

Abstract
Matrix Gla protein (MGP) is an inhibitor of vascular calcification but its mechanism of action and pathogenic role are unclear. This was examined in cultured rat aortas and in a model of vascular calcification in rats with renal failure. Both carboxylated (GlaMGP) and uncarboxylated (GluMGP) forms were present in aorta and disappeared during culture with warfarin. MGP was also released into the medium and removed by ultracentrifugation, and similarly affected by warfarin. In a high-phosphate medium, warfarin increased aortic calcification but only in the absence of pyrophosphate, another endogenous inhibitor of vascular calcification. Although GlaMGP binds and inactivates bone morphogenic protein (BMP)-2, a proposed mediator of vascular calcification through up-regulation of the osteogenic transcription factor runx2, neither warfarin, BMP-2, nor the BMP-2 antagonist noggin altered runx2 mRNA content in aortas, and noggin did not prevent warfarin-induced calcification. Aortic content of MGP mRNA was increased 5-fold in renal failure but did not differ between calcified and noncalcified aortas. Immunoblots showed increased GlaMGP in noncalcified (5-fold) and calcified (20-fold) aortas from rats with renal failure, with similar increases in GluMGP. We conclude that rat aortic smooth muscle produces both GlaMGP and GluMGP in tissue-bound and soluble, presumably vesicular, forms. MGP inhibits calcification independent of BMP-2-driven osteogenesis and only in the absence of pyrophosphate, consistent with direct inhibition of hydroxyapatite formation. Synthesis of MGP is increased in renal failure and deficiency of GlaMGP is not a primary cause of medial calcification in this condition.
Keywords: Calcification, Calcium, Carboxylation, Smooth Muscle, Vitamin K, Vascular, Warfarin
Introduction
Calcification of arterial smooth muscle (medial vascular calcification or Monckeberg arteriosclerosis) is a common finding in patients with advanced renal failure. The resulting decrease in vessel compliance is thought to contribute to the increased cardiovascular mortality in this condition. Medial calcification is distinct from intimal calcification, which is an inflammatory lesion associated with atherosclerosis. Although hyperphosphatemia is usually present in advanced renal failure and can clearly drive calcification, medial calcification cannot be ascribed solely to physicochemical effects of hyperphosphatemia because it also occurs in elderly individuals without renal failure or hyperphosphatemia. Abundant data indicate that deficiencies of inhibitory factors produced by vascular smooth muscle play an important role (1–5). One such factor is pyrophosphate (PPi), a direct inhibitor of hydroxyapatite formation (6) that prevents vascular calcification in vitro (1) and in vivo (7, 8). Absence of the enzyme responsible for synthesizing extracellular PPi leads to severe medial calcification both in mice (2) and humans (3). Deficiency of PPi resulting from increased hydrolysis by tissue-nonspecific alkaline phosphatase may contribute to vascular calcification in renal failure (9, 10).
Another inhibitor is matrix Gla protein (MGP),2 a vitamin K-dependent, γ-carboxylated protein present in bone and vascular smooth muscle (11). Rats treated with warfarin, a vitamin K antagonist, develop medial vascular calcification (5), and severe calcification occurs in mice with targeted deletion of the MGP gene (4). Humans with a mutant form of MGP (Keutel syndrome) also develop medial vascular calcification but it is not severe (12, 13). Calcification in MGP−/− mice is prevented by expression of MGP in smooth muscle but not liver (14), and warfarin induces calcification of cultured vascular smooth muscle cells (15), suggesting that MGP is a local inhibitor of vascular calcification.
The mechanism by which MGP inhibits medial calcification is not clearly established. Bone Gla protein (osteocalcin), a related protein, binds hydroxyapatite and inhibits its formation in vitro, and this inhibition is dependent on its γ-carboxylation (16). MGP also binds hydroxyapatite in vitro (17) and this is consistent with the fact that the content of MGP in calcified aortas is proportional to hydroxyapatite content (18) and that MGP is concentrated around hydroxyapatite deposits (19, 20). However, effects on hydroxyapatite formation are unknown. The MGP-fetuin complex in serum appears to prevent propagation of calcium phosphate precipitation (21) but the precise role of MGP in this is not clear. An alternative mechanism that has been proposed is the binding and inactivation of bone morphogenic protein 2 (BMP-2) (22, 23), a protein thought to drive osteogenic transformation of smooth muscle cells (24). However, the evidence that BMP-2 drives vascular calcification in a MGP-inhibitable manner is derived from studies in cultured vascular smooth muscle cells. Because these cells undergo a rapid and extensive phenotypic change in culture, their applicability to intact vascular smooth muscle is uncertain.
The role of MGP deficiency in vascular calcification in renal failure is equally unclear. Although increased amounts of MGP are seen at sites of medial calcification (19, 20, 25), most of this stains with antibodies directed against uncarboxylated rather than carboxylated MGP (25, 26). This has led to the hypothesis that carboxylation of MGP is impaired in states associated with medial calcification. However, this conclusion is based solely on immunohistochemistry, which can be influenced by nonspecific binding and variable exposure of epitopes, particularly if MGP is bound to hydroxyapatite. Also, immunohistochemistry cannot distinguish between intact MGP and fragments. Whether a deficiency of carboxylated MGP causes vascular calcification in renal failure remains unknown.
Despite the extensive investigation of MGP in vascular calcification, its synthesis and actions have never been studied in intact smooth muscle. In this study, a model of medial calcification in cultured rat aorta was used to examine the metabolism of MGP and its effect on calcification in the vessel wall. In addition, the role of MGP in pathologic calcification was examined in a model of vascular calcification in rats with renal failure by measuring the content of MGP mRNA and by assessing the content of carboxylated and uncarboxylated forms in aorta.
EXPERIMENTAL PROCEDURES
Aortic Culture
Aortic culture was performed as previously described (1). Briefly, aortas were removed under sterile conditions from male Sprague-Dawley rats weighing 150–300 g and adventitia was carefully removed. Adventitia was not completely removed as this leads to smooth muscle injury. The vessels were cut into 3–4-mm rings and placed in DMEM (Mediatech, Herndon, VA) without serum at 37 °C in a 5% CO2 incubator with medium changes every 3 days. The concentrations of calcium and phosphate in this medium were 1.8 and 0.9 mm, respectively. The vascular smooth muscle remains viable for at least 2 weeks under these conditions, continuing to transport ions and respond to hormones (27), and showing normal histology without apoptosis (1). To induce calcification, the phosphate concentration was increased to 3.8 mm and calf intestinal alkaline phosphatase was added at a concentration of 3.75 units/ml. To measure calcification, 0.05–0.1 μCi of 45Ca (PerkinElmer Life Sciences) was included in the culture medium. After 9 days, the aortic rings were washed 6 times in buffered salt solution and the remainder of the adventitia was removed under a dissecting microscope. The rings were extracted in 1 m HCl for 30 min and the extract was neutralized with an equal volume of 1 m NaOH. The rings were removed and dried, and radioactivity in the neutralized extract was counted by liquid scintillation. Results are expressed per dry weight of extracted aorta.
Measurement of MGP Protein
Aortas were frozen and then powdered in liquid nitrogen, and the powder homogenized in 300 μl of 4% sodium dodecyl sulfate, 0.5% Triton X-100, 0.5% sodium deoxycholate, 0.1 m dithiothreitol, 17% glycerol, 1.3% EDTA, 25 mm NaF, 5 mm sodium pyrophosphate, 75 mm NaCl, and 0.06 m Tris-HCl, pH 6.8, on ice. The homogenate was boiled for 5 min and then centrifuged at 14,000 × g for 10 min. Protein concentration was measured using the RC DC assay (Bio-Rad). Approximately 15–25 μl of supernatant (100 μg of protein) was electrophoresed on 15% polyacrylamide gels (unless otherwise indicated) using the Tris-Tricine buffer system (28). Proteins were transferred to an Immobilon PDVF membrane (Millipore, Bedford, MA) and incubated with antibodies in Tris-buffered saline without calcium. Culture medium was concentrated at least 10-fold using a Microcon 3-kDa cut-off filter (Millipore, Bedford, MA) and mixed with an equal volume of 0.1 m Tris-HCl, 8% SDS, 0.2 m dithiothreitol, and 24% glycerol prior to electrophoresis. Several antibodies were used to detect MGP. A monoclonal antibody directed against γ-carboxyglutamyl residues (nonspecific Gla antibody) was purchased from American Diagnostica (Stamford, CT). An affinity-purified polyclonal antibody specific for uncarboxylated bovine MGP was raised against a synthetic peptide containing residues 38–57 of the Gla-containing region of MGP where the 4 Gla residues were replaced with Glu residues and does not recognize Gla MGP (25). A polyclonal antibody directed against a portion of MGP that does not contain Gla residues was raised against a synthetic peptide comprising residues 6–37 (25) and was used to detect total MGP. Bands were identified with fluorescent secondary antibodies (Alexa Fluor 680; Invitrogen) and an infrared detector (LI-COR Biosciences, Lincoln, NE). Relative abundance was estimated by densitometry using software provided with the detector.
Quantitative Polymerase Chain Reaction
Aortas were frozen in liquid nitrogen, powdered, and then extracted with a guanidinium and phenol solution on ice with additional homogenization. Chloroform was added, and RNA was extracted by centrifugation. cDNA was synthesized using 2 μg of total RNA from each sample using a ThermoscriptTM cDNA Synthesis Kit (Invitrogen), and quantitative PCR was performed with an iCycler iQ Detection System using iQTM SYBR® Green Supermix (Bio-Rad). To avoid amplification of genomic DNA, primers were designed to flank at least one intron. Partial length cDNAs for QPCR standards were synthesized using a TOPO TA Cloning Kit (Invitrogen). Sequences of the primer pairs (F: forward; R: reverse) were as follow: MGP, AGGCAGACTCACAGGACACC (F), CATTTCTCCGGTTGGTGAAG (R); runx2, CTTCACAAATCCTCCCCAAG (F), GAGGCGGTCAGAGAACAAAC (R); osteopontin, CTGTCTCCCGGTGAAAGTG (F), GTCATCCGTTTCTTCAGAGG (R); osteocalcin, GCATTCTGCCTCTCTGACCT (F), CTCCAGGGGATCTGGGTAG (R); osterix, TCTCCATCTGCCTGACTCCT (F), TTCTTTGTGCCTCCTTTTCC (R); 18S RNA, GGGAGGTAGTGACGAAAAATA (F), TTGCCCTCCAATGGATCCT (R).
Enzyme Assays
Aortic rings were incubated in Hanks' buffered salt solution containing 1 mm CaCl2 and alkaline phosphatase and ectonucleotide pyrophosphorylase were measured spectrophotometrically as hydrolysis of p-nitrophenylphosphate and thymidine monophosphate p-nitrophenyl ester, respectively.
Renal Failure
Chronic renal failure and vascular calcification were produced by feeding adenine to rats (29, 30). This results in crystallization of 2,8-dihydroxyadenine in the tubules and interstitium with resulting interstitial inflammation and fibrosis and all the metabolic abnormalities of chronic renal failure (29). Rats were fed a 2.5% protein diet (Harlan Teklad, Madison, WI) with added sodium phosphate for final concentrations of 1.06% calcium and 0.92% phosphorus. Adenine was added at a concentration of 0.75% for a total of 4 weeks. Control rats were fed the same diet without adenine but the quantity was limited so that their weight loss matched that of the adenine-fed rats. Aortas were perfused with saline, harvested, and cleaned. A small section (3 mm) was removed for measurement of calcium and the remainder was frozen in liquid nitrogen for measurement of MGP or extraction of RNA as described above. The segments for calcium determination were dried, weighed, and extracted overnight in 1 m HCl. Calcium in the extract was measured colorimetrically by the cresolphthalein method (31). Plasma urea was measured colorimetrically by the urease-glutamate dehydrogenase method (ThermoDMA, Arlington, TX), plasma phosphate was measured colorimetrically by the molybdate method (32), and plasma calcium was measured by the cresolpthalein method (31).
Reagents
A monoclonal antibody directed against γ-carboxyglutamyl residues was purchased from American Diagnostica (Stamford, CT). Polyclonal antibodies specific for uncarboxylated bovine MGP (gluMGP) and a portion of MGP that does not contain Gla residues were raised against synthetic peptides and affinity purified as previously described (25). Human recombinant noggin and BMP-2 were obtained from R&D Systems (Minneapolis, MN).
Statistics
Statistical significance was determined by Student's two-tailed t test except as indicated otherwise.
RESULTS
Identification of MGP in aorta proved to be challenging because of its tissue binding and poor solubility. This required electrophoresis of samples containing large amounts of protein, often resulting in a number of extraneous bands. Immunoblotting with a monoclonal antibody directed against the γ-carboxyglutamyl (Gla) residue present in all γ-carboxylated proteins (33) recognized a 14-kDa protein in rat aortas that corresponded to GlaMGP purified from bone (Fig. 1A). The intensity of this band increased slightly when aortas were cultured with vitamin K1 and disappeared completely during culture with warfarin (Fig. 1B). The increase with vitamin K1 was not consistently observed, and large amounts of vitamin K1 only slightly reversed the effect of warfarin. None of the other bands observed on the immunoblots were affected by warfarin. An identical band was observed using an antibody directed specifically against the uncarboxylated form of MGP (GluMGP) and also disappeared after culture with warfarin (Fig. 1, C and D). This antibody occasionally recognized an additional band at ∼25 kDa that was not sensitive to warfarin and is of uncertain identity. Messenger RNA for MGP was also detected in rat aortas. The abundance increased ∼3.5-fold after 3 days in culture and was not affected by vitamin K1 or warfarin, or by raising the phosphate concentration (Fig. 2).
FIGURE 1.

MGP in rat aorta. A, immunoblot with anti-Gla antibody of extract from calcified aorta (right lane) and GlaMGP purified from bone (left lane). B, immunoblot with anti-Gla antibody of aortas cultured for 3 days in DMEM containing 10 μg/ml of vitamin K1, 25 μm warfarin, or 500 μg/ml of vitamin K1 + 25 μm warfarin. C, immunoblot with anti-GluMGP antibody of aortic extract. D, immunoblot with anti-GluMGP antibody of aortas cultured for 3 days in DMEM without or with 25 μm warfarin. Numbers on immunoblots indicate molecular mass standards in kilodaltons.
FIGURE 2.
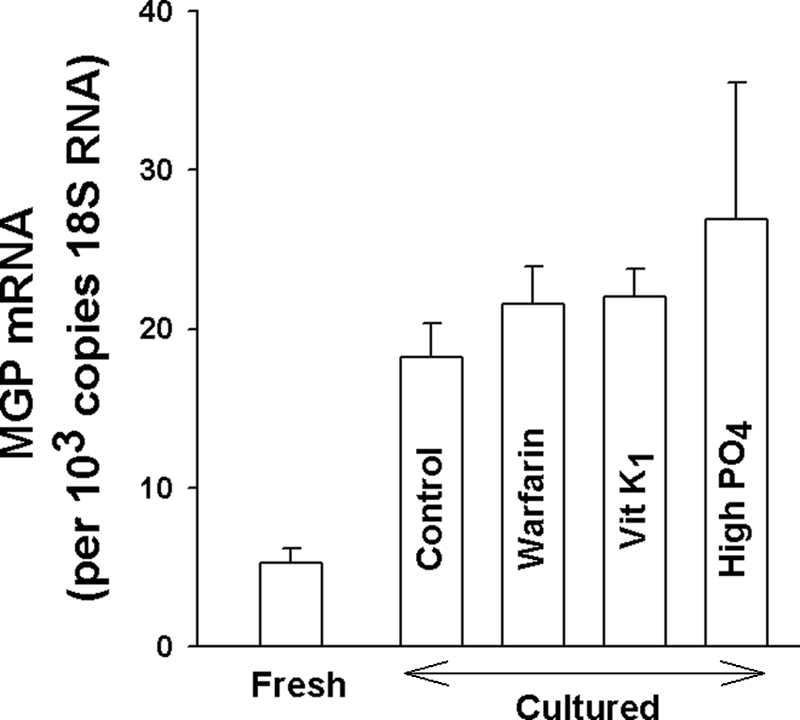
MGP mRNA in rat aorta. RNA was quantified by QPCR in fresh aortas (n = 14) and in aortas cultured in DMEM for 3 days without (n = 12) or with (n = 6) 10 μg/ml of vitamin K1, 25 μm warfarin (n = 6), or added phosphate to 3.8 mm (n = 7). There was no significant difference between mRNA content in any of the culture conditions.
Both forms of MGP were also present in the culture medium and showed the same response to warfarin (Fig. 3, A and B). Detection of MGP required that the medium be concentrated at least 10-fold. The apparent molecular weight of this band was the same as in aortic extracts, indicating that it is not a fragment of MGP. Based on the relative intensity of the staining compared with that in the aortic extract, the medium accounted for approximately half of the total MGP in the aortic cultures after 3 days. The additional band just above 20 kDa observed with the anti-Gla antibody was not affected by warfarin and remains unidentified. To determine the form of this “soluble” MGP, the culture medium was filtered through a 30-kDa cut-off cellulose filter (Amicon YM30, Millipore, Billerica, MA) and the filtrate was concentrated using a 3-kDa cut-off cellulose filter (Amicon PLCC). As shown in Fig. 3C, the medium MGP was retained by the 30-kDa filter and absent in the filtrate. The high molecular weight bands in the medium and retentate presumably represent protein aggregates that formed during concentration of the medium, but these were not routinely observed (Fig. 3, A and D, left-hand lanes). Most of the MGP was removed by ultracentrifugation and appeared in the pellet (Fig. 3D).
FIGURE 3.

MGP in culture medium. Aortas were incubated for 3 days in DMEM containing 10 μg/ml of vitamin K1, 25 μm warfarin, or 500 μg/ml of vitamin K1 + 25 μm warfarin. Medium was concentrated at least 10-fold using a 3-kDa cut-off filter prior to electrophoresis and sample volumes equivalent to the same amount of original medium were loaded in each lane for each blot. A, anti-Gla antibody. B, anti-GluMGP antibody. C, medium passed through a 30-kDa cut-off filter; anti-Gla antibody. D, medium centrifuged at 100,000 × g for 30 min; anti-Gla antibody. The pellet sample was derived from 3.5 times the volume of the medium or supernatant samples. Numbers indicate molecular mass standards in kilodaltons.
To examine the role of MGP in medial calcification, aortas were cultured in a high-phosphate medium for 9 days with and without warfarin (Fig. 4). Normal aortas do not calcify in this medium and warfarin had no effect. The small amount of 45Ca incorporated represents equilibration with existing aortic calcium (1). Because these aortas also make pyrophosphate, a potent inhibitor of vascular calcification, alkaline phosphatase was added to remove pyrophosphate as previously described (1). Under this condition, calcification was substantially increased by warfarin but was not reduced by vitamin K1 (not shown). Warfarin did not alter the activity of ectonucleotide pyrophosphorylase (Enpp1), the ectoenzyme that synthesizes pyrophosphate from extracellular ATP (0.96 ± 0.08 versus 1.12 ± 0.08 μmol/min/g in controls; n = 9) or tissue-nonspecific alkaline phosphatase, the ectoenzyme that hydrolyzes extracellular PPi (0.071 ± 0.011 versus 0.090 ± 0.007 μmol/min/g in controls; n = 6).
FIGURE 4.

Effect of warfarin on calcification of aortas in culture. Aortas were cultured for 9 days in DMEM containing 3.8 mm phosphate with or without 25 μm warfarin or 3.75 units/ml of calf intestinal alkaline phosphatase. Results are the mean ± S.E. of 7 rings.
Previous studies have proposed that MGP inhibits vascular calcification by binding and inactivating BMP-2 (22, 23), a protein thought to drive osteogenic transformation of smooth muscle cells (24). Thus, additional studies were performed to determine the role of BMP-2 and the downstream osteogenic transcription factor runx2. Runx2 mRNA was present at a very low level in freshly isolated aortas and increased during culture (Fig. 5A) but its content was not altered by warfarin, BMP-2, or the BMP-2 antagonist noggin (34), indicating that it is not under the control of MGP or BMP-2. Addition of noggin to aortic cultures did not decrease warfarin-induced calcification (Fig. 5B). Despite the increase in runx2 expression during culture, there was no detectable mRNA for osterix, another bone-specific transcription factor or for osteocalcin, a bone-specific protein, and expression of alkaline phosphatase decreased (data not shown).
FIGURE 5.

Role of BMP-2 in warfarin-induced calcification of cultured aortas. A, runx2 mRNA abundance in aortas cultured in DMEM for 3 days with or without 6.25 μm warfarin, 500 ng/ml of noggin, or 100 ng/ml of BMP2. Each bar is the mean ± S.E. of at least 3 aortas. B, calcification of aortas cultured for 9 days in DMEM containing 3.8 mm phosphate and 3.75 units/ml of alkaline phosphatase with or without 25 μm warfarin or 500 ng/ml of noggin. Results are mean ± S.E. of 12 aortic segments.
The role of MGP in vascular calcification in vivo was examined in rats with renal failure induced by feeding adenine. When fed a high-phosphate diet, some of these rats develop severe medial calcification of the aorta (35, 36). The variable calcification in this model (35, 36) does not track with the degree of renal failure or known calcification-related parameters including phosphate and is probably due to unknown genetic susceptibilities in this outbred strain. This is further evidence that vascular calcification in renal failure cannot be ascribed solely to hyperphosphatemia. However, this provided the opportunity to examine independently the effects of renal failure and calcification on aortic MGP. Also, some aortas were harvested at 16 days, after the development of renal failure but prior to the onset of calcification. Aortas were deemed calcified based on a calcium content >50 nmol/mg in a sample prior to processing for immunoblotting, a content that is not associated with staining for calcification by microscopy. Aortic calcium content and plasma chemistries are presented in Table 1. Although calcium content was significantly higher in aortas from rats with renal failure that were not considered to be calcified, the increase was minuscule compared with calcified aortas. This probably represents intracellular sequestering of calcium related to the hyperphosphatemia, which is also observed in cultured aortas (1). The plasma levels of calcium, phosphorus, or urea did not differ between rats with or without visible calcification. Immunoblotting revealed a substantial increase in total MGP (Fig. 6A) and both GlaMGP and GluMGP in noncalcified aortas from rats with renal failure compared with control aortas (Fig. 6B). Densitometry revealed that the increase in the MGP band in renal failure was similar for GlaMGP (4.6-fold) and GluMGP (3.9-fold). Calcified aortas showed an even greater increase in MGP (Fig. 6C), with the increase in band density again being similar for GlaMGP (20-fold) and GluMGP (17-fold).
TABLE 1.
Aortic calcium content and plasma chemistries in uremic and control rats
| Aortic calcium | Plasma calcium | Plasma phosphorus | Plasma urea | |
|---|---|---|---|---|
| nmol/mg | mm | mm | mm | |
| Control | 21.4 ± 3.0 | 2.02 ± 0.08 | 2.09 ± 0.08 | 8.4 ± 0.7 |
| Noncalcified uremic | 43.4 ± 3.1a | 1.43 ± 0.14a | 12.2 ± 1.2b | 57.5 ± 5.9b |
| Calcified uremic | 8079 ± 2236b | 1.79 ± 0.15 | 11.4 ± 0.92b | 64.2 ± 14.3b |
a p < 0.005 versus control.
b p < 0.0001 versus control.
FIGURE 6.

Effect of renal failure and calcification on MGP in aortas. A, immunoblot of aortas from rats with renal failure and control, pair-fed rats using a Gla-independent anti-MGP antibody. B, immunoblots of noncalcified aortas from rats with renal failure and aortas from control, pair-fed rats. The second lane from the left is from an aorta harvested at 16 days. C, immunoblots of calcified aortas and aortas from control, pair-fed rats. Numbers indicate molecular mass standards in kilodaltons. Each lane was loaded with 100 μg of protein.
To determine the mechanism for increased aortic MGP in renal failure, mRNA was measured in control aortas and in noncalcified and calcified aortas from rats with renal failure. Comparison of gene expression with and without renal failure required selection of a suitable reference gene. We studied the stability of the expression of 12 candidate reference genes in 5 uremic aortas (2 calcified) and 5 aortas from pair-fed control rats. For each candidate gene, expression levels were determined and then compared with each other gene (after logarithmic transformation). The standard deviation of ratios for each paired comparison was calculated. This yielded an M value for each gene representing its variability versus all the other genes, with the lowest M value indicating the most stable expression (37). Genes with highest M values were then excluded in a stepwise manner with recalculation of M values using the remaining genes, eventually yielding four of the most stable genes: calnexin, β-glucuronidase, ribosomal protein L13a, and cyclophylin B. Interestingly, β-actin, which is commonly used as a reference gene, had a very high M value and was the first gene eliminated. Initially, quantitative polymerase chain reaction (QPCR) data were normalized to the geometric mean of these four reference RNAs (38) but similar results were obtained when 18S RNA was used for normalization. Thus, for simplicity, 18S RNA was used to normalize all the QPCR data.
Changes in MGP gene expression in calcified and noncalcified aortas from rats with renal failure compared with control aortas are shown in Table 2. Osteopontin was included as a positive control because it is known to be up-regulated in vessels in renal failure (39) and further up-regulated in calcified vessels (40). Each group of aortas was compared with the same control aortas from rats without renal failure but in different reactions. There was a significant 5-fold increase in MGP mRNA in renal failure with no further increase in calcified aortas. Osteopontin mRNA content increased 4.5-fold in non-calcified aortas and 9.7-fold in calcified aortas, consistent with the changes in protein content previously observed (39, 40), indicating that the up-regulation of vascular osteopontin in renal failure and calcification is due to increased synthesis.
TABLE 2.
Changes in mRNA content in noncalcified and calcified aortas from rats with renal failure
Noncalcified and calcified aortas were compared to the same control aortas but in separate assays. Contents were normalized to 18S RNA. Results are means of 5 aortas.
| Non-calcified aortas |
Calcified aortas |
|||
|---|---|---|---|---|
| Copies/106 18S | p | Copies/106 18S | p | |
| MGP | ||||
| Control | 549 ± 118 | 437 ± 66 | ||
| Renal failure | 2770 ± 505 | 0.01 | 1266 ± 213 | 0.006 |
| Osteopontin | ||||
| Control | 418 ± 87 | 509 ± 40 | ||
| Renal failure | 1893 ± 323 | 0.009 | 4939 ± 1880 | 0.03 |
DISCUSSION
This is the first study of MGP metabolism and actions in intact arteries and the effects of renal failure and calcification. Previous studies have been limited to cultured vascular smooth muscle cells or to immunohistochemical detection in intact vessels. In this study, MGP was identified in extracts of rat aorta using an anti-Gla antibody. The fact that purified MGP yielded a band at the same apparent molecular weight confirmed its identity as MGP, with further confirmation provided by the fact that an anti-GluMGP yielded the same band in aortas. The identification of MGP in vascular smooth muscle is consistent with previous studies using immunohistochemistry (19, 20, 25) and demonstrates for the first time that the MGP is present in an intact form in vascular smooth muscle. The identification of uncarboxylated MGP as well in freshly isolated aortas indicates that some MGP is not carboxylated in normal rat aorta. The apparent absence of GluMGP in normal human arteries in a previous study (26) may be due to the lower sensitivity of immunohistochemical staining versus immunoblotting, differences in antibodies, or species differences. Because the aortic extracts contain intracellular MGP in addition to extracellular MGP, the GluMGP could represent intracellular MGP awaiting carboxylation. However, the finding of GluMGP in the culture medium indicates that some GluMGP is released from smooth muscle cells. The presence of GluMGP in normal aortas cannot be explained by a lack of antibody specificity because the anti-GluMGP antibody was previously shown not to react with GlaMGP (25). It is not possible with the techniques used here to determine the relative proportions of carboxylated and noncarboxylated MGP.
The selective disappearance of the 14-kDa band during culture with warfarin provides further confirmation of its identity as GlaMGP and also indicates that MGP is actively synthesized and degraded in vascular smooth muscle. Although the time course was not examined in detail, the rate of disappearance with warfarin was consistent with a half-life of 24 h or less. The disappearance from the aortas cannot be explained by loss into the medium because MGP in the medium also disappeared. This implies either extracellular proteolysis or reuptake and intracellular degradation. Surprisingly, the disappearance of GlaMGP with warfarin was accompanied by a decrease rather than an increase in uncarboxylated MGP, indicating that warfarin inhibited the synthesis of all MGP rather than just preventing its carboxylation. This phenomenon appears to be species-specific because it does not occur in vascular smooth muscle cells cultured from humans (15) but has also been described for prothrombin synthesis by rat liver (41). The underlying mechanism is unclear and could involve defective post-translational processing. It is also possible that GluMGP is rapidly degraded intracellularly. Aortic content of MGP is actually increased in rats treated with warfarin (18, 42) but this may be due to binding of MGP to hydroxyapatite. When warfarin was added to a model of aortic calcification in vitamin D-treated rats, the ratio of MGP to hydroxyapatite was 50% lower than with vitamin D treatment alone, indicating that warfarin does produce a relative deficiency of MGP (18). Both studies noted a preponderance of uncarboxylated MGP as opposed to the reduced synthesis of both GlaMGP and GluMGP in warfarin-treated aortas in culture. The presence of aortic MGP after warfarin treatment in vivo compared with the complete absence after in vitro treatment is probably explained by lower levels of warfarin obtained in vivo and the fact that vitamin K was also administered in vivo.
The rapid turnover of MGP in cultured aortas is consistent with the abundance of mRNA for MGP in rat aortas and the increase during culture noted in this study and a previous study (11). The content of MGP mRNA was not altered by warfarin in vitro, suggesting that MGP does not directly regulate its synthesis in vascular smooth muscle. MGP mRNA was also not affected by a high phosphate concentration, a frequent finding in renal failure and a factor shown to regulate other genes involved in calcification (43). The increased abundance of mRNA for MGP previously described in aortas from warfarin-treated rats (5) may have been due to the longer interval (5 weeks versus 3 days in culture) and the heavy calcification that was present, or to differences between in vivo and in vitro models. However, we did not observe an increase in MGP mRNA in calcified versus noncalcified vessels in renal failure. The results do not support the previous observation in cultured vascular smooth muscle cells that calcification reduces the abundance of MGP mRNA (44).
An additional finding was the significant quantities of MGP in culture medium, which was unexpected because MGP is an extremely insoluble protein (45). The apparent size indicates that the MGP in culture medium is not a breakdown product or precursor, and the retention by a 30-kDa cut-off filter indicates that the MGP is associated with a larger-sized complex. A fetuin-MGP complex as described in serum (21) is unlikely because there is no serum or plasma present in the aortic cultures. Furthermore, the removal of the MGP by ultracentrifugation indicates that it is in a particulate form, possibly associated with membrane vesicles as previously described in cultured vascular smooth muscle cells (46). Given the continual apoptosis in cultured cells, the release of vesicles is not surprising. The absence of apoptosis in cultured aortas (1) indicates that normal, quiescent vascular smooth muscle releases MGP in a soluble particulate form. What role this form has in inhibiting calcification remains to be determined.
Warfarin increased calcification of cultured aortas, indicating that MGP is a local inhibitor of calcification. This is consistent with the demonstration that restoration of MGP synthesis in smooth muscle but not liver rescues MGP-deficient mice from arterial calcification (14). However, the effect of warfarin on calcification was only observed when alkaline phosphatase was added to the medium to remove pyrophosphate, a potent inhibitor of calcification. Although this treatment completely removes pyrophosphate, we cannot rule out an effect of alkaline phosphatase on another factor. The inability of warfarin alone to produce calcification is unlikely to be due to residual MGP in the warfarin-treated aortas given the complete disappearance of MGP in immunoblots and the fact that warfarin markedly increased calcification in the presence of alkaline phosphatase. The results in culture suggest that deficiency of pyrophosphate but not MGP is sufficient to produce calcification and that deficiency of pyrophosphate is necessary for calcification in MGP deficiency. The relative importance of pyrophosphate and MGP are not clear from in vivo data since deficiency of either is sufficient to produce medial vascular calcification (2–5). Because aortic alkaline phosphatase activity decreases during culture,3 it is possible that smooth muscle pyrophosphate levels are lower in vivo and may permit calcification to occur in the absence of MGP. We could not find any evidence that warfarin alters pyrophosphate metabolism in cultured aortas.
MGP binds BMP-2 (22), a protein that can drive bone formation by inducing osteogenic transcription factors such as runx2. The resulting inactivation of BMP-2 has been proposed as the mechanism by which MGP inhibits vascular calcification based on studies in cultured cells (23, 47). However, the results in intact vessels do not support this. The quantity of runx2 mRNA was not affected by warfarin, BMP-2, or the BMP-2 antagonist noggin, and noggin did not abrogate the calcification induced by warfarin in vitro. The discrepancy may relate to the use of cultured cells in the previous studies. The lack of involvement of BMP-2 supports the possibility that MGP prevents medial calcification by directly binding hydroxyapatite and inhibiting its formation. This is consistent with the markedly increased content of MGP in calcified aortas and the fact that bone Gla-protein inhibits hydroxyapatite formation in vitro (16).
This is the first demonstration that the aortic content of MGP is increased in renal failure in the absence of calcification. The concurrent increase in MGP mRNA suggests that this occurs through increased gene transcription. A rigorous analysis was performed to select the optimal reference RNA for determining the effect of renal failure on gene expression. This has not been done in prior studies and it is of interest that β-actin, a commonly used reference gene, was inadequate for this purpose. The mechanism by which renal failure increases MGP mRNA is unclear but is probably not explained by the hyperphosphatemia that is invariably present in this model because phosphate concentration did not affect mRNA content in cultured aortas. Because it was necessary to use almost the entire aorta for immunoblotting or RNA extraction, only a very small segment was available to measure calcium content. Because aortic calcification can be heterogeneous and the segments assayed may not be representative of the entire aorta, a small amount of calcification cannot be ruled out. However, MGP was also increased in aortas after only 16 days of adenine feeding, well before any calcification occurs. The much greater abundance of MGP in calcified aortas without an increase in its mRNA is consistent with sequestering of MGP by the hydroxyapatite. The similar increase in the content of both GlaMGP and GluMGP is not consistent with the preponderance of GluMGP demonstrated by immunohistochemistry in calcified arteries (25, 26). The most likely explanation for this discrepancy is that binding of MGP to hydroxyapatite masks the Gla epitope from Gla-specific antibodies because binding of calcium to Gla residues produces a conformational change that can prevent antibody binding (33). Because the Gla residues probably contribute to the binding of MGP to hydroxyapatite (17), they are likely to be blocked by hydroxyapatite as well. Although the increased aortic synthesis of MGP in renal failure indicates that deficiency of GlaMGP is not a primary factor in vascular calcification in this model, the fact that mRNA expression does not increase further in calcified aortas could lead to a relative deficiency of MGP that could worsen calcification.
This work was supported, in whole or in part, by National Institutes of Health Grant DK069681 (to W. C. O.) and an Amgen Junior Faculty Award (to K. A. L.).
K. A. Lomashvili and W. C. O'Neill, unpublished data.
- MGP
- matrix Gla protein
- BMP
- bone morphogenic protein
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- QPCR
- quantitative PCR.
REFERENCES
- 1. Lomashvili K. A., Cobbs S., Hennigar R. A., Hardcastle K. I., O'Neill W. C. (2004) J. Am. Soc. Nephrol. 15, 1392–1401 [DOI] [PubMed] [Google Scholar]
- 2. Johnson K., Polewski M., van Etten D., Terkeltaub R. (2005) Arterioscler. Thromb. Vasc. Biol. 25, 686–691 [DOI] [PubMed] [Google Scholar]
- 3. Rutsch F., Vaingankar S., Johnson K., Goldfine I., Maddux B., Schauerte P., Kalhoff H., Sano K., Boisvert W. A., Superti-Furga A., Terkeltaub R. A. (2001) Am. J. Pathol. 158, 543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo G., Ducy P., McKee M. D., Pinero G. J., Loyer E., Behringer R. R., Karsenty G. (1997) Nature 386, 78–81 [DOI] [PubMed] [Google Scholar]
- 5. Price P. A., Faus S. A., Williamson M. K. (1998) Arterioscler. Thromb. Vasc. Biol. 18, 1400–1407 [DOI] [PubMed] [Google Scholar]
- 6. Meyer J. L. (1984) Arch. Biochem. Biophys. 231, 1–8 [DOI] [PubMed] [Google Scholar]
- 7. Fleisch H., Schibler D., Maerki J., Frossard I. (1965) Nature 207, 1300–1301 [DOI] [PubMed] [Google Scholar]
- 8. O'Neill W. C., Lomashvili K. A., Malluche H. H., Faugere M. C., Riser B. L. (2011) Kidney Int. 79, 512–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lomashvili K. A., Khawandi W., O'Neill W. C. (2005) J. Am. Soc. Nephrol. 16, 2495–2500 [DOI] [PubMed] [Google Scholar]
- 10. Lomashvili K. A., Garg P., Narisawa S., Millan J. L., O'Neill W. C. (2008) Kidney Int. 73, 1024–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hao H., Hirota S., Tsukamoto Y., Imakita M., Ishibashi-Ueda H., Yutani C. (1995) Arterioscler. Thromb. Vasc. Biol. 15, 1474–1480 [DOI] [PubMed] [Google Scholar]
- 12. Meier M., Weng L. P., Alexandrakis E., Rüschoff J., Goeckenjan G. (2001) Eur. Respir. J. 17, 566–569 [DOI] [PubMed] [Google Scholar]
- 13. Munroe P. B., Olgunturk R. O., Fryns J. P., Van Maldergem L., Ziereisen F., Yuksel B., Gardiner R. M., Chung E. (1999) Nat. Genet. 21, 142–144 [DOI] [PubMed] [Google Scholar]
- 14. Murshed M., Schinke T., McKee M. D., Karsenty G. (2004) J. Cell Biol. 165, 625–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schurgers L. J., Spronk H. M., Skepper J. N., Hackeng T. M., Shanahan C. M., Vermeer C., Weissberg P. L., Proudfoot D. (2007) J. Thromb. Haemost. 5, 2503–2511 [DOI] [PubMed] [Google Scholar]
- 16. Romberg R. W., Werness P. G., Riggs B. L., Mann K. G. (1986) Biochemistry 25, 1176–1180 [DOI] [PubMed] [Google Scholar]
- 17. Roy M. E., Nishimoto S. K. (2002) Bone 31, 296–302 [DOI] [PubMed] [Google Scholar]
- 18. Price P. A., Faus S. A., Williamson M. K. (2000) Arterioscler. Thromb. Vasc. Biol. 20, 317–327 [DOI] [PubMed] [Google Scholar]
- 19. Shanahan C. M., Cary N. R., Salisbury J. R., Proudfoot D., Weissberg P. L., Edmonds M. E. (1999) Circulation 100, 2168–2176 [DOI] [PubMed] [Google Scholar]
- 20. Spronk H. M., Soute B. A., Schurgers L. J., Cleutjens J. P., Thijssen H. H., De Mey J. G., Vermeer C. (2001) Biochem. Biophys. Res. Commun. 289, 485–490 [DOI] [PubMed] [Google Scholar]
- 21. Price P. A., Thomas G. R., Pardini A. W., Figueira W. F., Caputo J. M., Williamson M. K. (2002) J. Biol. Chem. 277, 3926–3934 [DOI] [PubMed] [Google Scholar]
- 22. Wallin R., Cain D., Hutson S. M., Sane D. C., Loeser R. (2000) Thromb. Haemost. 84, 1039–1044 [PubMed] [Google Scholar]
- 23. Boström K., Tsao D., Shen S., Wang Y., Demer L. L. (2001) J. Biol. Chem. 276, 14044–14052 [DOI] [PubMed] [Google Scholar]
- 24. Hruska K. A., Mathew S., Saab G. (2005) Circ. Res. 97, 105–114 [DOI] [PubMed] [Google Scholar]
- 25. Sweatt A., Sane D. C., Hutson S. M., Wallin R. (2003) J. Thromb. Hemost. 1, 178–185 [DOI] [PubMed] [Google Scholar]
- 26. Schurgers L. J., Teunissen K. J., Knapen M. H., Kwaijtaal M., van Diest R., Appels A., Reutelingsperger C. P., Cleutjens J. P., Vermeer C. (2005) Arterioscler. Thromb. Vasc. Biol. 25, 1629–1633 [DOI] [PubMed] [Google Scholar]
- 27. Jiang G., Cobbs S., Klein J. D., O'Neill W. C. (2003) Hypertension 41, 1131–1135 [DOI] [PubMed] [Google Scholar]
- 28. Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 29. Yokozawa T., Zheng P. D., Oura H., Koizumi F. (1986) Nephron 44, 230–234 [DOI] [PubMed] [Google Scholar]
- 30. Okada H., Kaneko Y., Yawata T., Uyama H., Ozono S., Motomiya Y., Hirao Y. (1999) Clin. Exp. Nephrol. 3, 82–88 [Google Scholar]
- 31. Moorehead W. R., Biggs H. G. (1974) Clin. Chem. 20, 1458–1460 [PubMed] [Google Scholar]
- 32. Cogan E. B., Birrell G. B., Griffith O. H. (1999) Anal. Biochem. 271, 29–35 [DOI] [PubMed] [Google Scholar]
- 33. Brown M. A., Stenberg L. M., Persson U., Stenflo J. (2000) J. Biol. Chem. 275, 19795–19802 [DOI] [PubMed] [Google Scholar]
- 34. Rosen V. (2006) Ann. N.Y. Acad. Sci. 1068, 19–25 [DOI] [PubMed] [Google Scholar]
- 35. Price P. A., Roublick A. M., Williamson M. K. (2006) Kidney Int. 70, 1577–1583 [DOI] [PubMed] [Google Scholar]
- 36. Lomashvili K. A., Monier-Faugere M. C., Wang X., Malluche H. H., O'Neill W. C. (2009) Kidney Int. 75, 617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spanakis E. (1993) Nucleic Acids Res. 21, 3809–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vandesompele J., DePreter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F. (2002) Genome Biol. 3, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bro S., Borup R., Andersen C. B., Moeller F., Olgaard K., Nielsen L. B. (2006) Arterioscler. Thromb. Vasc. Biol. 26, 570–575 [DOI] [PubMed] [Google Scholar]
- 40. Moe S. M., Duan D., Doehle B. P., O'Neill K. D., Chen N. X. (2003) Kidney Int. 63, 1003–1011 [DOI] [PubMed] [Google Scholar]
- 41. Zhang P., Suttie J. W. (1994) Blood 84, 169–175 [PubMed] [Google Scholar]
- 42. Schurgers L. J., Spronk H. M., Soute B. A., Schiffers P. M., DeMey J. G., Vermeer C. (2007) Blood 109, 2823–2831 [DOI] [PubMed] [Google Scholar]
- 43. Li X., Yang H. Y., Giachelli C. M. (2006) Circ. Res. 98, 905–912 [DOI] [PubMed] [Google Scholar]
- 44. Mori K., Shioi A., Jono S., Nishizawa Y., Morii H. (1998) FEBS Lett. 433, 19–22 [DOI] [PubMed] [Google Scholar]
- 45. Hackeng T. M., Rosing J., Spronk H. M., Vermeer C. (2001) Protein Sci. 10, 864–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reynolds J. L., Joannides A. J., Skepper J. N., McNair R., Schurgers L. J., Proudfoot D., Jahnen-Dechent W., Weissberg P. L., Shanahan C. M. (2004) J. Am. Soc. Nephrol. 15, 2857–2867 [DOI] [PubMed] [Google Scholar]
- 47. Zebboudj A. F., Shin V., Boström K. (2003) J. Cell. Biochem. 90, 756–765 [DOI] [PubMed] [Google Scholar]