

Abstract
The toroid-shaped nuclear protein export factor CRM1 is constructed from 21 tandem HEAT repeats, each of which contains an inner (B) and outer (A) α-helix joined by loops. Proteins targeted for export have a nuclear export signal (NES) that binds between the A-helices of HEAT repeats 11 and 12 on the outer surface of CRM1. RanGTP binding increases the affinity of CRM1 for NESs. In the absence of RanGTP, the CRM1 C-terminal helix, together with the HEAT repeat 9 loop, modulates its affinity for NESs. Here we show that there is an electrostatic interaction between acidic residues at the extreme distal tip of the C-terminal helix and basic residues on the HEAT repeat 12 B-helix that lies on the inner surface of CRM1 beneath the NES binding site. Small angle x-ray scattering indicates that the increased affinity for NESs generated by mutations in the C-terminal helix is not associated with large scale changes in CRM1 conformation, consistent with the modulation of NES affinity being mediated by a local change in CRM1 near the NES binding site. These data also suggest that in the absence of RanGTP, the C-terminal helix lies across the CRM1 toroid in a position similar to that seen in the CRM1-Snurportin crystal structure. By creating local changes that stabilize the NES binding site in its closed conformation and thereby reducing the affinity of CRM1 for NESs, the C-terminal helix and HEAT 9 loop facilitate release of NES-containing cargo in the cytoplasm and also inhibit their return to the nucleus.
Keywords: Nuclear Pore, Nuclear Transport, Protein Conformation, Protein Structure, Protein-Protein Interactions, Trafficking, X-ray Scattering
Introduction
Nuclear protein export is a fundamental function in all eukaryotes and is crucial for maintaining the appropriate nucleocytoplasmic distribution of a broad range of transcription factors, signaling molecules, and cell cycle regulators, as well as viral components such as the HIV Rev protein and the influenza NS2 protein. Transport takes place through nuclear pore complexes and is mediated primarily by the transport factor CRM1 (Xpo1p in yeast) in conjunction with the Ran GTPase (1–7). Proteins destined for export have nuclear export signal sequences (NESs)3 and bind cooperatively to CRM1 with RanGTP in the nucleus. This cargo-carrier complex is then equilibrated between the nucleus and cytoplasm through weak interactions between CRM1 and nuclear pore proteins that have characteristic repeating phenylalanine-glycine sequence motifs (FG-nucleoporins). When the cargo-carrier complex reaches the cytoplasm, RanGTP is removed through the action of RanBP1, RanGAP, and several nucleoporins. When RanGTP is removed, the affinity of CRM1 for the NES is reduced, leading to the release of the cargo, after which CRM1 is recycled to the nucleus (reviewed by 4–8). Energy for the cycle is ultimately provided by GTP hydrolysis on Ran that is stimulated by RanGAP.
CRM1 is a member of the β-karyopherin superfamily and is constructed from a tandem array of 21 HEAT repeats, each of which is based on two α-helices joined by loops of varying length (8–12). The molecule adopts an approximately toroidal conformation with one series of HEAT helices (the A-helix) located on the outer convex surface of the toroid, whereas the other (the B-helix) is located on its inner concave surface (8–12). The α-helix at the CRM1 C terminus (referred to as HEAT repeat 21B by Monecke et al. (10)) adopts different positions depending on whether or not RanGTP is bound. In the absence of RanGTP (9), the C-terminal helix projects across the central cavity of the CRM1 toroid, whereas when RanGTP is bound (10–12), the helix is displaced so that its tip sits near HEAT repeats 3 and 4 in the N-terminal region of the CRM1 toroid. Complexes of CRM1 with Snurportin (9, 10) and chimeras of Snurportin with a range of NESs (12) have shown that the NES-binding site is located on the outer surface of CRM1 in a hydrophobic groove formed between the A-helices of HEAT repeats 11 and 12. RanGTP, on the other hand, binds to the interior surface of the CRM1 toroid, making connections with the N-terminal half of CRM1, the central acidic loop HEAT repeat 9, and HEAT repeats 17 and 19 (10–12). Detailed structural studies of CRM1 with a range of different NESs bound has established a consensus for NESs and accounted for how CRM1 is able to recognize the range of different NESs observed (12). Because the binding and release of NESs in the nucleus and cytoplasm, respectively, is critical for nuclear protein export, it is important to understand the molecular mechanism by which the affinity of CRM1 for NESs is modulated. In the nucleus when RanGTP is bound, CRM1 has a higher affinity for NESs than in the cytoplasm when Ran has been removed. This change in affinity could be due to changes in the positions of the A-helices of HEAT repeats 11 and 12 that result from a contribution of local changes induced via an autoinhibitory mechanism of the HEAT 9 loop proposed by Koyama and Matsuura (11) and/or a larger, global change in CRM1 conformation as proposed by Dong et al. (13) and Monecke et al. (10). In addition to the role of the HEAT 9 loop in modulating the affinity of CRM1 for NESs, the C-terminal helix also influences NES binding. Although in the absence of RanGTP full-length CRM1 has weak affinity for NESs, deletion of the C-terminal helix increases this affinity to a level that, in pulldown binding studies, appeared comparable with that seen in the presence of RanGTP (13).
Here we use a combination of mutagenesis and small angle x-ray scattering (SAXS) to investigate the role of the C-terminal helix in greater detail and show that charged residues at its extreme C terminus are critical to its influence on the affinity of CRM1 for NESs. Deleting the last 9 residues of mouse CRM1 or mutating to alanine either the acidic residues in this region or a cluster of positive residues in the B-helix of HEAT repeat 12 (Lys-594, Arg-596, and Arg-597) increased the affinity of CRM1 for the PKI NES. Moreover, the behavior of a variant in which the charges of both clusters were reversed reverted to the affinity seen with wild type CRM1. SAXS indicated that the C-terminal helix was positioned across the CRM1 toroid and that none of the engineered variants generated a substantial conformational change in the molecule. Based on these results, we propose a model in which the C-terminal helix and the HEAT loop 9 induce local conformational changes that stabilize the closed conformation of the CRM1 NES binding site, which contributes to the release of NES-containing cargoes into the cytoplasm following the removal of Ran and to preventing the return of cargoes to the nucleus.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
S. cerevisiae Xpo1p was expressed from the pET30TB plasmid (a derivative of pET30a-TEV in which the BamHI site in the TEV sequence has been removed) that adds a His6-S tag followed by a TEV cleavage site to the N terminus (14). Mouse CRM1 was expressed as a TEV-cleavable His6-ZZ-tagged construct in a derivative of a pQE80 vector that was obtained as a kind gift from Dr. Dirk Görlich (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany). Mutations in CRM1/Xpo1p were introduced via overlapping PCRs with mutagenic primers using the Herculase II fusion system (Stratagene). Proteins were expressed in Escherichia coli BL21-CodonPlus(DE3)-RIL cells in ZYM-5052 autoinduction medium (15). A single colony of BL21-CodonPlus(DE3)-RIL cells that had been transformed with the desired plasmid was used to inoculate a starter culture of 10 ml of LB medium (that contained either 100 ng/ml ampicillin or 25 ng/ml kanamycin as appropriate). The culture was incubated overnight at 37 °C, shaking at 200 rpm, and was used to inoculate a large-scale 1-liter culture the following morning. Large scale cultures were grown in 2-liter conical flasks that contained ∼1 liter of ZYM-5052 medium. To prepare the final media, a stock of ∼1 liter of ZY medium (10 g of N-Z-amine AS and 5 g of yeast extract dissolved in 1 liter of MilliQ water) was supplemented with 20 ml of 50× 5052 solution (25% (w/v) glycerol, 2.5% (w/v) glucose, and 10% (w/v) α-lactose monohydrate), 20 ml of 50× M solution (1.25 m Na2HPO4, 1.25 m KH2PO4, 2.5 m NH4Cl, and 0.25 m Na2SO4), 2 ml of 1 m MgSO4, and 100 mg of either ampicillin or kanamycin as appropriate. Stocks of the 50× 5052 and 50× M solutions were sterile filtered and stored at 4 °C. The cultures were incubated at 37 °C, shaking at 200 rpm, for ∼6 h that allowed growth to an A600 ∼ 0.5, at which point the temperature was lowered to 20 °C for a further 18 h. The cells were harvested by centrifugation at ∼4,000 × g for 10 min at 4 °C, and cell pellets were stored at −20 °C. Frozen cell pellets were thawed at room temperature and resuspended in 10 ml of the appropriate lysis buffer per cell pellet from 1 liter of culture. A CompleteTM Mini EDTA-free protease inhibitor mixture tablet (Roche Applied Science) was added and dissolved in the lysis mixture. Cell lysis was carried out by high pressure cavitation using an EmulsiFlex C5 homogenizer (Avestin). Lysates were clarified by centrifugation at 45,000 × g for 20 min at 4 °C. Proteins were purified using a standard Ni-NTA resin (Qiagen) affinity purification protocol. Buffers typically contained 15 mm imidazole, pH 8.0, to prevent nonspecific binding of contaminant proteins. Clarified lysate containing soluble His6-tagged protein was mixed with the Ni-NTA resin and incubated at room temperature for 45 min. The resin was centrifuged at 1,500 rpm for 5 min at 4 °C and washed three times with 50 ml of binding buffer. Purified protein was eluted from the resin using binding buffer that contained at least 300 mm imidazole, pH 8.0. All of the protein constructs used in this work contained a tobacco etch mosaic virus (TEV) protease cleavage site. TEV cleavage was typically conducted overnight at 4 °C using a His6-tagged S219V variant of TEV that is less prone to autocatalysis and is more active at cold temperatures than wild type TEV protease (16). Following TEV cleavage, His6 tags and the His6-TEV S129V were removed from cleavage mixtures by a second incubation with fresh Ni-NTA resin. Gel filtration chromatography was carried out as a final protein purification step to remove contaminating proteins and CRM1 oligomers using a HiLoad Superdex 200 26/60 prep grade column (GE Healthcare). Protein fractions were analyzed using SDS-PAGE, and those that contained the target protein of sufficient purity (>95%) were pooled and concentrated using an Amicon-Ultra 15 centrifugal filter unit (Millipore) following the manufacturer's instructions. Proteins were concentrated to at least 50 mg/ml and stored at either 4 °C for immediate use or flash-frozen in liquid nitrogen and stored for longer term use at −80 °C. His-tagged-YFP-PKI-NES was prepared as described (11).
Binding Assays
Pulldown assays were used to assess the binding between proteins by immobilizing one protein on affinity resin via its affinity purification tag. A small aliquot (∼100 μl) of affinity resin (Ni-NTA or glutathione-Sepharose 4B (GE Healthcare)) was washed with binding buffer (typically 20 mm Tris-HCl, pH 7.4, 50 mm NaCl, 2 mm DTT). The protein that would act as bait in the binding assay was then loaded onto the beads. Either clarified lysate (∼0.5 ml) or purified protein was added to the resin and incubated at room temperature for 45 min. For GST pulldowns, a negative control was conducted in parallel by performing the assay on resin loaded with GST protein alone. For Ni-NTA pulldowns, resin without bait protein was used as a negative control for nonspecific binding to the beads. After incubation, beads were sedimented by centrifugation at 2,000 rpm for 3 min in a microcentrifuge and washed three times with 1 ml of binding buffer to remove excess bait protein. The prey proteins were added to the resin (in quantities that provided a molar excess to the bait protein), and the mixture was incubated for 45 min at room temperature. Unbound fractions were removed, and the resin was washed three times with 0.5 ml of binding buffer. After the final wash step, the beads were mixed with an equal volume of 2× SDS-PAGE loading dye and boiled at 95 °C to release bound proteins from the beads. Protein samples were analyzed by SDS-PAGE.
The binding of increasing concentrations of wild type and mutant CRM1 protein to 10–50 nm YFP-PKI-NES fusion protein (11) was measured by fluorescence anisotropy at 25 °C using a PerkinElmer LS55 fluorimeter equipped with a Hamilton Microlab titrator controlled by laboratory software. The reaction was followed in 20 mm Tris-HCl, pH 7.4, 50 mm NaCl, 5 mm DTT, 2 mm MgCl2. Excitation and emission wavelengths of 507 and 526 nm, respectively, were used with both excitation and emission slits set at 5 nm. Using a single-site binding model, the data were fitted to the equation,
 |
where F0 and F1 are the anisotropy in the absence of titrating protein and at saturation, respectively; [LT] and [PT] are the total concentrations of protein and YFP-NES, respectively; and Kd is the dissociation constant. The fraction of YFP-NES bound at each concentration of titratant, fb, was calculated using the fitted constants from the equation above using the following.
 |
The CRM1 variants gave excellent binding isotherms that were fitted well by the model (supplemental Fig. S1). However, low (∼15 μm) affinity of wild type CRM1 for the YFP-NES precluded our being able to attain saturation, which was taken to be the same as that observed for the variants.
Small Angle X-ray Scattering
SAXS data were collected at European Synchrotron Radiation Facility Beamline ID14-3 using a fixed wavelength of 0.931 Å. CRM1 or Xpo1p samples were diluted with the original gel filtration buffers to make a series of protein concentrations. The signal from protein scattering was collected in conjunction with a buffer sample measurement taken before and after each protein sample. For mouse CRM1, the detector was a Pilatus 1 m (Dectris), and the sample to the detector distance was 2.425 m. The momentum transfer range measured was 0.05 < s < 6 nm−1. The samples were exposed for 10 frames of 10 s each. For Saccharomyces cerevisiae Xpo1p, the detector was a VÅNTEC-2000 (Bruker AXS Ltd.), and the sample to the detector distance was 1.668 m. The momentum transfer range measured was 0.05 < s < 6 nm−1. The samples were exposed for 10 frames of 30 s each.
SAXS data were analyzed using the PRIMUS software suite (17). Each protein sample frame was inspected for radiation damage, and frames that contained significant damage were omitted. The buffer data were averaged from before and after the protein sample and subtracted from the merged protein dataset to provide a final curve for that protein concentration. The radius of gyration, Rg, was calculated from the slope of the Guinier region with output from the AUTORG program used as a guide to the best data range for this measurement. The pair distribution function, p(r), was calculated using the program GNOM that performs an indirect Fourier transform. An initial estimate of the Dmax was provided to GNOM, and the program was run several times to determine the best Dmax based on the shape of the p(r) function. The program CRYSOL was used to provide a direct comparison between the calculated scattering profile from a given Protein Data Bank coordinate file and an experimental SAXS profile (18). CRYSOL calculates the scattering, taking into account the hydration shell of the particle. It has been shown that no two unique protein structures have identical calculated scattering profiles (19). The final output provides a χ2 value to give a measure of the goodness of fit between the calculated and experimental scattering curves.
RESULTS
Acidic Residues at the C-terminal Helix Tip Modulate the Affinity of CRM1 for NESs in the Absence of RanGTP
Deletion of the C-terminal 44 residues of human CRM1 (that correspond to the C-terminal helix) has been shown to increase its affinity for NESs in the absence of RanGTP, which, in pulldown binding studies, appeared to be of a level comparable with that of wild type CRM1 in the presence of RanGTP (13). Sequence alignment of CRM1 from several species shows a strongly conserved pattern within the residues at the distal tip of the C-terminal helix, whereby a single basic residue is followed by a series of acidic residues (Fig. 1). In CRM1 from Homo sapiens, Mus musculus, and Xenopus laevis, there are four acidic residues, but the precise number can vary between three (in S. cerevisiae) and six (in Caenorhabditis elegans). Because there is no electron density corresponding to these acidic residues in any of the current crystal structures of CRM1 or Xpo1p, these residues are not present in any of the structural models (9–12).
FIGURE 1.

Alignment of CRM1 C-terminal helix sequences. The secondary structure of the human CRM1 chain from the CRM1-Snurportin complex (9) is shown above the sequences. The last residue for which there is clear electron density in the crystal structures is Pro-1061. Acidic and basic residues of the C-terminal helices are shown in red and blue, respectively. Note the conservation of acidic residues at the extreme C terminus.
In the crystal structure of the human CRM1-Snurportin complex (9), the C-terminal helix lies across the central cavity of the toroid, bringing the distal tip residues of the helix near to HEAT repeats 11 and 12. These HEAT repeats also form the primary binding site for NESs in a groove formed between their A-helices. The role of the acidic residues at the tip of the CRM1 C-terminal helix in modulating the affinity of CRM1 for NESs was probed using a series of deletion variants, which were evaluated for their affinity for immobilized GST-PKI NES (Fig. 2). Because it could be expressed to higher levels and appeared not to aggregate so extensively in solution, we employed mouse CRM1 rather than the human CRM1 employed previously (9). The sequences of these proteins are identical in the final 10 residues of the C-terminal helix (Fig. 1). CRM1 variants were generated in which the entire C-terminal helix was deleted (CRM1-1–1027; as in Dong et al. (13)); the distal half was deleted (CRM1-1–1040) so that it retained the base of the C-terminal helix, which in the CRM1-Snurportin-RanGTP (10) ternary complex makes contacts with residues in the inter-repeat loops of N-terminal HEAT repeats 3 and 4, or in which the last nine residues, that include the conserved acidic residues, were deleted (CRM1-1–1062). A CRM1 variant (CRM1-tipA) in which the charged residues of the final nine residues of CRM1 were replaced with alanines was also tested. All four of these mouse CRM1 variants bound the PKI NES with a higher affinity than wild type CRM1 (Fig. 2). Determination of binding constants using fluorescence anisotropy (Table 1) indicated that although deleting the entire C-terminal helix produced a larger increase in affinity for the PKI NES, simply removing the negative charge from the tip of the C-terminal helix caused over a 10-fold increase in affinity, consistent with the charged residues at the tip making a substantial contribution to the influence of the C-terminal helix on the affinity of CRM1 for NESs.
FIGURE 2.

Binding of CRM1 C-terminal helix variants to immobilized GST-PKI NES. A, whereas wild type CRM1 bound weakly to GST-PKI NES beads in pull-down experiments, all of the variant CRM1 constructs showed much stronger binding, consistent with the acidic residues at the extreme C terminus being critical. B shows the same proteins bound to the GST alone control resin.
TABLE 1.
Affinity of CRM1 and variants for YFP-PKI-NES determined using fluorescence anisotropy
Binding constants (Kd) were determined from three independent titrations using the data shown in supplemental Figure S1. The free energy change (ΔΔG) for each variant relative to wild-type CRM1 was calculated from the difference in binding constant.
| CRM1 protein | Kd | ΔΔG |
|---|---|---|
| μm | kcal/mol | |
| Wild-type | 15.6 ± 2.3 | |
| 1–1062 | 1.7 ± 0.1 | 1.3 ± 0.2 |
| VLV > AAA | 0.20 ± 0.03 | 2.6 ± 0.2 |
| 1–1027 | 0.30 ± 0.05 | 2.3 ± 0.2 |
| 1–1062 + VLV > AAA | 0.025 ± 0.002 | 3.8 ± 0.2 |
Mutation of Conserved Basic Residues in HEAT 12B Mimics Mutating the C-terminal Helix Tip
The important contribution to modulation of the affinity of CRM1 for the PKI NES made by the highly conserved patch of acidic residues at the tip of the C-terminal helix suggested that complementary positively charged residues on CRM1 might mediate an electrostatic interaction with this cluster. Examination of the electrostatic potential of the inner surface of CRM1 in the structure of the human CRM1-Snurportin complex (9), showed a basic patch located directly adjacent to where the C-terminal helix terminates (Fig. 3A). This patch is formed by a series of conserved basic residues, which sit at the ends of the B-helices of HEAT repeats 11 and 12 that are closest to the end of the C-terminal helix (Figs. 3B and 4). These conserved residues are Arg-553 and Arg-556 of HEAT 11B and Lys-594, Arg-596, and Arg-597 of HEAT 12B. Lys-590 is also conserved and has been shown to bind to residue Glu-429 from the HEAT 9 loop in the Xpo1p-RanGTP-RanBP1 crystal structure (11). In the mouse CRM1-Snurportin-RanGTP structure (10), Lys-590 binds to Glu-428 of the HEAT 9 loop rather than Glu-429, which instead binds to Tyr-155 of RanGTP. The proximity of the acidic C-terminal helix tip to the conserved basic patch located on the HEAT 11/12 B helices was consistent with an electrostatic interaction between these two sets of residues having the potential to contribute to the influence of the C-terminal helix of CRM1 on its affinity for NESs in the absence of RanGTP.
FIGURE 3.

The conserved basic patch on CRM1. A, electrostatic surface potential of human CRM1 from the CRM1-Snurportin crystal structure (9). Regions of acidic and basic potential are shown in red and blue, respectively. The basic patch on HEAT repeats 11B and 12B lies adjacent to the end of the C-terminal helix of CRM1. B, the HEAT 11B-12B regions of several species of CRM1 are aligned, showing the conservation of the six basic patch residues (in blue). HEAT repeats are indicated below the alignment.
FIGURE 4.

Conserved basic residues on HEAT 11B/12B of CRM1. The conserved basic residues (Arg-553, Arg-556, Lys-590, Lys-594, Arg-596, and Arg-597) shown in dark blue lie at the end of the HEAT 11B/12B helices closest to the end of the C-terminal helix (as modeled by Dong et al. (9)). The NES binds on the opposite side of the CRM1 backbone at the A helices of these HEAT repeats.
We tested this hypothesis by engineering a series of variants in the basic patch of mouse CRM1. If the C-terminal helix interacts with these residues to exert its effect on NES binding, then mutating the basic residues would be expected to have the same effect as mutating the C-terminal helix tip. The set of five conserved residues on the B helices of HEAT repeats 11 and 12 (Lys-590 was omitted because of its interaction with the HEAT 9 loop) was divided into two groups; group H11, consisting of Arg-553 and Arg-556 located on HEAT 11B, and group H12 consisting of Lys-594, Arg-596, and Arg-597 located on HEAT 12B. In both the group H11 and H12 constructs, the basic residues were mutated to alanine to provide an uncharged, small residue that would be unlikely to create an artifactual new interaction. A double mutant (denoted H11+H12), combined all five mutations in a single construct. In all three constructs (H11, H12, and H11+H12), the C-terminal helix region was not altered. The CRM1 basic patch variants were tested for their ability to bind immobilized GST-PKI NES from bacterial cell lysates, containing overexpressed CRM1 variants, in the absence of RanGTP (Fig. 5).
FIGURE 5.
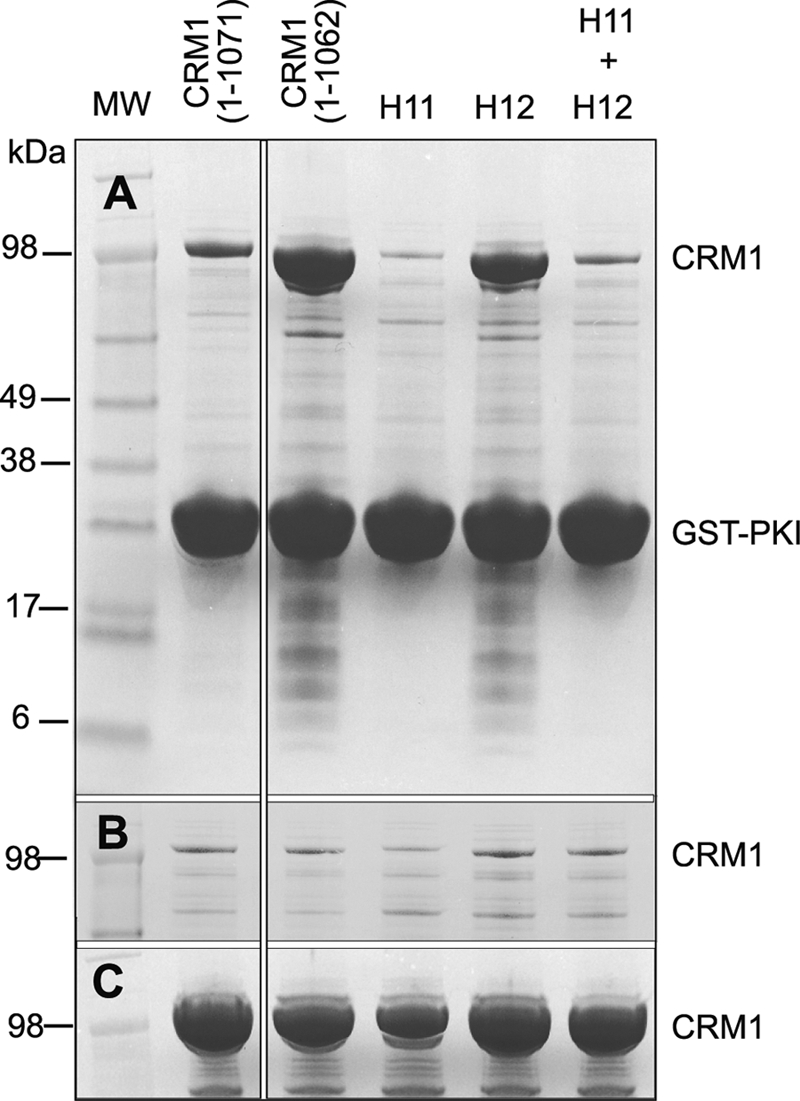
Binding of CRM1 basic patch variants to immobilized GST-PKI NES. A, pulldowns were carried out using cell lysates incubated with resin that had been preloaded with GST-PKI NES. B, none of the variants bound to GST alone. C, lysate samples incubated with Ni-NTA show that all of the lysates contained comparable amounts of CRM1. Variant H11 was R553A/R556A, whereas variant H12 was K594A/R596A/R597A.
Basic patch variant H11 showed a reduced affinity for the PKI NES compared with wild type CRM1 (1–1071), whereas variant H12 bound to the PKI NES with a much higher affinity than wild type CRM1, as did the C-terminal helix tip variant (1–1062) control (Fig. 5A). These results are consistent with only the basic residues of HEAT 12B (Lys-594, Arg-596, and Arg-597) contributing to a putative electrostatic interaction with the C-terminal helix tip. Interestingly, the combined mutant (H11+H12) reverted to the affinity seen in variant H11 alone. This suggested that mutating Arg-553 and Arg-556 inhibited PKI NES binding, because by itself it decreased the affinity for the PKI NES and also counteracted the positive effect of the H12 variant. It is possible that the H11 mutations may destabilize the conformation of the NES binding site. Indeed, examination of the mouse CRM1-Snurportin structure (9) shows that Arg-553 may be involved in an electrostatic interaction with Glu-510 on HEAT 11A that may contribute to the structural integrity of the NES cleft.
Reversing the Charges of Residues in the C-terminal Helix Tip and the HEAT 12B Basic Patch Restores Wild Type CRM1 NES Binding
Although mutating the charged residues of either the C-terminal helix tip or the H12 patch of residues each influenced the strength of NES binding, these findings do not necessarily show that these regions interact directly. To test the hypothesis that a direct interaction between the acidic residues of the C-terminal helix tip and the basic residues of the H12 group (Lys-594, Arg-596, and Arg-597) contributes to the inhibition of NES binding in the absence of RanGTP, a variant was engineered in which the charge of both groups of residues was reversed (residues 594–597 mutated from KCRR to ECDD and residues 1067–1071 mutated to KKMCR from EEMCD designated CRM1-Swap). Pulldown experiments using wild type CRM1 and CRM1-Swap in the both the absence and the presence of His6-S-RanΔC(1–180)-Q69L (Fig. 6A) demonstrated that swapping the charges between the C-terminal helix tip and the HEAT 12B basic patch residues restored the wild type behavior of the protein, whereby the binding of the GST-PKI NES to CRM1-Swap alone was weak but was strengthened by the addition of RanGTP. Consequently, the CRM1-Swap variant supports the hypothesis that the acidic residues of the C-terminal helix tip and the conserved basic residues of HEAT 12B form a direct electrostatic interaction that inhibits NES binding in the absence of RanGTP. Because the basic patch residues are located on the inner face of the CRM1 toroid, whereas the NES binding site is located on the outer surface, the putative interaction between the C-terminal helix tip residues and the basic patch residues probably influences NES binding via an allosteric effect analogous to the mechanism proposed for the inhibitory function of the HEAT 9 loop in Xpo1p (11). RanGTP binding could then disrupt this interaction (through displacing the C-terminal helix) and thereby contribute to the observed increase in affinity for NESs in these conditions.
FIGURE 6.

Binding of the Swap (A) and HEAT 9 loop (B) CRM1 variants to immobilized GST-PKI NES. Both wild type CRM1 (1–1071) and the CRM1-Swap variant bound to GST-PKI NES with a lower affinity than the tip deletion variant (1–1062). The addition of RanGTP significantly increased the affinity of wild type and CRM1-Swap for the NES. The HEAT 9 loop variant binds the NES with a higher affinity than wild type CRM1 in the absence of RanGTP, and the binding is comparable with that seen for the variant that combines the C-terminal helix tip deletion and HEAT 9 loop mutation. The variants all bound to the GST control with similar affinity (lower panel).
The C-terminal Helix and the HEAT 9 Loop Cooperate to Reduce the Affinity of CRM1 for NESs
The HEAT 9 loop hydrophobic residue variant shown to relieve the autoinhibitory effect of the S. cerevisiae Xpo1p HEAT 9 loop by Koyama and Matsuura (11) was engineered into mouse CRM1 by mutating the three hydrophobic residues, 430VLV432, to alanine. This variant resulted in a similar increase in affinity for the PKI NES in the absence of RanGTP to that seen when the C-terminal helix was removed (Fig. 6B and Table 1), confirming that the HEAT 9 loop has a similar effect on NES binding in M. musculus CRM1 as in S. cerevisiae Xpo1p (11). When the HEAT 9 loop variant (VLV > AAA) was combined with removal of the C-terminal helix tip (CRM1-1–1062), the affinity for the PKI NES was increased further, with the overall change in binding energy (ΔΔG) for the double mutant (3.8 ± 0.2 kcal/mol) being indistinguishable from the sum of the ΔΔG values (3.9 ± 0.2 kcal/mol) for the individual variants (Table 1). Moreover, the 25 nm affinity exhibited by this mutant was comparable with that observed in the presence of RanGTP (27). This result indicated that both the HEAT 9 loop and the C-terminal helix contribute to decreasing the affinity of CRM1 for the PKI-NES in the absence of RanGTP.
Mutations that Increase the Affinity of CRM1 for NESs Are Not Associated with a Large Global Conformational Change in CRM1
SAXS was used to examine the solution structure of CRM1 in the absence of bound ligands and to determine the position of the C-terminal helix under these conditions. SAXS was also used to investigate whether any of the mutations introduced in the C-terminal helix tip or the HEAT 12B basic patch altered the overall conformation of CRM1 and thereby establish whether the mutations increased the affinity of CRM1 for the NES via a global conformational change in CRM1 or via a more subtle local conformational effect near the NES binding cleft.
Previous SAXS studies (20) had indicated that S. cerevisiae Xpo1p had a toroidal conformation in solution analogous to that seen in crystal structures (9–12). However, the limited resolution of SAXS coupled with technical difficulties associated with generating models from SAXS data of toroidal objects (28) did not indicate the precise position of the C-terminal helix. The SAXS pattern we obtained from mouse CRM1 was very similar to that reported for Xpo1p, with a characteristic shoulder at s ≈ 0.8–1.2 nm−1. However, the availability of several crystal structures of CRM1 allowed more detailed interpretation of the SAXS pattern than was possible in the earlier study by simulating the SAXS patterns expected for different conformations. These comparisons indicated that the solution scattering for mouse CRM1 was best modeled by the conformation observed in the crystal structure of human CRM1 complexed with Snurportin (9), in which the C-terminal helix lies across the toroid (Fig. 7). Omitting the C-terminal helix from the model completely or using the conformation of CRM1 observed when it was complexed with Snurportin and RanGTP (in which the C-terminal helix does not lie across the toroid) gave models that fitted the observed SAXS scattering less well. Therefore, the SAXS data supported the hypothesis that in the absence of a bound NES and/or RanGTP, the C-terminal helix was arranged to lie across the toroid. This would place the extreme C terminus close to the B-helix of HEAT repeat 12, which is consistent with the mutagenesis data that indicated an electrostatic interaction between these two regions of CRM1.
FIGURE 7.

SAXS analysis of CRM1 and variants. A, Kratky plot of experimental mouse CRM1 SAXS data overlaid with simulated plots derived from crystal structures of human CRM1 (9), mouse CRM1 (10), and S. cerevisiae Xpo1p (11). B, simulated plots as in A overlaid with experimental S. cerevisiae Xpo1p SAXS data. C, effect on the Kratky plot of human CRM1 (9) of removing the density of the extended C-terminal helix from the crystal structure. D, χ2 values for the comparisons shown in the Kratky plots (A–C). The best fit was obtained between the mouse CRM1 experimental SAXS data and the structure of human CRM1 described by Dong et al. (9). Removing the C-terminal helix from this structure, in which it lies across the toroid, increases χ2 from 5 to 14.2.
The SAXS curves of the CRM1 C-terminal helix tip deletion variant (residues 1–1062) as well as the HEAT 12B basic patch variant and the variant in which charges were swapped were indistinguishable from the SAXS curve of the wild type mouse CRM1 (Fig. 8). These findings are consistent with these mutations not inducing a large global change in the conformation of the CRM1 backbone. In addition, the SAXS data obtained for the CRM1 variants indicated that these mutations did not change the position of the C-terminal helix from the position it occupied in wild type CRM1 in the absence of bound ligands. This is consistent with the putative C-terminal helix tip electrostatic interaction not being required to stabilize the C-terminal helix in its extended position. Further support for this hypothesis is provided by the human CRM1-Snurportin structure (9) that models the extended conformation of the C-terminal helix but does not show ordered electron density for the putative tip interaction.
FIGURE 8.

SAXS data for wild type CRM1 and C-terminal helix and basic patch variants. Shown are overlaid SAXS curves (A) and Kratky plots (B) of the three variants (CRM1-1–1062, CRM1 H12, and CRM1-Swap) and wild type CRM1. None of the mutations produced a significant change in the SAXS pattern, indicating that they did not generate a major conformational change in CRM1.
DISCUSSION
The mutagenesis and SAXS results presented here indicate that a major contribution to the modulation of the affinity of CRM1 for NESs by its C-terminal helix is mediated by a series of acidic residues located at the extreme C terminus of this helix. Moreover, these results are consistent with the influence of these residues being exerted through electrostatic interactions with a patch of basic residues located on the inner surface of the CRM1 toroid that has contributions from the B-helix of HEAT repeat 12. Mutating either the acidic C-terminal helix tip residues or the basic residues on HEAT 12B increased the affinity of CRM1 for the PKI NES in the absence of RanGTP, whereas reversing the charges of these two regions to reconstitute the putative electrostatic interaction between them restored the wild type binding behavior of CRM1. Importantly, RanGTP enhanced binding of the reversed charge variant to the NES just as effectively as wild type CRM1.
SAXS indicated that these mutations did not induce a major change in the overall CRM1 backbone conformation. Furthermore, simulations indicated that in solution the C-terminal helix was likely to be positioned across the CRM1 toroid so that its C terminus would be located close to the basic patch at HEAT 12B. These results support the hypothesis that mutations in either the C-terminal helix tip or the HEAT 12B basic patch altered the affinity of CRM1 for NESs by small local conformational changes near the NES binding site rather than through larger scale changes in the CRM1 molecule that were initially proposed to explain the influence of the C-terminal helix (13). The putative electrostatic interaction between the acidic residues of the C-terminal helix tip and the basic residues on HEAT 12B occurs on the inner surface of the CRM1 toroid, whereas the NES binding groove is located between the A-helices of HEAT repeats 11 and 12 on the outer surface. Because these two sites are not in direct contact with one another, an allosteric mechanism must be employed to communicate between them. Although further work will be required to establish the precise mechanism by which the effect of this interaction is propagated to the NES binding site, it is likely to occur by a local movement of the A-helices of HEAT repeats 11 and 12 analogous to that proposed (11) for the autoinhibitory effect of the HEAT 9 loop and also to the way in which RanGTP binding alters the affinity of importin-β for nucleoporins (21).
Our results indicate that the HEAT 9 loop and the C-terminal helix of CRM1 function together to prevent NES binding in the absence of RanGTP. Examination of the B helices of HEAT repeats 11 and 12 shows that the arrangement of the inner surface groove could accommodate simultaneous binding of both the HEAT 9 loop and the C-terminal helix tip. The HEAT 9 loop binds at the distal end of the groove (Fig. 9), and its interaction terminates at Lys-590. Residues Val-441, Leu-442, and Val-443 in the HEAT 9 loop pack closely against the B helices of HEAT repeats 11 and 12 that Koyama and Matsuura (11) propose generates a local conformational change that closes the external NES binding pocket on the A helices of these HEAT repeats. The conserved basic patch residues (Lys-594, Arg-596, and Arg-597) sit at the very proximal end of the B helix of HEAT 12 and are a full turn of the α-helix away from Lys-590, where the HEAT 9 loop interaction terminates. Thus, access of the C-terminal helix tip to the basic patch residues should not be hindered sterically by the presence of the HEAT 9 loop, provided it is arranged in the same conformation in unbound CRM1 as is seen in the Xpo1p-RanGTP-RanBP1 structure (11). In the presence of RanBP1, the HEAT 9 loop has been proposed (11) to drive the NES binding cleft toward its low affinity state, a process that is proposed to be promoted by a direct clash between the C terminus of RanGTP and the HEAT 9 loop (11). However, our results indicate that in unbound CRM1, where RanBP1 is not present, the additional energy provided by the putative C-terminal helix interaction at the HEAT 12B basic patch stabilizes the interaction of the HEAT 9 loop at HEAT repeats 11 and 12 and that both processes contribute to decreasing the affinity of CRM1 for NESs in the absence of RanGTP. Thus, whereas individually mutating the HEAT 9 loop or the C-terminal helix resulted in affinities of 0.2 ± 0.03 and 1.7 ± 0.1 μm, respectively, the combined mutant affinity increased to 25 ± 2 nm (Table 1), which was comparable with that observed in the presence of RanGTP. Moreover, the calculated ΔΔG for the combined mutant was indistinguishable from the sum of the individual ΔΔG values.
FIGURE 9.

The HEAT 11/12 B helices can accommodate binding of both the C-terminal helix tip and the HEAT 9 loop. Alignment of S. cerevisiae Xpo1p (11) and human CRM1 (9) indicates that the two proposed interactions at the HEAT 11/12 B helices (in cyan) cleft could occur simultaneously. The HEAT 9 loop is shown in the conformation that promotes closure of the NES cleft at the HEAT 11/12 A helices.
Regarding the implications for nuclear protein export, our data are consistent with a model in which both the HEAT 9 loop and C-terminal helix are responsible for modulating the affinity of CRM1 for NESs. Like other nuclear transport pathways (4, 6, 22), nuclear protein export relies on forming an export complex in the nucleus that is then equilibrated through nuclear pores with the cytoplasm where the complex is disassembled, thereby preventing the cargo returning to the nucleus. Transport factors such as CRM1 interact with nucleoporins to enable the cargo-carrier complex to move back-and-forth through nuclear pores and so equilibrate with the cytoplasm (6). Transport is driven by disassembling the cargo-carrier complex in the cytoplasm (through the action of RanGAP and RanBP1), which prevents the return of the cargo to the nucleus. Although the affinity of CRM1 for RanGTP in the absence of a cargo is only of the order of micromolar (23), the nuclear RanGTP concentration is generally higher than this (24), and so virtually all nuclear CRM1 should be complexed with RanGTP. When CRM1 is bound to RanGTP, both the HEAT 9 loop and the C-terminal helix are moved away from the NES binding site (10, 12), thereby enhancing the affinity of CRM1 for cargoes. The HEAT 9 loop moves to become bound to Ran, whereas the C-terminal helix moves to become located near the CRM1 N terminus at a site distant from HEAT repeats 11 and 12 (10, 12). When Ran is removed from the export complex in the cytoplasm as a result of the action of RanGAP and RanBP1, these two structural components are free to return to the positions in which they reduce the affinity for NESs and thereby facilitate cargo release. The C-terminal helix and the HEAT 9 loop could contribute to a ratchet-like mechanism that inhibits NESs rebinding to CRM1 after release in the cytoplasm and thus prevent the return of these cargoes to the nucleus. Such a function would be analogous to the way in which Nup2 and Nup50 have been proposed to function to prevent NLS-containing import cargoes rebinding to importin-α after they have been released in the nucleus (25, 26).
Fig. 10 represents a schematic illustration of how the HEAT 9 loop and the C-terminal helix are proposed to contribute to the nuclear protein export cycle. This model complements and extends the mechanism proposed by Koyama and Matsuura (11) in which it was envisaged that changes at the internal B helices of the NES binding site are propagated to the external binding site, thereby enabling the HEAT 9 loop to link the presence of RanGTP in the interior of the toroid with a change in the NES binding site on the outer surface. Our results, however, indicate that the HEAT 9 loop is not the only element that contributes to reducing the affinity of CRM1 for NESs in the absence of bound RanGTP. Alterations to the C-terminal helix of CRM1, whether they are large scale deletions or specific mutations that disrupt the putative electrostatic interaction at the tip of the C-terminal helix, also result in an increased affinity for NESs in the absence of RanGTP.
FIGURE 10.

Model for the function of the C-terminal helix and the HEAT 9 loop in modulating CRM1 cargo binding and release during the nuclear protein export cycle. The unbound state of CRM1 has a low affinity for the NES because of the combined autoinhibitory effect of the HEAT 9 loop and the C-terminal helix (A). In the nucleus, RanGTP binding sequesters the HEAT 9 loop and displaces the C-terminal helix (B), generating a high affinity state of CRM1 in which the open state of the NES binding site is favored. This, in turn, leads to NES binding and the formation of a ternary export complex (C). The ternary complex diffuses through nuclear pore complexes to the cytoplasm, where it is disassembled. RanGTP is removed by the action of RanGAP, RanBP1, and nucleoporins. In the absence of RanGTP (D), the HEAT 9 loop and the C-terminal helix can return to their autoinhibitory positions at the HEAT 11/12 B helices, facilitating NES release and the reversion of CRM1 to the low affinity state that then diffuses back to the nucleus where it can participate in a further export cycle. The proposed C-terminal tip electrostatic interaction is shown as a brown ellipse. HEAT repeats 11 and 12 of the NES binding site are modeled in red to indicate a closed state or green to indicate an open state. Dashed lines indicate regions in the Dong et al. (9) crystal structure, for which there was insufficient electron density to model the chain.
As illustrated in Fig. 10A, in isolated CRM1, both the HEAT 9 loop and the C-terminal helix bind on the internal surface of the toroid near the NES binding site and together reduce the affinity for NESs. The C-terminal helix lies across the CRM1 toroid in a conformation similar to that seen in the human CRM1-Snurportin complex (9), which is consistent with our SAXS data. This arrangement of the C-terminal helix allows the acidic residues at its distal tip to interact with the basic patch at HEAT 12B. The HEAT 9 loop sits in a conformation similar to that seen in the Xpo1p-RanGTP-RanBP1 complex (11) and stabilizes the closed state of the NES binding site by the same mechanism that is proposed for active dissociation of a bound NES (11).
The internal positioning of the C-terminal helix across the center of the CRM1 toroid, similar to the internal location of the HEAT 9 loop, renders it sensitive to the presence of RanGTP. When RanGTP binds to CRM1 (Fig. 10B), it interacts directly with the HEAT 9 loop, removing it from the HEAT 11/12 A-helices. Although the precise mechanism by which RanGTP displaces the C-terminal helix has not been established directly, it may involve a direct clash between these two chains as they compete for space within the interior of the CRM1 toroid. The repositioning of the C-terminal helix and HEAT loop 9 facilitates an opening of the NES binding site that allows the NES to bind with higher affinity (Fig. 10C). When the export cargo-carrier complex reaches the cytoplasm, Ran is removed through the action of RanGAP and RanBP1 (Fig. 10D). When RanGTP is no longer bound to CRM1, the C-terminal helix and HEAT loop 9 are able to return to their original autoinhibitory positions and, through their interactions with the B-helices of HEAT repeats 11 and 12, favor the propagation of a conformational change that tends to close the NES binding site, facilitating the release of the cargo and inhibiting its rebinding and return to the nucleus.
Although additional work will be required to define the precise mechanism by which each residue of the C-terminal helix contributes to modulating the affinity of CRM1 for NES, overall this model integrates the knowledge gained from the CRM1/Xpo1p crystal structures, the study of the HEAT 9 loop conducted by Koyama and Matsuura (11), and the study of the C-terminal helix presented here to provide a structural context for the modulation of binding and release of NES-containing cargoes by CRM1 that is crucial for the nuclear protein export cycle.
Supplementary Material
Acknowledgments
We are most grateful to our colleagues in Cambridge, especially Meindert Lamers, Kiyoshi Nagai, Neil Marshall, and Roger Williams, for many helpful comments and criticisms. We also thank Dr. Adam Round and Dr. Petra Pernot at European Synchrotron Radiation Facility Beamline ID14-3 in Grenoble for assistance with SAXS data collection, Dr. Yoshiyuki Matsuura for the YFP-PKI-NES construct, and Dr. Dirk Görlich (MPI Göttingen) for the generous gift of the mouse CRM1 expression construct.
This work was supported in part by a Wellcome Trust programme grant (to M. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- NES
- nuclear export signal
- SAXS
- small angle x-ray scattering
- Ni-NTA
- nickel-nitrilotriacetic acid
- TEV
- tobacco etch mosaic virus.
REFERENCES
- 1. Fornerod M., Ohno M., Yoshida M., Mattaj I. W. (1997) Cell 90, 1051–1060 [DOI] [PubMed] [Google Scholar]
- 2. Kutay U., Izaurralde E., Bischoff F. R., Mattaj I. W., Görlich D. (1997) EMBO J. 16, 1153–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stade K., Ford C. S., Guthrie C., Weis K. (1997) Cell 90, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 4. Weis K. (2003) Cell 112, 441–451 [DOI] [PubMed] [Google Scholar]
- 5. Kutay U., Güttinger S. (2005) Trends Cell Biol. 15, 121–124 [DOI] [PubMed] [Google Scholar]
- 6. Tran E. J., Wente S. R. (2006) Cell 125, 1041–1053 [DOI] [PubMed] [Google Scholar]
- 7. Strambio-De-Castillia C., Niepel M., Rout M. P. (2010) Nat. Rev. Mol. Cell Biol. 11, 490–501 [DOI] [PubMed] [Google Scholar]
- 8. Cook A. G., Conti E. (2010) Curr. Opin. Struct. Biol. 20, 247–252 [DOI] [PubMed] [Google Scholar]
- 9. Dong X., Biswas A., Süel K. E., Jackson L. K., Martinez R., Gu H., Chook Y. M. (2009) Nature 458, 1136–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Monecke T., Güttler T., Neumann P., Dickmanns A., Görlich D., Ficner R. (2009) Science 324, 1087–1091 [DOI] [PubMed] [Google Scholar]
- 11. Koyama M., Matsuura Y. (2010) EMBO J. 29, 2002–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Güttler T., Madl T., Neumann P., Deichsel D., Corsini L., Monecke T., Ficner R., Sattler M., Görlich D. (2010) Nat. Struct. Mol. Biol. 17, 1367–1376 [DOI] [PubMed] [Google Scholar]
- 13. Dong X., Biswas A., Chook Y. M. (2009) Nat. Struct. Mol. Biol. 16, 558–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsuura Y., Stewart M. (2004) Nature 432, 872–877 [DOI] [PubMed] [Google Scholar]
- 15. Studier F. W. (2005) Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 16. Kapust R. B., Tözsér J., Fox J. D., Anderson D. E., Cherry S., Copeland T. D., Waugh D. S. (2001) Protein Eng. 14, 993–1000 [DOI] [PubMed] [Google Scholar]
- 17. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 18. Svergun D. I., Barberato C., Koch M. H. (1995) J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 19. Sokolova A. V., Volkov V. V., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 865–868 [Google Scholar]
- 20. Fukuhara N., Fernandez E., Ebert J., Conti E., Svergun D. I. (2004) J. Biol. Chem. 279, 2176–2181 [DOI] [PubMed] [Google Scholar]
- 21. Bayliss R., Littlewood T., Stewart M. (2000) Cell 102, 99–108 [DOI] [PubMed] [Google Scholar]
- 22. Stewart M. (2007) Nat. Rev. Mol. Cell Biol. 8, 195–208 [DOI] [PubMed] [Google Scholar]
- 23. Bischoff F. R., Görlich D. (1997) FEBS Lett. 419, 249–254 [DOI] [PubMed] [Google Scholar]
- 24. Bischoff F. R., Ponstingl H. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 10830–10834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matsuura Y., Lange A., Harreman M. T., Corbett A. H., Stewart M. (2003) EMBO J. 22, 5358–5369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matsuura Y., Stewart M. (2005) EMBO J. 24, 3681–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paraskeva E, Izaurralde E., Bischoff F. R., Huber J., Kutay U., Hartmann E., Lührmann R., Görlich D. (1999) J. Cell Biol. 145, 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Volkov V. V., Svergun D. I. (2003) J. Appl. Crystallogr. 36, 860–864 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.