects viability in a nonspecific manner, Lipofectamine™ 2000, which is often employed as a source of efficient cationic liposomes for transfection, was used to conduct passive delivery of polymeric aptamers into different cells based on the charge interaction between the reagent and DNA segments of the conjugates. A range of 0.5 to 5 μL Lipofectamine™ 2000 was initially used per well to optimize the dose with different cell lines following the manufacturer's protocol. One μL was selected to mix with different polymeric aptamers for 15 min at room temperature. After that, the mixture was applied to CEM, K562 and Ramos cells, respectively, in a 96-well plate. Transfection was conducted for 6 h in the absence of serum, and the cells were incubated for 48 h in the presence of serum after removing the solution phase. Cell viability assay followed the methods described above.
Supplementary Material
Acknowledgment
The doxorubicin-resistant K562 cell line (K562/D) was kindly provided by Dr. Ruoping Tang and Prof. Troy A. A. Harkness in the Department of Anatomy and Cell Biology, College of Medicine, University of Saskatchewan. We acknowledge the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida. This work is supported by grants awarded by the National Institutes of Health (GM066137, GM079359 and CA133086), by the National Key Scientific Program of China (2011CB911000) and China National Grand Program (2009ZX10004-312) and by the National Natural Science Foundation of China (20975034),
Footnotes
Supporting Information Available: Results from fluorescence correlation spectroscopy, improved binding effect from PB10, internalization of Psgc8 and overview of cell viability after exposure to different kinds of aptamer components are available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Hermann T, Patel DJ. Science. 2000;287:820–825. doi: 10.1126/science.287.5454.820. [DOI] [PubMed] [Google Scholar]
- (2).Bunka DHJ, Stockley PG. Nature Reviews Microbiology. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]
- (3).Shangguan DH, Li Y, Tang ZW, Cao CZC, Chen WH, Mallikaratchy P, Sefah K, Yang CJ, Tan WH. Proc. Natl. Acad. U.S.A. 2006;103:11838–11843. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Fang XH, Tan WH. Accounts of Chemical Res. 2010;43:48–57. doi: 10.1021/ar900101s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wullner U, Neef I, Eller A, Kleines M, Tur MK, Barth S. Current Cancer Drug Targets. 2008;8:554–565. doi: 10.2174/156800908786241078. [DOI] [PubMed] [Google Scholar]
- (6).Wu YR, Sefah K, Liu HP, Wang RW, Tan WH. Proc. Natl. Acad. U.S.A. 2010;107:5–10. doi: 10.1073/pnas.0909611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Farokhzad OC, Cheng JJ, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R. Proc. Natl. Acad. U.S.A. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bagalkot VC, Farokhzad C, Langer R, Jon S. Nanomedicine-Nanotechnology Biology and Medicine. 2007;3:352–352. [Google Scholar]
- (9).McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Nat. Biothechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- (10).Eckford PDW, Sharom FJ. Chem. Rev. 2009;109:2989–3011. doi: 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- (11).Shewach DS, Kuchta RD. Chem. Rev. 2009;109:2859–2861. doi: 10.1021/cr900208x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sharma V, Pimnica-Worms D. Chem. Rev. 1999;99:2545–2560. doi: 10.1021/cr980429x. [DOI] [PubMed] [Google Scholar]
- (13).Farokhzad OC, Jon SY, Khadelmhosseini A, Tran TNT, LaVan DA, Langer R. Cancer Res. 2004;64:7668–7672. doi: 10.1158/0008-5472.CAN-04-2550. [DOI] [PubMed] [Google Scholar]
- (14).Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, Langer R, Farokhzad OC. Proc. Natl. Acad. Sci. U.S.A. 2008;105:2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chau Y, Padera RF, Dang NM, Langer R. Int. J. Cancer. 2006;118:1519–1526. doi: 10.1002/ijc.21495. [DOI] [PubMed] [Google Scholar]
- (16).Dong X, Mattingly CA, Tseng MT, Cho MJ, Liu Y, Adams VR, Mumper RJ. Cancer Res. 2009;69:3918–3926. doi: 10.1158/0008-5472.CAN-08-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Barza M, Luxenhoferb R, Zentela R, Kabanov AV. Biomaterials. 2009;30:5682–5690. doi: 10.1016/j.biomaterials.2009.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vlerken LEV, Duan Z, Little SR, Seiden MV, Amiji MM. Mol. Pharmaceutics. 2008;5:516–526. doi: 10.1021/mp800030k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mamot C, Drummond DC, Noble CO, Kallab VZ, Guo X, Hong KL, Kirpotin DB, Park JW. Cancer Res. 2005;65:11631–11638. doi: 10.1158/0008-5472.CAN-05-1093. [DOI] [PubMed] [Google Scholar]
- (20).Putnam D, Gentry CA, Pack DW, Langer R. Proc. Natl. Acad. Sci. U.S.A. 2001;98:1200–1205. doi: 10.1073/pnas.031577698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Breunig M, Lungwitz U, Liebl R, Goepferich A. Proc. Natl. Acad. Sci. U.S.A. 2007;104:14454–14459. doi: 10.1073/pnas.0703882104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Farrell D, Alper J, Ptak K, Panaro NJ, Grodzinski P, Barker AD. ACS NANO. 2010;4:589–594. doi: 10.1021/nn100073g. [DOI] [PubMed] [Google Scholar]
- (23).AshaRani PV, Mun GLK, Hande MP, Valiyaveettil S. ACS NANO. 2009;3:279–290. doi: 10.1021/nn800596w. [DOI] [PubMed] [Google Scholar]
- (24).Prasmickaite L, Hogset A, Selbo PK, Engesaeter BO, Hellum M, Berg K. British J. Cancer. 2002;86:652–657. doi: 10.1038/sj.bjc.6600138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Alemdaroglu FE, Herrmann A. Org. Biomol. Chem. 2007;5:1311–1320. doi: 10.1039/b617941j. [DOI] [PubMed] [Google Scholar]
- (26).Efimov VA, Buryakova AA, Chakhmakhcheva G. Nule. Aci. Res. 1999;27:4416–4426. doi: 10.1093/nar/27.22.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Erout MN, Troesch A, Pichot C, Cros P. Bioconjugate Chem. 1996;7:568–575. doi: 10.1021/bc960046a. [DOI] [PubMed] [Google Scholar]
- (28).Yang TH. Recent Patents on Mater. Sci. 2008;1:29–40. [Google Scholar]
- (29).Caulfield MJ, Hao, Qiao XJGG, Solomon DH. Polymer. 2003;44:1331. [Google Scholar]
- (30).Risbud MV, Bhonde RR. Drug Delivery. 2000;7:69–75. doi: 10.1080/107175400266623. [DOI] [PubMed] [Google Scholar]
- (31).Sefah K, Tang ZW, Shangguan DH, Chen H, Lopez-Colon D, Li Y, Parekh P, Martin J, Meng L, Phillips JA, Kim YM, Tan WH. Leukemia. 2009;23:235–244. doi: 10.1038/leu.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lakowicz JR. Principles of Fluorescence Spectroscopy. Edition 3 Springer; 2006. p. 806. [Google Scholar]
- (33).Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. ACS Chem. Bio. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- (34).Johnson R, Kopečková P, Kopeček J. Bioconjugate Chem. 2009;20:129–137. doi: 10.1021/bc800351m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. J. Am. Chem. Soc. 2002;124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- (36).Kolonko EM, Kiessling LL. J. Am. Chem. Soc. 2008;130:5626–5627. doi: 10.1021/ja8001716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liu J, Kova PK, Buhler P, Wolf P, Pan H, Bauer H, Elsasser-Beile U, Kopecek J. Mol. Pharmaceutics. 2009;6:959–970. doi: 10.1021/mp8002682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Xiao Z, Shangguan D, Cao Z, Fang X, Tan W. Chem. Eur. J. 2008;14:1769–1775. doi: 10.1002/chem.200701330. [DOI] [PubMed] [Google Scholar]
- (39).Smyth MJ, Krasovskis E, Sutton R, Johnstone RW. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7024–7029. doi: 10.1073/pnas.95.12.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ferrao PT, Frost MJ, Siah SP, Ashman LK. Blood. 2003;102:4499–4503. doi: 10.1182/blood-2003-01-0083. [DOI] [PubMed] [Google Scholar]
- (41).Waite CL, Sparks SM, Uhrich KE, Roth CM. BMC Biotechnology. 2009;9:10. doi: 10.1186/1472-6750-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Patil ML, Zhang M, Betigeri S, Taratula O, He H, Minko T. Bioconjugate Chem. 2008;19:1396–1403. doi: 10.1021/bc8000722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.