

Abstract
An appealing genome-wide association study design compares one large control group against several disease samples. A pioneering study by the Wellcome Trust Case Control Consortium that employed such a design has identified multiple susceptibility regions, many of which have been independently replicated. While reusing a control sample provides effective utilization of data, it also creates correlation between association statistics across diseases. An observation of a large association statistic for one of the diseases may greatly increase chances of observing a spuriously large association for a different disease. Accounting for the correlation is also particularly important when screening for SNPs that might be involved in a set of diseases with overlapping etiology. We describe methods that correct association statistics for dependency due to shared controls, and we describe ways to obtain a measure of overall evidence and to combine association signals across multiple diseases. The methods we describe require no access to individual subject data, instead, they efficiently utilize information contained in P-values for association reported for individual diseases. P-value based combined tests for association are flexible and essentially as powerful as the approach based on aggregating the individual subject data.
Keywords: GWAS, shared controls, meta-analysis, multiple testing, combining correlated P-values
INTRODUCTION
For a number of genetic associations, the Wellcome Trust Case Control Consortium (WTCCC) study [The Wellcome Trust Case Control Consortium, 2007] has established “guilt beyond a reasonable doubt” [Altshuler and Daly, 2007], promising a refreshing change from the widespread concern about the abundance of “freely associating” studies with dismal rates of replication [Cohen, 1999]. Several WTCCC associations were successfully replicated by independent studies, and the study had quickly accumulated over 700 citations by the end of 2008. Thus, the shared controls design employed by the WTCCC proved to be successful. A “News and Views” article in Nature described the design as an “instructive approach to large-scale genomic scans of this type, showing that a set of common controls can be used for a variety of diseases with relatively little loss of analytical power” [Bowcock, 2007]. The WTCCC study design used 3,000 shared controls for the seven studied diseases with about 2000 cases per disease, typed for about 500K SNPs.
The WTCCC article raised a concern about the usage of shared controls, related to the potential for misclassification: some of the shared controls may have a disease of interest, and some will develop it in the future. Other concerns with the design are related to a possibility of confounding of the case-control status with factors that affect genotyping quality (e.g., possibility of spurious results due to “plate effects”). These problems are inherently difficult to avoid.
An important issue that we focus on here is statistical: the fact of reusing a control group while testing for genetic association with different diseases creates a correlation between the results. Fortunately, various problems that stem from this fact can be addressed and taken into account efficiently when only summary data, such as association P-values, are available. Here we mainly focus on three important statistical consequences of using shared controls in association studies.
For any given SNP, the shared control design induces a correlation between association test statistics for different diseases. This correlation may lead to a substantial increase in the rate of spurious associations: an observation of a small P-value for one of the diseases greatly increases chances of finding a small P-value for a disease that used the same sample of controls. Thus, if a SNP is selected based on a small association P-value, correlation must be taken into account while computing P-values for any other diseases. We provide methods that not only quantify chances of observing a small P-value, but also give an appropriate correction, by explicitly incorporating the correlation.
Evaluation of an overall evidence for association of a SNP with multiple, related diseases must also account for the correlation. We provide two ways to combine P-values for etiologically similar diseases into an overall P-value. One of these approaches does not suffer a loss of power when the association direction “flips” between diseases, while the other approach gains power by capitalizing on the assumption of a similar association effect among diseases.
There is a multiple testing problem due to reporting P-values for several diseases at a SNP, embedded into the issue of testing multiple SNPs in a GWAS. Accounting for the correlation allows one to obtain an appropriate P-value for a given disease, adjusted for having tested multiple diseases. We investigate the effect of the correlation on the multiple testing correction and give practical recommendations for obtaining an disease-specific P-value for a SNP, adjusted for the number of diseases tested at that SNP.
The methods we will describe are not only efficient and broadly applicable, but also straightforward to apply: they make it possible for a researcher to conduct meaningful analysis while only having access to P-values, sample sizes, and in some cases, knowledge of which allele confers susceptibility (“the effect direction”).
METHODS
When a control sample is reused, association test statistics are no longer independent. In the absence of association, the asymptotic correlation for common types of chi-square association statistics (such as allelic trend test and genotypic tests) does not depend on the total sample size, or on the allele frequencies at a genetic locus. This correlation depends only on the ratio of the number of shared controls in the two studies (N0) to the number of cases, which is denoted by N1 for the first, and by N2 for the second disease group (study). For common association tests, such as the chi-square test for allele frequency differences, and the allelic trend test, the correlation is, asymptotically,
| (1) |
This correlation also holds for multiple degrees of freedom chi-square statistics. When the control samples overlap only partially, sharing N0 individuals, with N01 and N02 individuals being distinct, the correlation becomes
| (2) |
A more general expression when both case and control samples overlap and the corresponding details are given in Appendix A. Strictly, these expressions are asymptotic, however they are nearly exact in practice, for sample sizes as small as one hundred. A square root of this correlation (for a signed version of the statistics) was recently reported by Lin and Sullivan, by considering a logistic regression model [Lin and Sullivan, 2009]. When two studies share all of the controls (N01 = N02 = 0), as in the WTCCC study, then (2) reduces to (1). For several disease samples, the correlation structure is represented by the matrix of correlations, {ρij}. Dunnett’s correlation [Dunnett, 1955] derived in the context of analysis of variance is the square root of the correlation in (1). If the two case sample sizes are the same, N1 = N2, then . Thus, when there is no association in reality, a decrease in the N0/N1 ratio drives the correlation value toward 1, regardless of the total sample size, N0 + N1.
1. Conditional and adjusted P-values
The first problem created by the aforementioned correlation is best illustrated with a graph. The histogram in Figure 1A is a histogram of P-values obtained by an application of the trend test to multiple SNPs using simulated samples of sizes N0 = 3000 and N1 = 2000 under no association. Only SNPs with P-values below 0.01 were retained, thus the resulting distribution is uniform on 0 to 0.01. Next, the sample of controls was reused to conduct trend tests for the second disease (again, under no association) with independently sampled cases (N2 = 2000), using only the retained set of SNPs. The distribution of P-values in the second histogram now appears badly skewed with a large excess of small P-values. Can the proportion of P-values that are smaller than some α-level (type-I error) be evaluated theoretically? More importantly, can an observed value of P1 be used to adjust P2 so that the histogram of adjusted set of P2 is uniform on 0 to 1? The answer to both questions is “yes”.
Figure 1. Distribution of P-values under no association.

(A) P1: histogram of P-values for the first disease, while retaining P1 ≤ 0.01. (B) P2: histogram of P-values for the second disease. (C) Corrected P2, calculated by equation (B-5).
We derive the type I error rate and the conditional P-value correction from the joint distribution of the chi-square association statistics for k diseases, . This probability can be represented in terms of a multivariate normal cumulative distribution function (CDF) of the same dimension. Using this representation, the conditional P-value can be derived, Pr(Pj ≤ pj | {P−j} = {p−j}), where {P−j} denotes a set of random P-values, excluding that for disease j, and lowercase p is the observed value of the corresponding random variable. Details of this calculation are given in Appendix B.
2. Overall association with multiple diseases
In exploration of SNPs involved in multiple diseases with overlapping etiologies, the WTCCC took an approach of pooling cases of related diseases together and contrasting the resulting sample against the control group. Two P-value based methods will be considered here. First, we will describe a method for obtaining results of such a pooled analysis when only association P-values for separate diseases are available. Our approach appropriately incorporates the correlation between P-values due to shared controls, and does not require access to the genotype data. It is similar to a meta-analysis method in Lin and Sullivan [Lin and Sullivan, 2009]. Our method is simple and most powerful when the same allele is associated with every disease. It is plausible that, although the same SNP is involved in related diseases, the association direction is different for different diseases. This heterogeneity may be a consequence of either genuine difference in the underlying mechanism, or it may reflect differences in haplotype structure between disease samples [Zaykin and Shibata, 2008]. The pooled analysis would lack power in this situation because the associations with opposite directions would cancel one another. Therefore, we also propose a second method that can combine heterogeneous association signals across diseases in a stratified manner. Both alternative ways to perform the combination analysis are valuable, and the choice between the two should be determined by a researcher, depending on the respective assumptions.
Combining homogeneous effects: inverse normal method
For the pooled analysis with the assumption that the same allele is associated with every disease, the inverse normal transformation is applied first, with weights :
| (3) |
| (4) |
“Effect” here refers to the direction of association for either one of the two alleles at a SNP (e.g. minor allele). Next, these Z-scores are combined as
| (5) |
where Rij are the correlations for the signed statistics:
| (6) |
The combined P-value, pc is obtained as follows:
| (7) |
In this approach, the effect direction for one of the alleles is incorporated into calculation in order to approximate the value of an overall statistic that would have been obtained directly from the pooled data. Such statistic is approximated here by first combining one-sided statistics that depend on the effect direction (equation 4), and then by converting the result to a two-sided P-value (equation 7). The inverse normal transformation used here is a natural choice because of an asymptotically normal distribution of the one-sided statistic. It is possible to use a different transformation, such as a chi-square. However, with non-symmetric transformations, the step given by equation (4) would have to be carried out twice, for each tested effect direction. Therefore, the step in equation (7) would yield two combined two-sided P-values. The minimum of these two would have to be doubled, due to a multiple-testing penalty. Fortunately, in the case of the normal transformation the two P-values are the same, and no penalty is necessary. The choice of the weights ( ) is motivated by the goal of approximating a statistic that pools raw data, (Zraw), in a way analogous to the WTCCC analysis. The factor accounts for the fact that the variance of the unweighted score depends on the correlation structure: the conditional variance of Zi becomes equal to one when the weights are set to be equal to that factor. As the relative size of the control sample size increases, the correlation decreases, and the weights wi approach . It is the optimal weighting under independence of Zi’s, because it makes the value of the combination statistic as close as possible to that of the combined statistic based on the pooled data, Zraw. Numerator of Zraw is a mean difference for the entire data set, T̄, and denominator is its standard error, ST̄. In terms of the standard deviation, ST, and the sample size nT, we have . Consider an operation that is inverse to pooling the data. If the sample is split into two parts, then the respective statistics are . We can re-write Zraw in terms of these two means for sub-samples:
The weighted statistic is
While ST can only be approximated by using SX and SY, we can recover the numerators for the two terms in Zraw by choosing .
Combining heterogeneous effects: inverse chi-square method
Two-tailed P-values obtained with the above approach correspond to results of analysis where all case data are pooled and then contrasted against the control group. One pitfall of this approach is that it is possible for association direction to differ among the combined diseases. Simple pooling would result in cancellation of association effects, and the ensuing lack of power. In this case we would like to combine correlated two-sided P-values directly. One approach is to consider one of the usual transformations of P-values, such as Fisher’s −2 log(P), or the inverse normal transformation [Kost and McDermott, 2002]. However, such approaches do not recover the original joint distribution, and their accuracy at small α-levels is suspect. For example, the inverse normal transformation of chi-square P-values yields normally distributed scores that are not jointly normal. Therefore, we suggest that the association test statistic should be recovered from the P-value with the inverse of its distribution. We combine dependent chi-square scores obtained with the transformation, where Ψ−1(·) denotes the inverse chi-square distribution with one degree of freedom (assuming that P-values were obtained with a one degree of freedom association statistic). As in (6), denote the matrix of correlations {Rij} by R. As before, the weights for the underlying multivariate normal scores are . The variance for the vector of weighted scores is V = diag(w) R diag(w)T, with the eigenvalues {λi}. For k diseases, the sum of weighted correlated chi-squares, , is equal to the weighted sum of independent chi-squares, where weights are the eigenvalues {λi} [Box, 1954a]. Simple methods exist for approximating the distribution of the sum of weighted chi-squares by a scaled chi-square distribution. We evaluated two such methods: one based on approximating the scale and the degrees of freedom by functions of eigenvalues of the correlation matrix [Box, 1954b]; and a different method based on matching of the first two moments [Kost and McDermott, 2002]. Neither of these simple methods provided sufficient accuracy in the extreme tail of the distribution for GWAS applications, therefore we implemented a more sophisticated approach. The distribution of the weighted sum of independent chi-squares, (equivalently, that of the sum of weighted dependent statistics, can be represented by an infinite series and evaluated to a high precision by considering a large number of terms [Kotz et al., 1967; Ruben, 1962]. We translated Farebrother’s modification of Ruben’s algorithm [Farebrother, 1984] into C++ and implemented it as a function for the popular statistical package R [R Development Core Team, 2009]. The function inputs sample sizes and P-values and evaluates the combined two-sided P-value. We verified via simulating the actual distribution that this method provides accurate P-values in the tail at least as small as 1 × 10−9.
3. Multiple testing adjustment for disease-specific P-values
Disease-specific P-values may have to be adjusted for having tested multiple diseases at a SNP. The adjustment can be derived from the joint distribution of the chi-square association statistics for k diseases, . If chi-square association statistics were independent, the Bonferroni-corrected P-value would be p* = 1 − (1 − p)k. This can be written in terms of the multivariate normal density, φ(·), as
| (8) |
where the mean vector is zero (μ = 0), the correlation R is an identity matrix, and the limits are , where Ψ−1 denotes the one degree of freedom inverse chi-square CDF. With the shared control design, statistics are correlated, and the entries in R are given by (2) instead. Then formula (8) gives the distribution of the maximum of correlated chi-square statistics, . This approach is also the basis for Dunnett’s critical values in the analysis of variance design where several treatments are compared with a reference group [Dunnett, 1955].
SIMULATION STUDIES
We evaluated precision and performance of our analytical approaches with a series of simulation experiments.
1. Simulation setup for conditional P-values
Table 1 and Figure 1 are designed to demonstrate the influence of correlation due to usage of shared controls on the distribution of P-values, and the ability of the conditional adjustment approach to appropriately correct this distribution. The empirical probabilities in Table 1 were obtained by generating trinomial samples of genotypes from populations in Hardy-Weinberg equilibrium, and conducting the Cochran-Armitage test. The control sample in each simulation was shared between the studies (diseases), while the case samples were obtained independently. Samples where the first P-value exceeded α1 were discarded. For the remaining samples, P -values for the second disease (P2) were recorded. Table 1 gives proportions of P2 that were found to be less than or equal to several values of α2, as well as the theoretical values obtained by equation (B-2). The number of simulations was at least 100,000 for each row in Table 1, not counting simulations that resulted in rejected samples. If controls were not shared, the entries in Table 1 would all be around α2. However, due to the correlation, the numbers (that represent the actual type-I error) are expected to be inflated. “Setting 4” in the table assumes that the first disease association is known to be genuine: our approach allows to utilize case/control allele frequencies, assuming that they are estimated well. In this case, we assumed a multiplicative genotypic risk for the simulations, penetrances of 0.041 and 0.048 for the two SNP alleles, and the population allele frequency of 0.155. Case and control genotype frequencies were obtained given these parameters by the Bayes rule. Multinomial samples of genotypes for simulations were obtained repeatedly, using these case and control genotype frequencies. The disease prevalence in this model is 1.8%; the expected risk allele frequencies are 0.155 (in controls) and 0.175 (in cases). This is modeling allele frequencies for a novel Crohn’s disease association in the WTCCC study (SNP rs2542151), as estimated by an independent replication study [Todd et al., 2007]. Figure 1A was constructed by generating trinomial samples of genotypes for cases and controls under no association, and by retaining only those samples where the trend test P-values for the first disease were smaller than 0.01. Samples of controls from that subset were reused, and genotype samples for the cases were sampled again to produce P-values in Figure 1B. Figure 1C was constructed by applying equation (B-3) to pairs of P-values for the two diseases, obtained by the same type of simulations.
Table 1.
Monte-Carlo probabilities Pr(P2 ≤ α2 | P1 ≤ α1) for the Cochran-Armitage test, and those obtained by the analytical approach (equ. B-2)
| Method | α2 | ||||
|---|---|---|---|---|---|
| 0.001 | 0.01 | 0.05 | 0.1 | 0.2 | |
| Setting 1 | |||||
| Simulations | 0.010 | 0.062 | 0.190 | 0.298 | 0.449 |
| Analytical | 0.011 | 0.062 | 0.193 | 0.300 | 0.450 |
|
| |||||
| Setting 2 | |||||
| Simulations | 0.038 | 0.157 | 0.370 | 0.505 | 0.660 |
| Analytical | 0.037 | 0.157 | 0.368 | 0.502 | 0.656 |
|
| |||||
| Setting 3 | |||||
| Simulations (a) | 0.011 | 0.077 | 0.248 | 0.381 | 0.555 |
| Simulations (b) | 0.010 | 0.074 | 0.247 | 0.383 | 0.556 |
| Simulations (c) | 0.011 | 0.075 | 0.245 | 0.379 | 0.554 |
| Simulations (d) | 0.013 | 0.075 | 0.249 | 0.382 | 0.552 |
| Analytical | 0.011 | 0.078 | 0.247 | 0.381 | 0.555 |
|
| |||||
| Setting 4 | |||||
| Simulations | 0.011 | 0.066 | 0.203 | 0.312 | 0.462 |
| Analytical | 0.011 | 0.066 | 0.200 | 0.309 | 0.461 |
Simulations: empirical probabilities by application of the Cochran-Armitage test to simulated samples (based on 100000 simulations).
Analytical: probabilities obtained by equation (B-2).
Setting 1: α1 = 0.01, N0=3000, N1=N2=2000; allele frequency = 0.2.
Setting 2: α1 = 10−4, N0=3000, N1=N2=2000; allele frequency = 0.2.
Setting 3: α1 = 0.05, (a) N0=300, N1=400, N2=500; allele frequency = 0.2;. (b) N0=300, N1=400, N2=500; allele frequency = 0.05; (c) partial overlap of control samples: N0 = 300, N01 = 127, N02 = 0, N1 = 996, N2 = 796, allele frequency = 0.05; (d) partial overlap of control samples: N0 = 300, N01 = 100, N02 = 100, N1 = 1500, N2 = 1643, allele frequency = 0.01.
Setting 4: α1 = 10−7, allele frequencies: q1=0.175, q0=0.153, N0=3000, N1= N2=2000.
2. Simulation setup for combined P-values
Type-I error and power for the combination methods were evaluated via a similar type of simulations. Power simulations assumed a multiplicative risk model. The population value of allele frequency for the low risk allele was 0.15. The penetrance value for the low-susceptibility allele was 0.30. The relative risk value varied between different simulation settings to ensure about 90% power for a test with the best power characteristics. Risk values and sample sizes used in each simulation setting are given in the legends of Tables 3 and 4. Assuming Hardy-Weinberg equilibrium, population genotype frequencies in cases and controls are obtained by the standard application of the Bayes rule and depend on the penetrance values of the genotypes and the allele frequency in the entire population. We used multinomial sampling with these phenotype-specific frequencies as parameters to obtain samples of cases and controls. Because we used a multiplicative model of risk, the distinction between typing a causal variant and typing a proxy SNP that tags a mutation via linkage disequilibrium is inconsequential with regard to the purpose of these simulations: under the proxy model the induced risks at the proxy SNP remain multiplicative [Zheng et al., 2009].
Table 3.
Power results for the combined analysis
| Setting | Method | ||
|---|---|---|---|
| Chi-square on raw data | Inverse normal | Inverse chi-square | |
| Power at α = 0.05 | |||
| Setting 1 | 0.915 | 0.917 | 0.912 |
| Setting 2 | 0.907 | 0.908 | 0.753 |
| Setting 3 | 0.604 | 0.603 | 0.904 |
| Setting 4 | 0.854 | 0.855 | 0.913 |
| Setting 5 | 0.269 | 0.264 | 0.977 |
| H0 (no association) | 0.0493 | 0.0492 | 0.0497 |
Setting 1: The same effect for all diseases (log-relative risk γ = 0.115); ; N0 = 3000; N1, …, N10 = 2000.
Setting 2: The same effect for all diseases (log-relative risk γ = 0.1); large N0/Ni ratio: N0 = 10000; N1, …, N10 = 500.
Setting 3: The same effect direction with effect size heterogeneity; disease-specific log-relative risks: {γi} = {0.26/i}; N0 = 3000; N1, …, N10 = 2000.
Setting 4: The same effect direction with effect size heterogeneity and large N0/Ni ratio: N0 = 10000; N1, …, N10 = 500; {γi} = {0.31/i}.
Setting 5: The same effect magnitude with the sign flipped in 3 out of 10 diseases; |γ| = 0.108; N0 = 3000; N1, …, N10 = 2000.
Table 4.
Power results for the combined analysis – variable sample sizes
| Setting | Method | ||
|---|---|---|---|
| Chi-square on raw data | Inverse normal | Inverse chi-square | |
| Power at α = 0.05 | |||
| Setting 1 | 0.917 | 0.919 | 0.915 |
| Setting 2 | 0.997 | 0.997 | 0.994 |
| Setting 3 | 0.607 | 0.607 | 0.852 |
| Setting 4 | 0.982 | 0.983 | 0.998 |
| Setting 5 | 0.282 | 0.270 | 0.970 |
| H0 (no association) | 0.0499 | 0.0498 | 0.0493 |
Setting 1: The same effect for all diseases (log-relative risk γ = 0.115); ; N0 = 3000; N1, N2, …, N10 = 400, 800, …, 4000.
Setting 2: The same effect for all diseases (log-relative risk γ = 0.1); large N0/Ni ratio: N0 = 10000; N1, N2, …, N10 = 400, 800, …, 4000.
Setting 3: The same effect direction with effect size heterogeneity; disease-specific log-relative risks: {γi} = {0.26/i}; N0 = 3000; N1, N2, …, N10 = 400, 800, …, 4000.
Setting 4: The same effect direction with effect size heterogeneity and large N0/Ni ratio: N0 = 10000; N1, N2, …, N10 = 400, 800, …, 4000; {γi} = {0.31/i}.
Setting 5: The same effect magnitude with the sign flipped in 3 out of 10 diseases; |γ| = 0.108; N0 = 3000; N1, N2, …, N10 = 400, 800, …, 4000.
3. Simulation setup for multiplicity-adjusted P-values
Table 5 was constructed to investigate conservativeness of a simple Bonferroni correction when there is a correlation between k test statistics (i.e. k diseases). Values in Table 5 were obtained by simulating k-variate equicorrelated normal vectors, with correlation ρ = 0.5 and collecting the value of the largest absolute value (X). For each k, 108 X values were drawn to build a sample from the distribution of the largest absolute value statistic. If h is the (1 − α) empirical quantile of X, then one can compute p = 1 − Ψ (h2) and p* = 1 − (1 − p)k, where Ψ denotes the one degree of freedom chi-square CDF. As correlation approaches zero, p* would approach α (in a large number of simulations), otherwise p* values, that are given in the table for ρ = 0.5, should be larger than α. The discrepancy between p* and α represents the effect of doing a simple Bonferroni correction (i.e., the effect of ignoring the correlation). For small k, we checked the simulated values by direct integration using equation (8).
Table 5.
Change in per-disease significance values when the correlation (ρ = 0.5) is ignored
| Dimension (k) | True P-value
|
||||
|---|---|---|---|---|---|
| 0.05 | 0.01 | 1E-3 | 1E-4 | 1E-5 | |
| 3 | 0.055 | 0.0106 | 1.03E-3 | 1.01E-4 | 1.01E-5 |
| 10 | 0.064 | 0.0116 | 1.11E-3 | 1.03E-4 | 1.04E-5 |
| 50 | 0.082 | 0.0137 | 1.12E-3 | 1.07E-4 | 1.05E-5 |
| 100 | 0.093 | 0.0150 | 1.23E-3 | 1.11E-4 | 1.06E-5 |
| 1000 | 0.150 | 0.0216 | 1.55E-3 | 1.30E-4 | 1.17E-5 |
| 10000 | 0.253 | 0.0335 | 2.11E-3 | 1.59E-4 | 1.33E-5 |
Entries in the table show increase in the P-value adjusted with the Bonferroni correction, compared to the “true” P that accounts for correlation between P-values for k diseases. For example, the true P=0.05 becomes 0.055 when there are three diseases.
RESULTS
Genome wide association approaches have proved to be capable of identifying novel variants that influence susceptibility to complex diseases. Moreover, these approaches have highlighted genetic loci involved in etiological overlap between diseases with shared pathogenesis, such as cardiovascular and autoimmune diseases. To find loci of etiological overlap was one of the explicit goals of the WTCCC study. One of the conclusions of the WTCCC study is that there appears to be a number of novel associations involved in diseases with common etiology. The WTCCC approach was to combine samples of cases across diseases with possible common etiologies. Finding of a strong association at a particular SNP in the combined sample, accompanied by a substantial associations of that SNP with individual diseases can be used as evidence of an involvement of the SNP variants in overlapping disease mechanisms.
1. Conditional P-values
The WTCCC study reported a novel association at the PTPN2 gene, a regulator of inflammatory response. The SNP rs2542151 showed a strong association (P = 4.6 × 10−8) with Crohn’s disease (CD), as well as with type-1 diabetes (T1D), with P = 1.9 × 10−6. Further, WTCCC study reported a weaker association with rheumatoid arthritis (RA), with P = 0.019). The combined trend test P value for all three diseases was significant with P = 9 × 10−8. These findings supported the hypothesis of overlapping pathways in the pathogenesis of these inflammatory diseases.
The weaker RA association has come to attention because of an observation of strong associations with two other inflammatory diseases, particularly with CD. However, correlation between P-values due to the shared control sample greatly inflates chances of observing a small RA P-value. Conditioning only on the CD P-value, and assuming no association for CD, the corrected RA P-value is 0.39. After taking into account both the T1D and the CD P-values, and assuming no association for these two diseases, the corrected RA P-value becomes 0.71. Note that this calculation, assuming the null hypothesis of no association, does not depend on allele frequency (equ. B-5). The associations with CD and T1D have been independently replicated and are likely genuine [Todd et al., 2007; Franke et al., 2008; Parkes et al., 2007]. Our approach allows one to incorporate these independent estimates of case-control allele frequency differences into the calculation (cf equation B-5), thus arriving at a smaller RA P-value, 0.037. In general, even when there is an association at one of the diseases, there is still considerable bias in the distribution of P-values at the other disease, as long as SNPs are selected based on the magnitude of P-values for the first disease.
The approach we developed provides an analytical way of quantifying the type-I error and gives a conditional P-value correction. A simulation study was designed to evaluate precision of the analytical approach. We considered a situation where a subset of SNPs is selected based on a P-value. The histogram of P-values in Figure 1A was obtained by an application of the trend test to samples of genotypes under no association, using sample sizes of the WTCCC study. SNPs with P-values below 0.01 were selected, producing a uniform histogram on 0 to 0.01. Selected SNPs were re-tested for association with the second disease. The resulting histogram (in the middle) shows a substantial increase in the proportion of spuriously small P-values. An application of equation (B-3) corrects the distribution, bringing it back to uniform (graph on the right). Similar results were obtained for conditioning on more than a single disease (data not shown). The actual empirical proportions of P-values for a few levels are given in Table 1 (Setting 1, first row). The table quantifies bias illustrated by Figure 1B. For example, 0.190 of P-values in the graph were smaller than 0.05. The next row (“Analytical”) shows the proportion obtained theoretically (by equation B-2). The last pair of rows (“Setting 4”) shows that there is still bias even when there is a genuine association with the first disease: there is still an increase in the type-I error at the second disease, for which there is no association. In all cases, α-levels obtained via simulations are nearly identical with the analytical results, confirming that the formulas on which the conditional P-value correction is based are highly accurate. The simulation approach is also highly computer-intensive, since the majority of P-values (P1 > α1) need to be discarded.
Rows for “Setting 3” (c,d) give results for the case of a partial overlap of control samples. Sample sizes for the partial overlap were chosen in such a way that the theoretical correlation between association statistics matched the value computed for the complete sharing (“Setting 3” a,b). Table 1 values for complete and the partial sharing are very similar, confirming that the conditional (and the joint) distribution of P-values is driven by the correlation. Because the asymptotic correlation depends on the ratios of sample sizes, these ratios are important, rather than a particular design (i.e. complete vs. partial overlap). We also varied the population allele frequency in these simulations (“Setting 3”) to confirm that results do not depend on a particular frequency. In all settings, simulation values in Table 1 agree well with the analytical computation, even in the case when the frequency is as small as 0.01.
2. Overall association with multiple diseases
Next, we applied our P-value combination approach to the signals in the autoimmune disease group of the WTCCC study. Table 2 shows results of the inverse chi-square method for a group of SNPs that show a strong association in two or all three diseases, as well as a reversal of association direction between diseases. In such situations, the WTCCC analysis which aggregated case samples into one group, may not be powerful. The “Pooled” column under the P-value heading lists P-values of that method, confirming that the signal is being lost due to the flip in the association direction. In contrast to that, the inverse chi-square method yields very small P-values in support for an overall association. The last column of the table gives gene names for the SNPs. In all cases, the listed genes appear to be involved in pathogenesis of autoimmune diseases. SNPs rs206015, rs9391858, and rs438475 reside in the genes had been independently found to be associated with autoimmune diseases [Tazi-Ahnini et al., 2003; Duvefelt et al., 2004; Feng et al., 2009]. Eleven out of 22 SNPs listed in the table are confirmed associations with T1D [Barrett et al., 2009].
Table 2.
Combined analysis of the WTCCC data
| SNP | Effect | χ2 statistic | P-value | Gene | |||||
|---|---|---|---|---|---|---|---|---|---|
| T1D | RA | CD | T1D | RA | CD | Pooled | Inverse-χ2 | ||
| rs2227127 | 0.26 | −0.14 | −0.07 | 184.66 | 50.22 | 12.89 | 0.180 | 2.95E-51 | HLA-DQA2 |
| rs9461799 | −0.25 | 0.14 | 0.07 | 175.82 | 50.12 | 12.21 | 0.203 | 2.86E-49 | TAP2 |
| rs9469240 | −0.26 | 0.13 | 0.07 | 175.59 | 44.74 | 12.17 | 0.152 | 4.25E-48 | TAP2 |
| rs9296044 | 0.25 | −0.12 | −0.08 | 167.22 | 33.97 | 14.44 | 0.117 | 1.31E-44 | HLA-DQB2 |
| rs9296043 | 0.25 | −0.11 | −0.07 | 167.35 | 31.59 | 13.97 | 0.088 | 4.65E-44 | HLA-DQB1 |
| rs1051336 | −0.20 | 0.12 | 0.03 | 138.60 | 60.88 | 4.46 | 0.127 | 3.41E-42 | HLA-DRA1 |
| rs206015 | 0.08 | −0.10 | −0.04 | 42.53 | 47.99 | 10.53 | 0.083 | 5.09E-14 | NOTCH4 |
| rs9276435 | −0.11 | 0.11 | 0.03 | 43.58 | 50.74 | 4.61 | 0.407 | 8.71E-14 | HLA-DQA1 |
| rs9391858 | 0.11 | −0.06 | −0.04 | 68.37 | 15.17 | 8.90 | 0.626 | 4.75E-13 | C6ORF10 |
| rs3177928 | 0.10 | −0.06 | −0.05 | 59.13 | 15.86 | 10.71 | 0.921 | 3.61E-12 | HLA-DRA1 |
| rs438475 | −0.08 | 0.07 | 0.03 | 36.61 | 20.21 | 5.60 | 0.707 | 3.52E-09 | NOTCH4 |
| rs4713466 | −0.05 | 0.06 | 0.04 | 14.72 | 16.07 | 6.95 | 0.228 | 5.73E-06 | HCP5 |
| rs2233967 | 0.05 | −0.06 | −0.03 | 15.92 | 16.35 | 4.72 | 0.336 | 6.98E-06 | PSORS1C1 |
| rs1894407 | −0.09 | 0.04 | 0.05 | 25.17 | 4.15 | 6.68 | 0.786 | 8.80E-06 | TAP2 |
| rs6908994 | 0.05 | 0.05 | −0.05 | 12.26 | 9.15 | 13.38 | 0.145 | 1.50E-05 | MHC |
| rs9295957 | 0.05 | 0.05 | −0.06 | 9.91 | 8.54 | 15.57 | 0.257 | 2.03E-05 | MHC |
| rs3128921 | −0.10 | 0.09 | −0.01 | 30.12 | 25.07 | 0.46 | 0.496 | 2.50E-08 | MHC |
| rs3128930 | 0.09 | −0.09 | 0.02 | 26.27 | 24.74 | 1.14 | 0.504 | 7.28E-08 | HLA-DPB1 |
| rs2844463 | −0.06 | 0.06 | −0.02 | 21.71 | 25.81 | 3.09 | 0.415 | 1.20E-07 | BAT3 |
| rs429916 | 0.05 | −0.06 | −0.01 | 17.16 | 29.29 | 0.17 | 0.658 | 3.92E-07 | HLA-DOA |
| rs1367731 | 0.06 | −0.08 | −0.01 | 18.27 | 24.77 | 0.33 | 0.551 | 1.00E-06 | HLA-DOA |
| rs2233969 | 0.08 | −0.05 | −0.03 | 28.99 | 10.30 | 3.85 | 0.916 | 1.01E-06 | C6orf10 |
Comparison of WTCCC P-values obtained by pooling case data for three autoimmune diseases with P-values obtained with the inverse chi-square method. “Effect” columns list case-control allele frequency differences.
The inverse chi-square test combines association statistics with no regard to which allele is associated. One might want to combine signals while hypothesizing that there is a common association direction among diseases with a similar etiology. Ideally, such method should mimic results of an analysis where individual data for the disease samples are pooled and contrasted against the control sample. The inverse normal method provides such a test. Figure 2 illustrates correspondence between the “ideal test” P-value (where individual WTCCC disease samples are pooled) with the combined P-value, where individual P-values for association are combined by our inverse normal approach. The graphs show an excellent agreement between the two P-values. In general, discrepancies between the two P-values are rare and seem to be confined to cases where there are flips in association direction between diseases (data not shown).
Figure 2. Correspondence of combined P-values and “true” P-values in WTCCC data for 10,000 random SNPs.

“True” P-values, computed from Zraw are on the y-axis, plotted against P-values combined by the inverse normal method with different weightings (right graph: −log10 (P) plot).
We have suggested two methods for obtaining an overall P-value that combines signals across several diseases that are thought to have a common etiology. The inverse normal method capitalizes on the assumption that the same allele confers susceptibility in every disease. Thus, we expect that when this assumption is true, the inverse normal method would be more powerful than the inverse chi-square method. Conversely, the inverse chi-square method should gain power over the inverse normal when there is a flip in the direction of association in some of the diseases. Table 3 shows power values for signals combined across k = 10 diseases, as well as proportions of rejections under no association (type I error). A value k = 3 was also tried, but the change in k did not appear to have a noticeable effect on the pattern of power values (data not shown). In all cases, power values for the chi-square test that uses pooled individual data are almost exactly the same as those for the inverse normal method. This corroborates findings by Lin and Zeng that proper meta-analysis based on summary statistics is as efficient as analysis based on individual participant data [Lin and Zeng, 2010]. As expected, the inverse normal method has a greater power than the inverse chi-square, when the population relative risk values are the same for all diseases. However, the power gain is sufficiently large only when the statistics approach independence (i.e. when the ratio of the control to the case sample size is large). At the WTCCC values (≈ 3/2 sample size ratio) there is a very little difference between the power values. Moreover, the inverse chi-square method has a greater power than the inverse normal when there is a substantial heterogeneity between disease-specific relative risks, even when the susceptibility allele is the same allele for all diseases. As with the WTCCC data, the inverse chi-square method has a clear power advantage when association signs are reversed in some of diseases. Table 4 presents a similar comparison of the methods for the case of very different sample sizes for disease groups. Pattern of power values in this case agrees with the previous scenario, where sample sizes for all disease groups are the same. Figures 3, 4 show the effect of using different weightings on performance of the inverse normal statistic in simulated data under H0. Graphs plot the “true” P-values obtained with the trend test on pooled data against the combined P-value, using different weightings. The optimal weighting is with the weights . As N0/Ni increases, the correlation approaches zero, and approaches . Given an appropriate weighting, there is a very close correspondence of combined P-values with P-values that are based on pooling individual data and conducting a single trend test. This gives additional support to findings of Lin and Zeng who advocated meta-analysis based on summary data [Lin and Zeng, 2010]. Further, our findings suggest that when the weighted inverse normal method is applied for meta-analysis of independent P-values, is a more appropriate weighting than Ni. Less efficient weighting by the sample size is being commonly used and has been previously advocated for meta-analysis of independent P-values [Whitlock, 2005].
Figure 3. Effect of different weighting on the combined analysis; large N0/Ni ratio.

“True” P-values, computed with the trend test on pooled data are on the y-axis, plotted against P-values combined with the inverse normal method, using different weightings; N0 = 5000; Ni = 100, 200, …, 700.
Figure 4. Effect of different weighting on the combined analysis; small N0/Ni ratio.

“True” P-values, computed with the trend test on pooled data are on the y-axis, plotted against P-values combined with the inverse normal method, using different weightings; N0 = 200; Ni = 100, 200, …, 700.
3. Multiple testing adjustment for disease-specific P-values
Lastly, there is an issue of significance of association for a given disease. With k diseases tested at a SNP, there is a multiple testing problem. If the P-values were independent, the adjusted value would take the usual form, p* = 1 − (1 − p)k, which is the distribution function of the minimum P-value. In the presence of correlation, we need to consider the distribution of the largest statistic among k correlated chi-square statistics (Methods section). Realistic values of correlation are not likely to be great, however, and we suggest that ignoring the correlation and doing the Bonferroni adjustment instead is not likely to affect analysis to an appreciable degree. When the control sample size is at least as large as the size of the largest case group, the largest correlation between the signed (non-squared) statistics is 0.5. Table 5 shows changes in per-disease significance levels for correlated chi-square tests when the correlation is ignored. Calculations assumed the common correlation value of 0.5 for the underlying normal scores. With three diseases, the Bonferroni correction would result in a P-value that is equal to 0.055 when the “true” P-value (that takes into account the correlation) is 0.05. The table shows that at smaller significance levels, which are more relevant in the context of GWAS, the consequence of ignoring the correlation is negligible. We considered values of k as high as 10000, to illustrate the fact that even at large k there is little change in P-values at small significance levels.
DISCUSSION
In this manuscript, we describe methods for combining and adjusting P-values in the context of the shared controls design. We suggest that P-value based analysis provides powerful means of inference for association studies that reuse control individuals.
First problem that we consider is the conditional P-value adjustment. This type of adjustment arises specifically in GWAS, when a particular SNP comes in the spotlight because of a significant P-value for a particular disease. A scenario that we consider is when case samples for several etiologically related diseases are contrasted against a common control group. Suppose that a small P-value is observed for one disease, significant at the GWAS level. Suppose that next we notice that the association P-value for a related disease at that SNP is 0.01. This value by itself would not stand out among the GWAS results. However, if these two P-values were independent, we could claim that there is support for a hypothesis of common etiology. With the shared controls design, P-values are no longer independent. Chances of observing a second P-value as small as 0.01 or smaller is no longer 1%, and can be considerably higher. Thus, without taking the correlation into account we would arrive at a spurious conclusion. Or approach allows to quantify just how likely it is to observe a small P value, given observations of small P-values for one or several related diseases and leads to a conditional P-value adjustment.
Next, we consider the problem of combining association signals across several diseases. We find that weighted versions of P-value combination methods that take into account correlation due to shared individuals are as powerful as analysis that aggregates individual genotype information. These methods are especially useful in meta-analytic applications. Association signals can be combined across distinct diseases with similar, genetically mediated etiology. The inverse chi-square method that is robust in the presence of either association heterogeneity or association direction reversal is especially useful. Alternatively, P-values obtained for a single disease and several independent case samples, contrasted against the same control group, can be combined to ascertain an overall strength of association. The proposed inverse normal method is most appropriate for this situation. The P-value combination methods described here are useful in broader contexts than just the shared controls design. These methods can be applied whenever asymptotically normal statistics, or their squared versions are used, and the correlation between the statistics is either known, or when the tests are independent. The idea of combining several two-sided P-values in a meta-analytic application by first converting them to one-sided, combining, and converting the result back to a two-sided P-value was considered previously. Overall and Rhoades (1986) considered such approach based on the Fisher combination method [Overall and Rhoades, 1986]. However, because the effect direction cannot be chosen beforehand, one would have to consider both directional hypotheses in turn with that approach, then compute two one-sided P-values, and then double the minimum of the two. Doubling is nothing more than the Bonferroni penalty, which results in a conservative test. In fact, the resulting P-value can be greater than one. With the inverse normal approach that we advocate, two combined Z-scores are identical but opposite in sign. Thus, our approach avoids the penalty, and gives a single two-sided combined P-value. Moreover, we find that in most cases, the resulting P-value is nearly the same as that provided by the overall statistic, based on pooling raw data. Our weighting approach for combined P-values gives an improvement over a previously suggested weighting by the study size for combining independent tests [Whitlock, 2005]. We suggest that for the independent tests, the optimal weighting is by the square root of the study size, that is, the weights should be proportional to the inverse of the standard error.
The last issue is that of multiple testing. Whenever tests are combined with the goal of obtaining a consensus evidence in support of a common hypothesis, there is a possibility that a significant result is driven by just one very small P-value. If P-values for k diseases are combined, one might be interested in examining individual P-values, adjusted for having made k tests. In general, taking into account the correlation between these tests results in a less conservative penalty than that provided by a simple Bonferroni adjustment. However, we find that when the shared control group is at least as large as the largest case group, the improvement over the Bonferroni adjustment is negligible, especially at small significance levels that are appropriate in the context of GWAS. Thus, we recommend that when the multiplicity adjustment is made based on testing association for k diseases, the Bonferroni adjustment is sufficient.
Recently, Lin and Sullivan described ways to perform meta-analysis by combining individual records as well as summary statistics which also allow for shared study subjects [Lin and Sullivan, 2009]. Their approaches and the approaches we describe are mutually complementary, building toward a statistical framework for comprehensive analysis of genetic data with overlapping subjects. The P-value based approach is efficient, but it is also useful because of its simplicity and broad applicability. Most of the analysis described here only requires access to association P-values and knowledge of sample sizes. The inverse normal approach has an additional obvious requirement for knowledge of the association direction: with this approach, one would not want P-values to reinforce the combined result, unless the respective effect directions are in agreement.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113. David Umbach, Gang Zheng and two anonymous reviewers provided valuable comments.
APPENDIX A
CORRELATION BETWEEN ASSOCIATION STATISTICS
We appeal to asymptotic normality of the one-sided version of common association statistics, such as the Cochran-Armitage statistic or chi-square statistics for testing the difference between two binomial proportions. Under population HWE, these statistics are themselves asymptotically equivalent [Guedj et al., 2008]. We start by establishing correlation between two differences of binomial proportions, (q01 − q11), (q02 − q12). Here, q11, q12 denote estimated allele frequency in two sample of cases, and q01, q02 denote estimated allele frequency in corresponding samples of controls. Next, assume that samples partially overlap, i.e. share N0S controls, and N1S cases, and that the population allele frequency is q, under the absence of association. The numbers of distinct controls in studies 1 and 2 are denoted by N01 and N02. For the numbers of distinct cases the notation is N11 and N12. Due to the presence of shared individuals, there is covariance between two differences, and it depends on the allele frequency:
However, allele frequencies cancel out in the corresponding correlation:
| (A-1) |
This corresponds to the correlation derived for a signed statistic in a logistic regression model by Lin and Sullivan [Lin and Sullivan, 2009].
The main focus of this paper is on the situation when only controls are reused. The formula (A-1) simplifies, and to facilitate notation, we drop the “S” subscript. Thus, from now on, N0 will denote the number of shared controls, and N1, …, Nk will denote the sample sizes of cases for diseases (or studies) 1 through k. With partially overlapping controls, the correlation formula becomes:
Next, we are interested in the correlation between asymptotically normal test statistics,
, and in the correlation between their squares, where STi stands for the standard deviation and N denotes sample size. Sample sizes are set to be equal to simplify notation, which is not essential for the argument. Denote
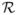 = Cor(q01 − q11, q02 − q12). Under no association,
= Cor(q01 − q11, q02 − q12). Under no association,
These approximations utilize the asymptotic normality of T̄i in computing the variance
, first order Taylor series approximations for variances and covariances of functions of random variables, and the fact that the square of a zero-mean bivariate normal pair with correlation
has the correlation
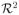 .
.
This result extends to the correlation of two chi-square statistics for 2 × C contingency tables that share one of the rows (samples). One such statistic is the case-control association statistic based on the counts of the three SNP genotype classes. Each chi-square statistic can be written in the form that depends on the sum of C squared frequency differences for the two rows,
where q̄i is the pooled frequency for the column i, and mij are sample sizes for the cell (i, j). Let the shared row be the row zero, with the sum N0 = Σ m0i. Without loss of generality, assume the sample is shared completely. Then, for any given term i of the sum above, the covariance between the two tables, considering only that term, is 2(qi(1 − qi)/N0)2, where qi is the population frequency under the H0. There is also covariance between the terms within a table, as well as covariance between the terms i, j; i ≠ j between the two tables. In the equation below, the former contributes to the covariance (the second part of the numerator); and the later to the variance (the denominator)
Thus, the correlation is equal to
.
APPENDIX B
CONDITIONAL P-VALUES AND TYPE-I ERROR: DERIVATIONS
Allowing for association with disease 1, we denote the standardized mean allele frequency difference by
| (B-1) |
where q0, q1 are population allele frequencies in controls and the disease 1 cases. Under the null hypothesis of no association, η1 = 0. First, we derive the distribution for the P-values in study 2 (P2) given that the P-value in study 1 (P1) was smaller than a given value (henceforth denoted by α1). Denote the joint bivariate normal cumulative distribution function (CDF) evaluated at X = x, Y = y by , where η1, 0 are the mean parameters, the variance is 1, and the correlation parameter is . Denote the corresponding conditional CDF of X evaluated at x, given Y = y by . Denote the one-dimensional normal CDF, with the variance 1, mean η1, and evaluated at x by Φ(x; η1). The corresponding density is denoted by φ (x; η1). Denote the one degree of freedom chi-square CDF, with the non-centrality λ, evaluated at x by Ψλ (x). The corresponding density is denoted by ψλ (x). The quantiles are denoted by the inverses, e.g. Ψ−1(x). The conditional probability that the second study P-value is less than or equal to α2 is then given by
| (B-2) |
where
The sums define regions over which the CDF should be evaluated, after taking square roots of the squared statistics. Under the null hypothesis (η1 = 0), M is just α1. As ratios N0/N1, N0/N2 increase, J approaches Pr(P1 ≤ α1) Pr(P1 ≤ α2). The probability in (B-2) gives the type-I error for the test of association with the second disease. For example, if the nominal significance level is α, then in the context of a GWAS with L tests, one can define α1 to be the Bonferroni threshold, i.e. α1 = α/L. The value α1 needs to be pre-defined, although the observed value of P2, i.e. P2 = p2 can be used in place of α2. In practice, one would like to have a way to plug in the actual P-values for the two diseases, to obtain a new P-value, corrected for correlation due to shared controls. This is accomplished with the following formula, obtained by differentiating the distribution in (B-2) with respect to the first dimension.
| (B-3) |
where
The conditioning can be extended to many P-values in two ways. The first way proceeds as before using the fact that the probability can be represented in terms of a multivariate normal CDF of the same dimension. This method has an advantage in that the effect directions do not need to be known. However, there is a disadvantage in that the formulas become increasingly complicated as k increases. The second method requires knowledge of which of the two alleles at a SNP is positively associated with a particular disease. To derive a general formula for k P-values, Pr(Pk ≤ pk | P1 = p1, P2 = p2, …, Pk−1 = pk−1), we utilize the symmetry of the normal distribution. First, the P-values, pi, are transformed to Z-scores by equations (3), (4).
The “effect” is defined as the observed direction of association for the minor allele. The scale factors for Zi’s are , where the matrix R has elements as given by equation 6. The covariance matrix of Zi, C = diag(w) R diag(w)T, can be partitioned as
Under the hypothesis of no association for disease k, the conditional distribution of Zk has the variance 1 and the mean , where Z(−k) is the vector of Zi’s with Zk omitted, and the means (η) are the standardized frequency differences, computed as in (B-1), i.e.
| (B-4) |
Under the hypothesis of no association for any of the diseases, η = 0. Then
| (B-5) |
When k = 2, this probability is the same as (B-3), which was derived directly for two-tailed statistics. Because of the symmetry of the normal transformation, the choice of which allele defines the direction is inconsequential in this approach, because the result is being converted back to a two-tailed P-value. Utilization of the allele effect simply allows one to keep track of switches in the direction of association between Zi’s.
The values η(−k) would usually be set to zero, thus assuming no association for any of the diseases. Only when some of the associations are believed to be genuine, allele frequency estimates, obtained from large independent studies, can be plugged in into (B-4). Although this would result in a less conservative adjustment, the utility of allowing for non-zero means appears limited.
Footnotes
Software implementing the methods described here is available at the NIEHS website, (http://www.niehs.nih.gov/research/atniehs/labs/bb/staff/zaykin/index.cfm), or by a request to D.V.Z.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- Altshuler D, Daly M. Guilt beyond a reasonable doubt. Nat Genet. 2007;39:813–815. doi: 10.1038/ng0707-813. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS The Type 1 Diabetes Genetics Consortium. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type I diabetes. Nature Genetics. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowcock AM. Genomics: guilt by association. Nature. 2007;447:645–646. doi: 10.1038/447645a. [DOI] [PubMed] [Google Scholar]
- Box GEP. Some theorems on quadratic forms applied in the study of analysis of variance problems, I. Effect of inequality of variance in the One-Way classification. The Annals of Mathematical Statistics. 1954a;25:290–302. [Google Scholar]
- Box GEP. Some theorems on quadratic forms applied in the study of analysis of variance problems, II. Effects of inequality of variance and of correlation between errors in the two-way classification. The Annals of Mathematical Statistics. 1954b;25:484–498. [Google Scholar]
- Cohen B. Freely associating. Nat Genet. 1999;22:1–2. doi: 10.1038/8702. [DOI] [PubMed] [Google Scholar]
- Dunnett CW. A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc. 1955;50:1096–1121. [Google Scholar]
- Duvefelt K, Anderson M, Fogdell-Hahn A, Hillert J. A NOTCH4 association with multiple sclerosis is secondary to HLA-DR*1501. Tissue Antigens. 2004;63:13–20. doi: 10.1111/j.1399-0039.2004.00135.x. [DOI] [PubMed] [Google Scholar]
- Farebrother RW. Algorithm AS 204: The distribution of a positive linear combination of χ2 random variables. Applied Statistics. 1984;33:332–339. [Google Scholar]
- Feng BJ, Sun LD, Soltani-Arabshahi R, Bowcock AM, Nair RP, Stuart P, Elder JT, Schrodi SJ, Begovich AB, Abecasis GR, Zhang XJ, Callis-Duffin KP, Krueger GG, Goldgar DE. Multiple loci within the Major Histocompatibility Complex confer risk of psoriasis. PLoS Genetics. 2009;5:e1000606. doi: 10.1371/journal.pgen.1000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, Schuldt D, Nikolaus S, Rosenstiel P, Krawczak M, Schreiber S. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–715. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- Guedj M, Nuel G, Prum B. A note on allelic tests in case-control association studies. Annals of Human Genetics. 2008;72:407–409. doi: 10.1111/j.1469-1809.2008.00438.x. [DOI] [PubMed] [Google Scholar]
- Kost JT, McDermott MP. Combining dependent P-values. Statistics and Probability Letters. 2002;60:183–190. [Google Scholar]
- Kotz S, Johnson NL, Boyd DW. Series representations of distributions of quadratic forms in normal variables. I. Central case. The Annals of Mathematical Statistics. 1967;38:823–837. [Google Scholar]
- Lin D, Zeng D. Meta-analysis of genome-wide association studies: no efficiency gain in using individual participant data. Genetic Epidemiology. 2010;34:60–66. doi: 10.1002/gepi.20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DY, Sullivan PF. Meta-analysis of genome-wide association studies with overlapping subjects. Am J Hum Genet. 2009;85:862–872. doi: 10.1016/j.ajhg.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall JE, Rhoades HM. Beware of a half-tailed test. Psychological Bulletin. 1986;100:121–122. [PubMed] [Google Scholar]
- Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, Drummond H, Lees CW, Khawaja SA, Bagnall R, Burke DA, Todhunter CE, Ahmad T, Onnie CM, McArdle W, Strachan D, Bethel G, Bryan C, Lewis CM, Deloukas P, Forbes A, Sanderson J, Jewell DP, Satsangi J, Mansfield JC, Consortium WTCC, Cardon L, Mathew CG. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2009. [Google Scholar]
- Ruben H. Probability content of regions under spherical normal distributions, IV: The distribution of homogeneous and non-homogeneous quadratic functions of normal variables. The Annals of Mathematical Statistics. 1962;33:542–570. [Google Scholar]
- Tazi-Ahnini R, Cork MJ, Wengraf D, Wilson AG, Gawkrodger DJ, Birch MP, Messenger AG, McDonagh AJG. Notch4, a non-HLA gene in the MHC is strongly associated with the most severe form of alopecia areata. Human genetics. 2003;112:400–403. doi: 10.1007/s00439-002-0898-9. [DOI] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, Lowe CE, Szeszko JS, Hafler JP, Zeitels L, Yang JHM, Vella A, Nutland S, Stevens HE, Schuilenburg H, Coleman G, Maisuria M, Meadows W, Smink LJ, Healy B, Burren OS, Lam AAC, Ovington NR, Allen J, Adlem E, Leung HT, Wallace C, Howson JMM, Guja C, Ionescu-Tirgoviste C, GET1FIN, Simmonds MJ, Heward JM, Gough SCL, Dunger DB, Wicker LS, Clayton DG Consortium TWTCC. Robust associations of four new chromosome regions from genome-wide analyses of type I diabetes. Nat Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. Journal of Evolutionary Biology. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- Zaykin DV, Shibata K. Genetic flip-flop without an accompanying change in linkage disequilibrium. Am J Hum Genet. 2008;82:794–796. doi: 10.1016/j.ajhg.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Joo J, Zaykin D, Wu C, Geller N. Robust tests in genome-wide scans under incomplete linkage disequilibrium. Statistical Science. 2009;24:503–516. doi: 10.1214/09-sts314. [DOI] [PMC free article] [PubMed] [Google Scholar]