

Abstract
GPR56 is an adhesion G protein-coupled receptor that plays a key role in cortical development. Mutations to GPR56 in humans cause malformations of the cerebral cortex, but little is known about the normal function of the receptor. We found that the large N terminus (NT) of GPR56 is cleaved from the rest of the receptor during processing but remains non-covalently associated with the seven-transmembrane region of the receptor, as indicated by coimmunoprecipitation of the two GPR56 fragments from both transfected cells and native tissue. We also found that truncation of the GPR56 NT results in constitutive activation of receptor signaling, as revealed by increased GPR56-stimulated signaling upon transfection of HEK-293 cells with truncated GPR56, greatly enhanced binding of β-arrestins by truncated GPR56 relative to the full-length receptor, extensive ubiquitination of truncated GPR56, and cytotoxicity induced by truncated GPR56 that could be rescued by cotransfection of cells with β-arrestin 2. Furthermore, we found that the GPR56 NT is capable of homophilic trans-trans interactions that enhance receptor signaling activity. On the basis of these findings, we suggest a model of receptor activation in which the large N terminus of GPR56 constrains receptor activity but N-terminal interactions (GPR56 NT with an extracellular ligand and/or GPR56 NT homophilic trans-trans associations) can remove this inhibitory influence of the N terminus to activate receptor signaling.
Keywords: Adhesion, Brain, Development, G Protein-coupled Receptors (GPCR), G Proteins, Neural Stem Cell, Neurodevelopment, Signal Transduction
Introduction
During the development of the cerebral cortex, neuronal precursors proliferate in the ventricular and subventricular zones that line the cerebral cavity and then migrate outward to make connections with other neurons. Given the billions of cells involved and the requirements for temporal and spatial precision, it is perhaps not surprising that many different types of problems can arise during this process. Abnormalities in cortical development can lead to a range of distinct neurodevelopmental disorders, some of which are caused by mutations to a single gene. For example, bilateral frontoparietal polymicrogyria is a condition in which patients exhibit profound cognitive abnormalities and seizures because of disordered cortical connectivity in the frontoparietal area. Bilateral frontoparietal polymicrogyria is an autosomal recessive syndrome that results from mutations in the orphan receptor GPR56 (1). Thus, insights into the natural function of GPR56 might shed light on the specific pathology underlying bilateral frontoparietal polymicrogyria and also lead to new insights about the fundamental mechanisms controlling cortical development.
GPR56 is a member of the adhesion family of G protein-coupled receptors (GPCRs)2, which are characterized by extremely large extracellular N termini (NT) exhibiting homology to adhesion proteins (2). There are approximately 30 adhesion GPCRs, all of which are still considered to be orphan receptors. Almost all members of the adhesion GPCR family possess an N-terminal region known as a “GPCR proteolytic site” or GPS domain. For several members of the family, it has been shown that the N terminus is clipped within the GPS domain during receptor processing but then remains associated with the seven-transmembrane (7TM) region of the receptor (3). There is good evidence that the GPS domain cleavage is autocatalytic and occurs during receptor processing (4), but the significance of this cleavage event is not understood, and very little in general is known about the mechanisms of activation for adhesion GPCRs.
Transfection of GPR56 into HEK-293 cells has been reported to result in activation of Gα12/13 and Rho (5) as well as stimulation of transcriptional regulators downstream of Rho, including the serum response element (5) and β-catenin (6). Furthermore, stimulation of GPR56 in cultured neural precursor cells with an activating antibody results in decreased cellular migration (5). These observations in cultured cells are consistent with in vivo studies demonstrating that GPR56 knockout mice exhibit a cobblestone-like malformation of the cerebral cortex resulting from overmigration of neural precursor cells (7). These findings suggest a model in which GPR56 expressed in neural precursor cells plays a key role in sensing an extracellular signal that cues the cells to stop and differentiate. If GPR56 is non-functional, as in humans with GPR56 mutations or mice lacking GPR56, then neural precursor cells fail to stop in the proper locations, and cortical development becomes disordered.
In this study, we sought to explore the role of the GPR56 N terminus in receptor activation. Because an N-terminal antibody can stimulate GPR56 signaling (5), our initial hypothesis was that removing the N terminus of the receptor should result in a receptor that lacks signaling activity. Surprisingly, we instead found evidence that truncation of the GPR56 N terminus results in enhanced constitutive activity of the receptor. Moreover, we found that homophilic interactions between GPR56 N termini can influence receptor signaling. These observations suggest a new model for the mechanism of activation for GPR56.
EXPERIMENTAL PROCEDURES
Cell Culture
For all cell-based assays, HEK-293 cells (ATCC) were cultured and maintained in DMEM containing 10% FBS and 1% penicillin/streptomycin at 37 °C with 5% CO2. Transfections were performed by incubating cells with Lipofectamine 2000 (Invitrogen) and cDNA for 4 h in serum-free DMEM, then stopping transfection with complete media. Experiments were performed 24–48 h post-transfection.
Antibodies
Antibodies against HA (Roche), FLAG (Sigma-Aldrich), β-arrestin 2 (Sigma-Aldrich), c-myc (Sigma-Aldrich), and biotinylated GPR56 N terminus (R&D Systems) were purchased from the manufacturers. The anti-GPR56 C-terminal antibody was developed by Orbigen, Inc. via injection of rabbits with a peptide (CSNSDSARLPISSGSTSSSRI) derived from the GPR56 C terminus, followed by affinity purification using the same peptide that was used as the immunogen.
Plasmids
N-terminal FLAG-GPR56 was a gift from Christopher Stipp (University of Iowa). Untagged human GPR56 wild-type was subcloned into pcDNA 3.1. The FLAG-tagged ΔNT mutant was cloned into pcDNA 3.1 by creating primers starting with an N-terminal FLAG epitope followed by the human GPR56 sequence starting at amino acid 343. Similarly, an untagged GPR56ΔNT mutant was made by creating primers using the human GPR56 sequence starting at amino acid 343. FLAG-NT GPR56 and c-myc-NT GPR56 constructs were created in the pCMV 2B plasmid and correspond to human GPR56 amino acids 1–342. The HA-Ubiquitin construct was a gift from Keqiang Ye (Emory University), and the GFP-β-arrestin2 construct was a gift from Jeffrey Benovic (Thomas Jefferson University). GST-RBD (Addgene, Inc.), HA-β-arrestin2 (Addgene, Inc.), and HA-Rho (Missouri S&T cDNA Resource Center) were all obtained commercially.
Western Blotting
Samples were resolved by SDS-PAGE on 4 to 20% Tris-glycine gels, followed by transfer to nitrocellulose membranes. The membranes were incubated in blocking buffer (2% nonfat dry milk, 50 mm NaCl, 20 mm HEPES, and 0.1% Tween 20) for 30 min and then incubated with primary antibody for 1 h at room temperature. Next, the membranes were washed three times in blocking buffer and incubated with HRP-conjugated secondary antibody for 30 min, washed three times more, and finally visualized via ECL reagent followed by exposure to film. When using the biotinylated GPR56 N-terminal primary antibody, the ABC kit (VECTASTAIN) was used to visualize immunoreactive bands in lieu of secondary antibody.
Rho Activation Assay
HEK-293 cells were transfected with HA-Rho (Addgene, Inc.) and either pcDNA 3.1, GPR56 wild-type, or GPR56ΔNT. After 24 h, cells were scraped and resuspended in 500 μl lysis buffer (1% Triton X-100, 150 mm NaCl, 25 mm HEPES, 10 mm MgCl2, 1 mm EDTA, and 2% glycerol). Cells were incubated in lysis buffer for 30 min at 4 °C and then cleared by high-speed centrifugation. Soluble lysates were incubated for 30 min with 30 μl of GST-Rhotekin binding domain (GST-RBD) coupled to glutathione-agarose beads. Beads were washed twice with lysis buffer, resuspended in 60 μl 2× sample buffer, and boiled for 10 min. Total Rho levels were first determined through Western blotting of cell lysates with anti-HA antibody to normalize levels before determining active Rho levels. Active Rho was visualized by standard Western blot analysis procedure, probing the normalized GST-RBD samples for HA.
Coimmunoprecipitation
HEK-293 cells were transfected with various constructs to be assessed for ability to coimmunoprecipitate. After 24 h, cells were scraped and resuspended in 500 μl lysis buffer (1% Triton X-100, 150 mm NaCl, 25 mm HEPES, 10 mm MgCl2, 1 mm EDTA, and 2% glycerol). Cells were incubated in lysis buffer for 30 min at 4 °C and then cleared by high-speed centrifugation. Soluble lysates were incubated for 60 min with 30 μl of protein A/G beads with corresponding antibody (to protein being immunoprecipitated). Beads were washed three times with lysis buffer then resuspended in 60 μl 2× sample buffer and boiled for 10 min. Coimmunoprecipitation was detected by standard Western blot analysis procedure. Rat kidney was substituted for transfected HEK-293 cells using the same protocol. For ubiquitination studies, HEK-293 cells were transfected with HA-Ubiquitin and either empty vector, GPR56 wild-type, or GPR56ΔNT mutant. Cells were lysed as described and immunoprecipitated with protein A/G beads coupled to HA antibody. A Western blot analysis procedure was used to detect GPR56 using the GPR56-CT antibody.
Coculturing Experiments
HEK-293 cells were transfected with desired plasmids and incubated at 37 °C (called “base cells”). After 24 h, different plates of HEK-293 cells (transfected depending on the experiment) were resuspended using 0.25% trypsin and complete media and plated on top of base cells and incubated at 37 °C. After 24 h, cells were lysed and used for coimmunoprecipitation or Rho activation experiments.
Cytotoxicity
HEK-293 cells were transfected with pcDNA 3.1, GPR56 wild-type, or GPR56ΔNT in the presence or absence of HA-β-arrestin 2. After transfection, a media sample was taken from each plate of cells every 24 h for 72 h. Cytotoxicity was assessed by measuring lactate dehydrogenase levels in the media samples using the CytoTox 96 cytotoxicity assay kit (Promega).
Confocal Microscopy
HEK-293 cells transiently transfected with GFP-β-arrestin 2, plus or minus wild-type GPR56 or GPR56ΔNT, were plated onto poly-D-lysine-coated chamber slides (Biocoat), allowed to attach overnight, and fixed at room temperature in 2% paraformaldehyde. The cells were incubated with the anti-GPR56-CT antibody (1:500) for two h at 37 °C, washed extensively, and then incubated with Alexa Fluor goat anti-rabbit 546 secondary (1:250) for 1 h at 37 °C. Slides were viewed using the ×63 objective of an LSM 510 META confocal microscope (Carl Zeiss, Inc., Thornwood, NY), and images were acquired with a constant setting for comparison across conditions using Zeiss LSM software.
Cell Surface Biotinylation
HEK-293 cells were transfected with GPR56 wild-type or GPR56ΔNT constructs. After 24 h, cells were washed and incubated for 2 h at 4 °C in 2 mm Sulfo-NHS-LC-Biotin (Thermo Scientific) in PBS to biotinylate surface proteins. After biotinylation, cells were washed, scraped, and resuspended in lysis buffer (1% Triton X-100, 150 mm NaCl, 25 mm HEPES, 10 mm MgCl2, 1 mm EDTA, and 2% glycerol). Cells were incubated in lysis buffer for 30 min at 4 °C with end-over-end agitation and then cleared by high-speed centrifugation. Soluble lysates were incubated with streptavidin beads (Pierce) for 2 h at 4 °C to pull down surface biotinylated proteins. Beads were washed three times and resuspended in sample buffer. Surface expression of GPR56 and GPR56ΔNT was assessed by analyzing these samples via Western blotting.
Cell Surface Luminometry
HEK-293 cells were transfected with pcDNA3.1, FLAG-GPR56, FLAG-GPR56ΔNT, or FLAG-NT and incubated at 37 °C for 24 h. After 24 h, each HEK-293 plate was split into triplicate 35-m dishes and incubated at 37 °C for 12 h. After 12 h, cells were washed and fixed with 4% paraformaldehyde. Fixed cells were then blocked with 2% dry nonfat milk in PBS for 30 min. Cells were then incubated for 2 h at room temperature with FLAG-HRP primary antibody (1:1000). After washing three times (2 ml each wash), luminescence was measured using a TD 20/20 luminometer (Turner Designs).
Statistical Analysis
All statistical analyses were performed on single comparisons with Student's t test using GraphPad Prism software (GraphPad Software, Inc., San Diego, CA).
RESULTS
GPR56 Is Processed into Two Fragments That Remain Associated
We developed a polyclonal anti-GPR56 antibody to visualize the C terminus of the receptor. A representative Western blot analysis utilizing this antibody to detect GPR56 in transfected HEK-293 cells is shown in Fig. 1A. The specificity of the antibody is evident from the fact that no immunoreactive bands are detectable in untransfected HEK-293 cells (first lane). In cells transfected with GPR56, several different processed forms of the receptor are evident. The prominent band at 45 kDa represents the 7TM region following GPS domain cleavage to remove the large N terminus, as this 45 kDa band was observed in cell surface biotinylation experiments to be the main surface-expressed fragment of the receptor (supplemental Fig. S1A) and is precisely the predicted size of the 7TM region following cleavage at the GPS domain. The handful of lower molecular weight bands are presumably derived from additional cleavage events, and the higher molecular weight bands probably represent GPR56 not yet processed at the GPS domain and/or unresolved receptor oligomers. None of these species were found in the plasma membrane as determined by cell surface biotinylation (supplemental Fig. S1A). GPR56 from these transfected cells was solubilized in 1% Triton X-100, immunoprecipitated with the C-terminal antibody, and visualized on Western blot analyses using a commercially available antibody to detect the GPR56 NT. The GPR56 NT was visualized in these Western blot analyses as an approximately 75-kDa band that, upon deglycosylation, decreased in size to 37 kDa, consistent with past reports (8, 9). As shown in Fig. 1B, robust coimmunoprecipitation of the cleaved GPR56 NT was observed with the 7TM region of the receptor, suggesting that the two fragments of the receptor remain associated in cells even following cleavage at the GPS domain. Similar experiments were performed on endogenous GPR56 in rat kidney, a tissue where GPR56 is highly expressed (10). Fig. 1C shows the expression of GPR56 in rat kidney using the GPR56 C-terminal antibody. The main band at just under 50 kDa and the processed forms at smaller sizes were very similar to the pattern of bands observed in the GPR56-transfected HEK-293 cells. Immunoprecipitation with the C-terminal antibody resulted in robust coimmunoprecipitation of the endogenous N-terminal fragment (Fig. 1D).
FIGURE 1.

The cleaved N terminus of GPR56 associates with the 7TM region of the receptor. A, lysates from HEK-293 cells transfected with empty vector (EV) or GPR56 were probed with anti-GPR56 C-terminal antibody. B, the same antibody was used to immunoprecipitate (IP) the GPR56 7TM region from solubilized lysates. The IP samples were then probed with the GPR56 N-terminal antibody (n = 3). C, rat kidney tissue was solubilized and probed for GPR56 expression using the GPR56 C-terminal antibody. The mock lane (MK) was loaded with buffer only. D, IP was performed with the GPR56 C-terminal antibody, and IP samples were probed with the GPR56 N-terminal antibody. Mock (MK) in this case represents the IP protocol performed with beads but no antibody (n = 3). WB, Western blotting.
Removal of the GPR56 NT Enhances GPR56-mediated Stimulation of Rho Activity and Induces Receptor Ubiquitination
Previous reports have shown that transfection of GPR56 into HEK-293 cells results in stimulation of Gα12/13 to activate downstream Rho and β-catenin signaling (5, 6). To explore the importance of the GPR56 NT for the signaling activity of the receptor, we created a truncated GPR56 construct lacking the N terminus up to the GPS domain (Fig. 2A). Cell surface biotinylation experiments (supplemental Fig. S1A) and cell surface luminometry (supplemental Fig. S1B) revealed that wild-type GPR56 and the ΔNT mutant were found in the plasma membrane at roughly comparable levels. The truncated receptor was expressed in HEK-293 cells and assessed for its ability to stimulate Rho activity relative to the wild type. As shown in Fig. 2, B and C, transfection of the cells with wild-type GPR56 resulted in significant increases in Rho activity, consistent with previous findings (5). Strikingly, transfection with the ΔNT mutant enhanced Rho signaling to an even greater extent than transfection with the wild-type GPR56. These findings suggest that the ΔNT mutant might have enhanced constitutive activity. To further decipher the activity levels of wild-type versus truncated mutant GPR56, we examined the ubiquitination state of each receptor because many GPCRs undergo extensive ubiquitination upon prolonged activation (11–14). As shown in Fig. 2D, GPR56ΔNT was found to be heavily ubiquitinated, whereas ubiquitination of the wild-type receptor was barely detectable.
FIGURE 2.

N-terminal truncation enhances GPR56-mediated signaling and receptor ubiquitination. A, schematic diagram showing the location of the N-terminal GPR56 truncation. The numbers in parentheses indicate the amino acid positions of the N terminus, the GPS domain starting position, and the C terminus for each construct. B, quantification of active RhoA via pull-down with GST-RBD. Total Rho was first normalized between samples before probing for active Rho. Active Rho levels were compared with empty vector-transfected cells. *, p < 0.05; n = 10. WB, Western blotting. C, top panel, Western blot analysis of active RhoA pull-down with GST-RBD beads from HEK-293 cells transfected with empty vector (EV), wild-type GPR56, or the ΔNT mutant. Bottom panel, Western blot analysis of total RhoA levels from HEK-293 cells transfected with empty vector, GPR56, and GPR56ΔNT. D, ubiquitination of wild-type versus truncated GPR56. Full-length GPR56 or GPR56ΔNT were transfected into HEK-293 cells with HA-Ubiquitin (HA-Ub). Immunoprecipitation (IP) was performed with anti-HA antibodies and immunoprecipitates were probed via Western blot analysis with anti-GPR56-CT antibodies to visualize ubiquitinated GPR56. The data shown are representative of three independent experiments.
Truncation of the GPR56-NT Enhances Receptor Interactions with β-Arrestin 2
The signaling and ubiquitination studies suggested that GPR56ΔNT may be a constitutively active receptor. If this were the case, then one additional prediction would be that this truncated receptor should exhibit enhanced interactions with β-arrestins, a family of regulatory proteins that are known to interact with active GPCRs to tone down G protein-mediated signaling (15) and exhibit especially robust associations with constitutively active receptors (16, 17). As shown in Fig. 3A, wild-type GPR56 could be detected in complex with β-arrestin 2 in coimmunoprecipitation experiments, which is consistent with observations that this receptor has some level of activity when transfected into HEK-293 cells. Strikingly, however, the ΔNT mutant exhibited a massive increase in coimmunoprecipitation with β-arrestin 2 relative to the wild-type receptor (Fig. 3A, last lane, more than 10-fold increase in β-arrestin 2 associations was observed for the ΔNT mutant relative to wild-type in six independent experiments). Reciprocal coimmunoprecipitation experiments in which anti-GPR56 immunoprecipitates were probed for β-arrestin 2 also showed significant increases in β-arrestin 2 associations with the ΔNT mutant over wild-type GPR56 (Fig. 3B, last lane). The enhanced interaction of the ΔNT mutant with β-arrestin 2 was also observed in confocal microscopy experiments (Fig. 3, C–K). β-arrestin 2-GFP was found to be evenly distributed throughout the cell when transfected into HEK-293 cells by itself (Fig. 3C). However, cotransfection with GPR56 resulted in enrichment of β-arrestin 2-GFP in a perinuclear compartment (Fig. 3F), and cotransfection with the ΔNT mutant resulted in an even more dramatic targeting of β-arrestin 2 to the perinuclear region (Fig. 3I), where it exhibited strong colocalization with the internalized receptor (Fig. 3K). Such targeting of arrestins and internalized receptors to perinuclear endosomes has been observed for many GPCRs upon prolonged periods of receptor activation (18–20). Thus, these data provide further evidence for the idea that removal of the N terminus induces constitutive activation of GPR56.
FIGURE 3.

β-arrestin 2 binds avidly to GPR56ΔNT and attenuates ΔNT-induced cytotoxicity. A, HEK-293 cells were transfected with GPR56 or GPR56ΔNT in the absence or presence of HA-β-arrestin 2 (HA-β-arr2). Immunoprecipitation (IP) was performed with HA antibody coupled to agarose beads. Coimmunoprecipitation of GPR56 was detected by Western blotting (WB) with the anti-GPR56-CT antibody. EV, empty vector. B, HEK-293 cells were transfected with HA-β-arrestin 2 and either empty vector, GPR56, or GPR56ΔNT. Immunoprecipitation was performed with the anti-GPR56-CT antibody and protein A/G agarose. Coimmunoprecipitation of β-arrestin 2 was detected by Western blotting with anti-HA antibody. C–K, wild-type GPR56 and GPR56ΔNT promote β-arrestin 2 cellular redistribution and perinuclear aggregation. β-Arrestin 2-GFP expressed alone was distributed evenly throughout HEK-293 cells (C and E), but co-expression with GPR56 promoted translocation of β-arrestin 2 to the perinuclear region where it colocalized with the receptor (H). Translocation of β-arrestin 2 to the perinuclear region was even more dramatic upon coexpression with GPR56ΔNT (K). DAPI staining is shown in E, H, and K. These data are representative of four independent experiments.
Overexpression of GPR56ΔNT Induces Cell Death That Can Be Rescued by Coexpression of β-Arrestin 2
In addition to enhancing signaling, undergoing extensive ubiquitination, and interacting robustly with β-arrestins, another hallmark of constitutively active GPCRs is causing toxicity in the cells in which they are expressed (21, 22). Thus, we performed cytotoxicity tests on HEK-293 cells transfected with wild-type GPR56 versus the ΔNT mutant. Overexpression of wild-type GPR56 failed to induce any toxicity at any of the time points examined (Fig. 4). Transfection of the ΔNT mutant also did not result in any evident cytotoxicity at 24 h post-transfection, the time point at which all of the signaling studies described above were performed. However, overexpression of the ΔNT mutant did cause a significant increase in cell death at 48 h and an even larger increase in cell death at 72 h. Furthermore, this cytotoxicity induced by overexpression of the ΔNT mutant could be greatly attenuated by coexpression of β-arrestin2, which would be expected to boost the arrestin-to-receptor ratio in the cells and thereby drive arrestin associations with the overactive receptors to dampen down their activity. Taken together with the signaling data, ubiquitination data and β-arrestin 2 interaction data, these findings suggest that truncation of the GPR56-NT results in constitutive receptor activation.
FIGURE 4.
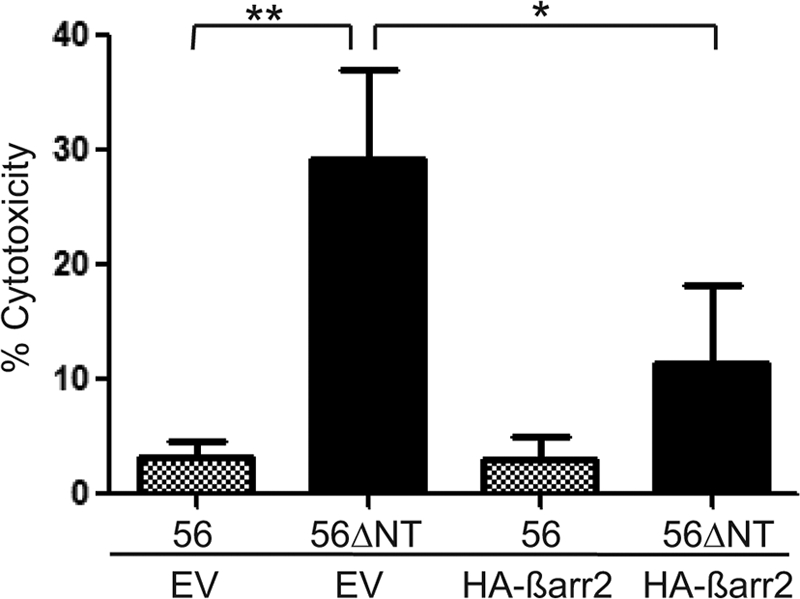
β-arrestin 2 attenuates GPR56ΔNT-stimulated cytotoxicity. Cytotoxicity induced by GPR56 or GPR56ΔNT expression in HEK-293 cells was determined by lactate dehydrogenase secretion in the media at 72 h post-transfection. A significant difference was observed between GPR56 and GPR56ΔNT (**, p < 0.01; n = 6). Cotransfection of HA-β-arrestin 2 resulted in a significant reduction in GPR56ΔNT-dependent cytotoxicity (*, p < 0.03; n = 3), but had no effect on toxicity in cells transfected with the GPR56 wild type. EV, empty vector.
GPR56 Trans-Trans N-terminal Interactions Enhance Receptor Signaling
If it is true that the GPR56-NT associates with the 7TM region of the receptor to constrain receptor activity, then GPR56 NT binding partners might conceivably alter the conformation of the GPR56 NT to alleviate this inhibitory influence on receptor signaling. In this regard, it is of interest to note that the adhesion GPCRs Celsr2 and Celsr3 have been reported to undergo homophilic N-terminal interactions in a trans-trans fashion (e.g. interactions of the same receptor type on adjacent cells), with these interactions strongly influencing receptor activity (23). It is not known if such trans-trans interactions represent a general mechanism for controlling the activity of adhesion GPCRs or if, instead, this mechanism is unique to Celsr2/3. Because our data described above revealed the importance of the GPR56 N terminus in controlling receptor activity, we examined whether GPR56 might be capable of trans-trans interactions via its N-terminal domain.
A FLAG-tagged GPR56-NT construct (lacking the seven-transmembrane and intracellular domains) was created and transfected into HEK-293 cells. A similar GPR56 NT construct was also created, containing a c-myc tag in place of the FLAG tag. Both FLAG- and Myc-GPR56-NT expressed well in cells with only minimal amounts of the expressed GPR56 NT protein secreted into the medium (supplemental Fig. S1C). Moreover, cell surface luminometry experiments showed that the secreted GPR56 NT was mainly found associated with the extracellular face of the plasma membrane, explaining its absence from the media (supplemental Fig. S1B). The GPR56 NT may be tethered to the cell membrane through interaction with known GPR56-binding partners such as the matricellular protein transglutaminase 2 and/or the transmembrane proteins CD9 and CD81 (24, 25). To investigate GPR56 N-terminal interactions, the two differentially tagged GPR56 NT constructs were separately transfected into HEK-293 cells, which were cocultured for 24 h before coimmunoprecipitation experiments were performed. As shown in Fig. 5A, the two GPR56 NT proteins were found to robustly interact with each other even though they were expressed in different sets of cells. Interestingly, removal of the C-terminal third of the Myc-GPR56-NT (114 amino acids removed) resulted in complete abrogation of the interaction with FLAG-GPR56-NT, thereby mapping the essential region for GPR56 NT-NT association as between amino acids 228 and 342 (Fig. 5A, third lane).
FIGURE 5.

GPR56 NT-NT interactions enhance GPR56-mediated signaling. A, coimmunoprecipitation (IP) between FLAG-GPR56-NT and Myc-GPR56-NT. HEK-293 cells were separately transfected with FLAG-NT, empty vector (EV), Myc-NT, or Myc-NTΔ228–342. After 24 h, FLAG-NT transfected cells were cocultured with empty vector- (first lane), Myc-NT- (second lane), or Myc-NTΔ228–342 (third lane)-transfected cells. The next day, cells were lysed, and immunoprecipitation was performed with protein A/G beads coupled to c-myc antibody, followed by Western blotting (WB) for FLAG-NT. B, HEK-293 cells transfected with FLAG-NT (FL-NT) were cocultured with HEK-293 cells transfected with empty vector (first and third lanes), GPR56 wild-type (second lane), or GPR56ΔNT mutant (fourth lane). After coculturing for 24 h, immunoprecipitation was performed with protein A/G beads coupled to GPR56-CT antibody, followed by Western blotting for FLAG-NT. In parallel experiments, FLAG-NT-expressing cells were grown separately, harvested, and then mixed with lysates from cells transfected with empty vector (fifth lane) or GPR56 wild-type (sixth lane) prior to immunoprecipitation with anti-GPR56-CT antibodies. The data shown are representative of three to five independent experiments for the various conditions. C, schematic drawing of coculturing technique. HEK-293 cells were transfected with HA-RhoA plus empty vector or GPR56 wild-type. These base cells were then cocultured for 24 h with cells expressing EV, GPR56, or FLAG-NT. The activity of HA-Rho in the base cells was then measured and expressed as fold over EV/EV cells. D, quantification of active Rho in coculturing experiments. Also included is the Western blot analysis of active and total Rho. A significant difference in GPR56-mediated RhoA activation was seen for base cells cocultured with cells expressing GPR56 (*, p < 0.05; n = 3) and FLAG-NT (*, p < 0.04; n = 3).
Because we found that the N terminus of GPR56 could interact with other GPR56 N termini, we further examined whether such NT-NT interactions might occur in the context of the full-length receptor. Cells expressing FLAG-GPR56-NT were cocultured with cells expressing untagged full-length GPR56, and potential GPR56 trans-trans interactions were examined via a coimmunoprecipitation approach. As shown in Fig. 5B, immunoprecipitation of the full-length GPR56 (from the base cells in this experiment) using our GPR56-CT antibody resulted in robust coimmunoprecipitation of the FLAG-GPR56-NT from the cocultured cells. Interestingly, no coimmunoprecipitation was observed between the GPR56 ΔNT mutant and FLAG-GPR56-NT upon coculturing of cells transfected with these two constructs (Fig. 5B, fourth lane), revealing the importance of the N-terminal domain for GPR56 trans-trans interactions. Additionally, no coimmunoprecipitation was observed when GPR56- and FLAG-GPR56-NT-transfected cells were cultured separately and then solubilized and mixed together (Fig. 5B, last two lanes), demonstrating that the NT-NT interactions are not artifacts of post-solubilization aggregation. These data reveal that GPR56 is capable of trans-trans interactions via its N-terminal domain.
We next examined whether the GPR56 trans-trans interactions that we observed in the coculturing experiments might influence GPR56-mediated Rho activation. HEK-293 cells transfected with either empty vector, full-length GPR56, or FLAG-GPR56-NT were cocultured with base cells transfected with either HA-Rho alone or HA-Rho/full-length GPR56, and the activation state of HA-Rho in the base cells was assessed (Fig. 5C). Coculturing with any of the differentially transfected cells had no effect on the activity of HA-Rho in base cells transfected with HA-Rho alone. However, in base cells transfected with HA-Rho/full-length GPR56, coculturing with cells expressing GPR56 or FLAG-GPR56-NT resulted in a significant potentiation in GPR56-mediated HA-Rho activation in the base cells (Fig. 5D). These data provide evidence that GPR56 trans-trans N-terminal interactions can enhance GPR56-mediated Rho signaling.
DISCUSSION
GPR56 regulates the migration of neural precursor cells in vitro (5) and in vivo (7, 26), but little is known about the mechanism of activation for GPR56. On the basis of our findings here, we propose that removal of the GPR56 N terminus can result in receptor activation. This idea is based on four lines of evidence: 1) Transfection of cells with the GPR56 ΔNT mutant results in significantly enhanced activation of Rho signaling relative to wild-type GPR56, 2) the ΔNT mutant is much more heavily ubiquitinated than wild-type GPR56, 3) the ΔNT mutant associates much more avidly than wild-type GPR56 with β-arrestins, which preferentially bind to active receptors, and 4) expression of the ΔNT mutant is toxic for cells in a manner that is rescued by coexpression of β-arrestins. All of these phenomena (enhanced signaling activity, increased ubiquitination, enhanced binding of β-arrestins, and toxicity to cells) are characteristic of constitutively active GPCRs. Thus, we propose that removal of the GPR56 N terminus results in greatly enhanced constitutive signaling activity of the receptor.
N-terminal truncations do not typically result in constitutive activation of GPCRs. For example, truncations to the N termini of β2-adrenergic (27), α1D-adrenergic (28), CB1 cannabinoid (29), GPR37 orphan (30), or μ opioid (31) receptors can cause alterations in receptor trafficking and/or ligand binding properties but do not result in constitutive receptor activation. The only GPCRs that are known to be activated by N-terminal removal are the four members of the protease-activated receptor family (32) and the thyrotropin receptor (33, 34). Protease-activated receptor and thyrotropin receptor cleavage by extracellular proteases is believed to be a key step in the natural mechanism of activation for these receptors. Similarly, we propose that relief of the inhibitory influence of the GPR56 NT on the signaling activity of the receptor may be a key step in the mechanism of activation for GPR56. Because GPS domain cleavage is believed to be autoproteolytic (4), GPR56 does not require the action of an exogenous protease to achieve separation of its N-terminal region from the rest of the receptor. However, the two halves of GPR56 remain non-covalently associated for some period of time following GPS domain cleavage, and our data suggest that this association constrains the signaling activity of the 7TM region of the receptor. We propose that interaction of the GPR56 NT with an extracellular ligand results in either the release of the GPR56 NT from the 7TM region or a conformational change in the GPR56 NT that relieves the inhibitory influence of the GPR56 NT and thereby allows for receptor activation (Fig. 6).
FIGURE 6.

Schematic diagram of the proposed mechanism of activation for GPR56. A, the GPR56 NT is cleaved from the 7TM region of the receptor, but the two halves of the receptor remain non-covalently associated, and the receptor is largely inactive. B, the GPR56 N terminus engages an adhesive ligand, possibly another GPR56 N terminus, in a trans-trans fashion to induce receptor activation. GPR56-NT interactions with the adhesive ligand might physically disrupt NT associations with the GPR56 7TM region or simply alter NT conformation to enhance the functional activity and G protein coupling (G) of the 7TM region.
If GPR56 is activated by extracellular interactions that either remove the NT or alter its conformation, then it is clearly a point of major interest to identify the extracellular ligand(s) of the receptor. GPR56 has been shown to associate via its N-terminal region with the extracellular matrix protein transglutaminase 2 (24) and also shown to form complexes with the tetraspanins CD9 and CD81 via undetermined domains (25), but these associations have not been shown to have any effect on receptor activity. The only protein that is known to enhance GPR56-mediated signaling is an anti-GPR56 antibody that binds to the N terminus of the receptor (5). Similarly, N-terminal antibodies have been shown to activate the adhesion GPCR EMR2 (35), and an N-terminal-binding toxin (latrotoxin) has been shown to activate the adhesion GPCR latrophilin 1 (36). Despite the artificial nature of these antibody and toxin treatments, these studies are of significant interest in that they reveal the importance of N-terminal binding partners for receptor activation. A less artificial example of N-terminal engagement leading to adhesion GPCR activation comes from work on Celsr2 and Celsr3, which have been shown to be activated via homophilic N-terminal interactions in a trans-trans fashion (23). Similarly, Flamingo, the Celsr homolog in Drosophila, has been shown to undergo homophilic trans-trans interactions that are critical for the development of planar cell polarity (37, 38). In our studies, we found that the N terminus of GPR56 is capable of homophilic trans-trans interactions and that these associations promote receptor activation. It remains to be determined whether these GPR56/GPR56 associations truly activate the receptor or simply make the receptor permissive for signaling (for example, by creating a binding site for an as-yet-unidentified ligand). It also remains to be determined whether NT-NT interactions are a common feature for other members of the adhesion GPCR family beyond Celsr2/3 and GPR56.
In summary, we have found that removal of the GPR56 N terminus results in constitutive activation of receptor signaling. This observation may be relevant to the natural mechanism of GPR56 activation. If migrating cells that express GPR56 encounter other cells presenting a ligand that binds to the GPR56 NT, this could either dislodge the GPR56 NT from the 7TM region of the receptor or at least alter the conformation of the GPR56 NT to remove its inhibitory influence on receptor signaling. In this way, GPR56 could be activated to stimulate Rho signaling, thereby inducing actin cytoskeleton remodeling and the inhibition of cellular migration. Furthermore, our data reveal that the GPR56 N terminus is capable of homophilic trans-trans interactions that enhance receptor activity. Taken together, these findings shed light on the regulation of GPR56 signaling activity and may also provide general insights concerning the activation mechanisms for other members of the adhesion GPCR family.
Supplementary Material
Acknowledgments
We thank Xianhua Piao (Harvard University), John Northup (National Institutes of Health), Ana Monteiro (Emory University), Nicole Brown (Emory University), Ayush Kishore (Emory University), and T. J. Murphy (Emory University) for advice and discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R21-NS063029 and R01-NS072394 (to R. A. H.) and Training Grant T32-GM008602 (to K. J. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- GPCR
- G protein-coupled receptor
- GPS
- GPCR proteolytic site
- CT
- C terminus
- NT
- N terminus
- 7TM
- seven-transmembrane
- RBD
- Rhotekin binding domain.
REFERENCES
- 1. Piao X., Hill R. S., Bodell A., Chang B. S., Basel-Vanagaite L., Straussberg R., Dobyns W. B., Qasrawi B., Winter R. M., Innes A. M., Voit T., Ross M. E., Michaud J. L., Déscarie J. C., Barkovich A. J., Walsh C. A. (2004) Science 303, 2033–2036 [DOI] [PubMed] [Google Scholar]
- 2. Fredriksson R., Gloriam D. E., Höglund P. J., Lagerström M. C., Schiöth H. B. (2003) Biochem. Biophys. Res. Commun. 301, 725–734 [DOI] [PubMed] [Google Scholar]
- 3. Yona S., Lin H. H., Siu W. O., Gordon S., Stacey M. (2008) Trends Biochem. Sci. 33, 491–500 [DOI] [PubMed] [Google Scholar]
- 4. Lin H. H., Chang G. W., Davies J. Q., Stacey M., Harris J., Gordon S. (2004) J. Biol. Chem. 279, 31823–31832 [DOI] [PubMed] [Google Scholar]
- 5. Iguchi T., Sakata K., Yoshizaki K., Tago K., Mizuno N., Itoh H. (2008) J. Biol. Chem. 283, 14469–14478 [DOI] [PubMed] [Google Scholar]
- 6. Shashidhar S., Lorente G., Nagavarapu U., Nelson A., Kuo J., Cummins J., Nikolich K., Urfer R., Foehr E. D. (2005) Oncogene 24, 1673–1682 [DOI] [PubMed] [Google Scholar]
- 7. Li S., Jin Z., Koirala S., Bu L., Xu L., Hynes R. O., Walsh C. A., Corfas G., Piao X. (2008) J. Neurosci. 28, 5817–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin Z., Tietjen I., Bu L., Liu-Yesucevitz L., Gaur S. K., Walsh C. A., Piao X. (2007) Hum. Mol. Genet. 16, 1972–1985 [DOI] [PubMed] [Google Scholar]
- 9. Ke N., Ma H., Diedrich G., Chionis J., Liu G., Yu D. H., Wong-Staal F., Li Q. X. (2008) Biochem. Biophys. Res. Commun. 366, 314–320 [DOI] [PubMed] [Google Scholar]
- 10. Huang Y., Fan J., Yang J., Zhu G. Z. (2008) Mol. Cell. Biochem. 308, 133–139 [DOI] [PubMed] [Google Scholar]
- 11. Shenoy S. K., McDonald P. H., Kohout T. A., Lefkowitz R. J. (2001) Science 294, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 12. Marchese A., Raiborg C., Santini F., Keen J. H., Stenmark H., Benovic J. L. (2003) Dev. Cell 5, 709–722 [DOI] [PubMed] [Google Scholar]
- 13. Martin N. P., Lefkowitz R. J., Shenoy S. K. (2003) J. Biol. Chem. 278, 45954–45959 [DOI] [PubMed] [Google Scholar]
- 14. Shenoy S. K. (2007) Circ. Res. 100, 1142–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reiter E., Lefkowitz R. J. (2006) Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 16. Mhaouty-Kodja S., Barak L. S., Scheer A., Abuin L., Diviani D., Caron M. G., Cotecchia S. (1999) Mol. Pharmacol. 55, 339–347 [DOI] [PubMed] [Google Scholar]
- 17. Ferrari S. L., Bisello A. (2001) Mol. Endocrinol. 15, 149–163 [DOI] [PubMed] [Google Scholar]
- 18. Zhang J., Barak L. S., Anborgh P. H., Laporte S. A., Caron M. G., Ferguson S. S. (1999) J. Biol. Chem. 274, 10999–11006 [DOI] [PubMed] [Google Scholar]
- 19. Innamorati G., Le Gouill C., Balamotis M., Birnbaumer M. (2001) J. Biol. Chem. 276, 13096–13103 [DOI] [PubMed] [Google Scholar]
- 20. Lelouvier B., Tamagno G., Kaindl A. M., Roland A., Lelievre V., Le Verche V., Loudes C., Gressens P., Faivre-Baumann A., Lenkei Z., Dournaud P. (2008) J. Neurosci. 28, 4336–4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Miura S., Karnik S. S. (2000) EMBO J. 19, 4026–4035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dale L. B., Bhattacharya M., Anborgh P. H., Murdoch B., Bhatia M., Nakanishi S., Ferguson S. S. (2000) J. Biol. Chem. 275, 38213–38220 [DOI] [PubMed] [Google Scholar]
- 23. Shima Y., Kawaguchi S. Y., Kosaka K., Nakayama M., Hoshino M., Nabeshima Y., Hirano T., Uemura T. (2007) Nat. Neurosci. 10, 963–969 [DOI] [PubMed] [Google Scholar]
- 24. Xu L., Begum S., Hearn J. D., Hynes R. O. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 9023–9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Little K. D., Hemler M. E., Stipp C. S. (2004) Mol. Biol. Cell. 15, 2375–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koirala S., Jin Z., Piao X., Corfas G. (2009) J. Neurosci. 29, 7439–7449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dixon R. A., Sigal I. S., Rands E., Register R. B., Candelore M. R., Blake A. D., Strader C. D. (1987) Nature 326, 73–77 [DOI] [PubMed] [Google Scholar]
- 28. Hague C., Chen Z., Pupo A. S., Schulte N. A., Toews M. L., Minneman K. P. (2004) J. Pharmacol. Exp. Ther. 309, 388–397 [DOI] [PubMed] [Google Scholar]
- 29. Andersson H., D'Antona A. M., Kendall D. A., Von Heijne G., Chin C. N. (2003) Mol. Pharmacol. 64, 570–577 [DOI] [PubMed] [Google Scholar]
- 30. Dunham J. H., Meyer R. C., Garcia E. L., Hall R. A. (2009) Biochemistry 48, 10286–10297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Muller I., Sarramegna V., Milon A., Talmont F. J. (2009) Appl. Biochem. Biotechnol. [DOI] [PubMed] [Google Scholar]
- 32. Traynelis S. F., Trejo J. (2007) Curr. Opin. Hematol. 14, 230–235 [DOI] [PubMed] [Google Scholar]
- 33. Zhang M., Tong K. P., Fremont V., Chen J., Narayan P., Puett D., Weintraub B. D., Szkudlinski M. W. (2000) Endocrinology 141, 3514–3517 [DOI] [PubMed] [Google Scholar]
- 34. Quellari M., Desroches A., Beau I., Beaudeux E., Misrahi M. (2003) Eur. J. Biochem. 270, 3486–3497 [DOI] [PubMed] [Google Scholar]
- 35. Yona S., Lin H. H., Dri P., Davies J. Q., Hayhoe R. P., Lewis S. M., Heinsbroek S. E., Brown K. A., Perretti M., Hamann J., Treacher D. F., Gordon S., Stacey M. (2008) FASEB J. 22, 741–751 [DOI] [PubMed] [Google Scholar]
- 36. Lelianova V. G., Davletov B. A., Sterling A., Rahman M. A., Grishin E. V., Totty N. F., Ushkaryov Y. A. (1997) J. Biol. Chem. 272, 21504–21508 [DOI] [PubMed] [Google Scholar]
- 37. Carreira-Barbosa F., Kajita M., Kajita M., Morel V., Wada H., Okamoto H., Martinez Arias A., Fujita Y., Wilson S. W., Tada M. (2009) Development 136, 383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Usui T., Shima Y., Shimada Y., Hirano S., Burgess R. W., Schwarz T. L., Takeichi M., Uemura T. (1999) Cell 98, 585–595 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.